Exploring the Metabolism of (+)-[18F]Flubatine In Vitro and In Vivo: LC-MS/MS Aided Identification of Radiometabolites in a Clinical PET Study †

 ,
,
Abstract
:
1. Introduction
2. Results
2.1. Radiosynthesis of (+)-[18F]1
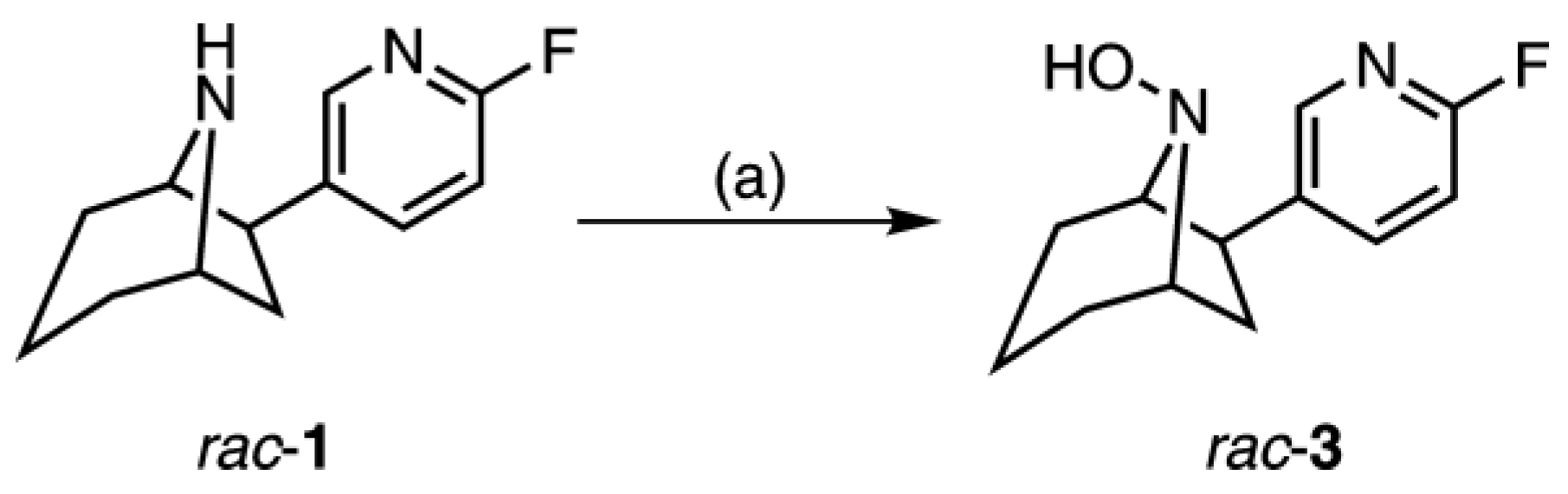
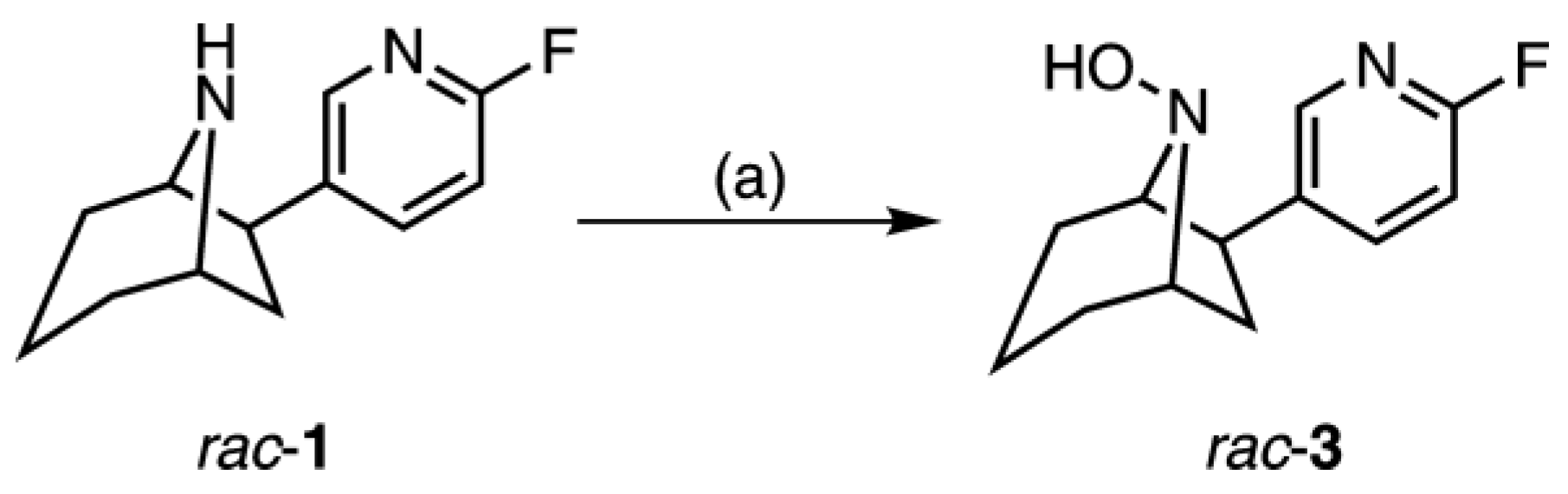
2.2. Synthesis of rac-8-Hydroxy-flubatine (rac-3)
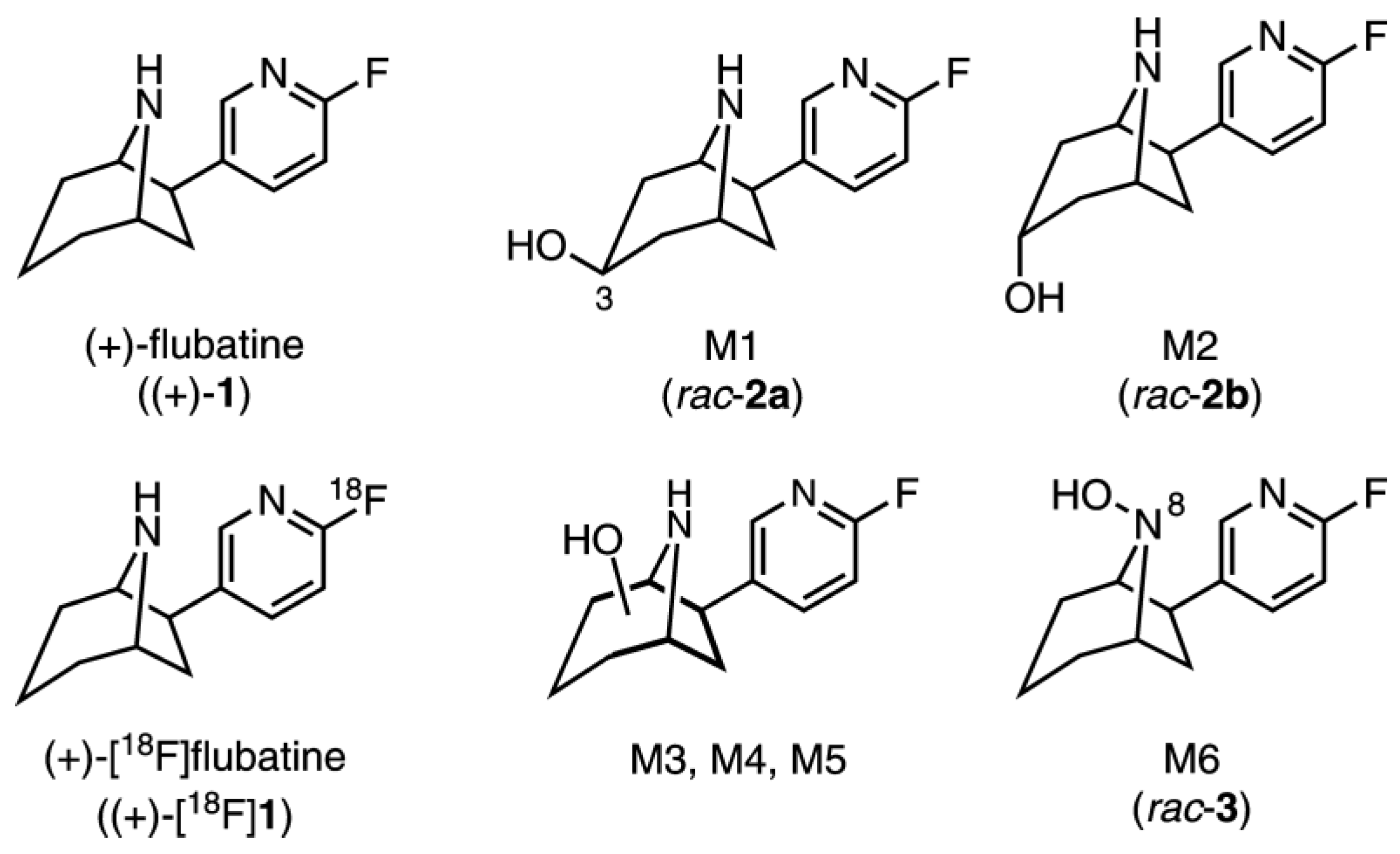
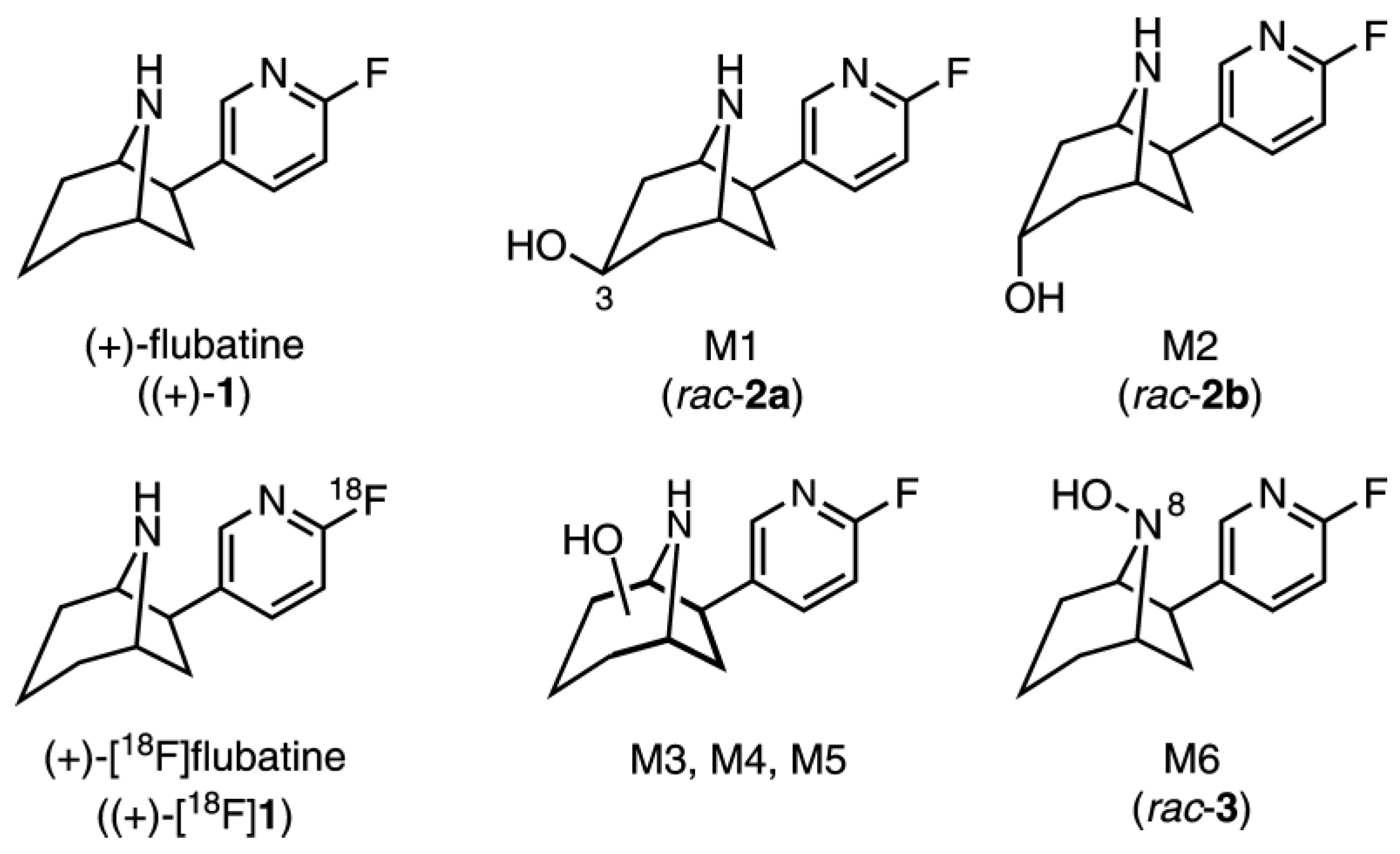
2.3. Investigation of (+)-1 in Pig–Identification of Phase I and Phase II Metabolites by LC-MS/MS
2.4. In Vitro Glucuronidation of (+)-[18F]1, (+)-1, (−)-1, and rac-3 by HLM
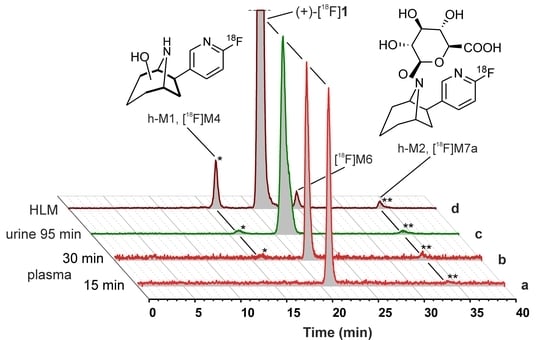
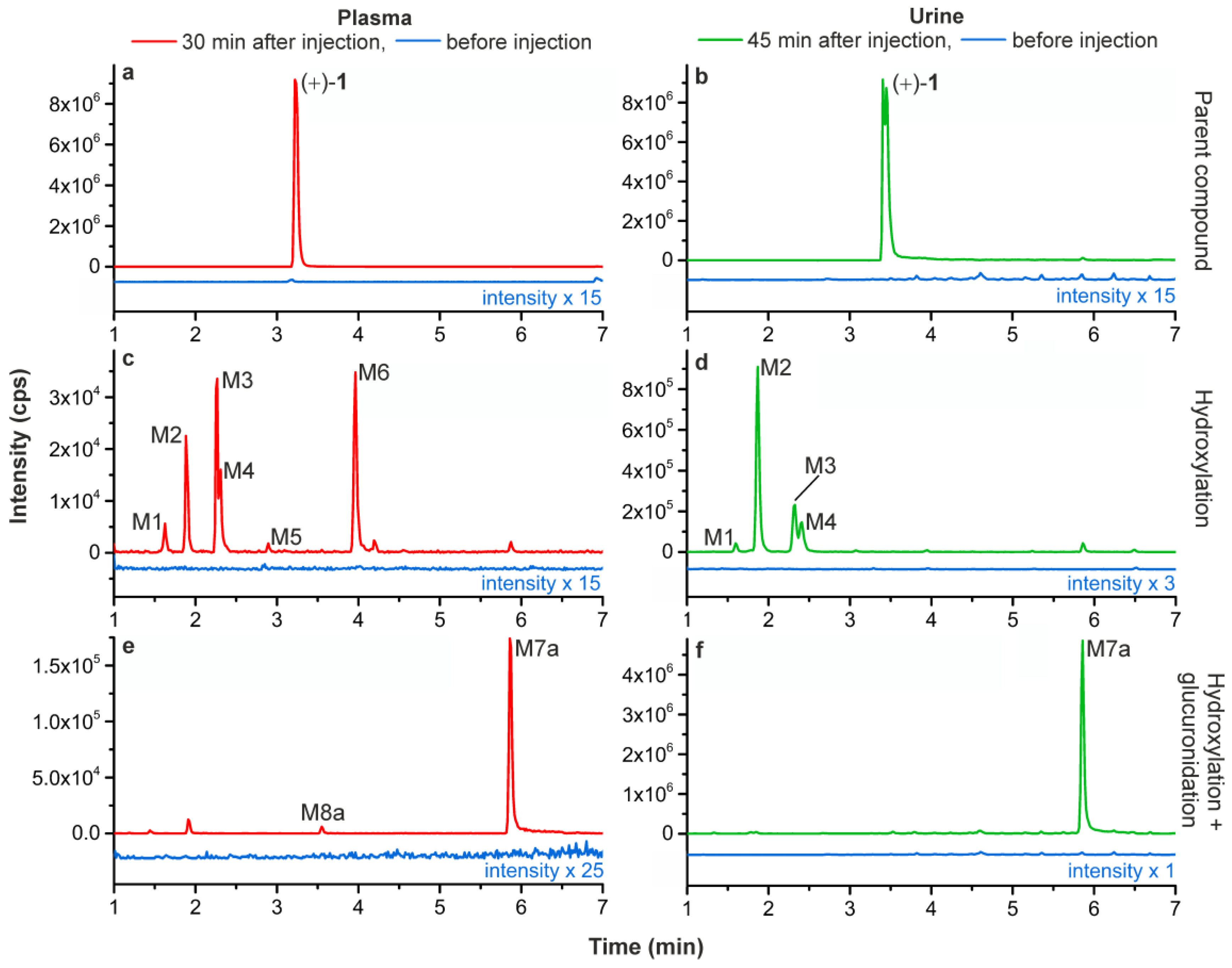
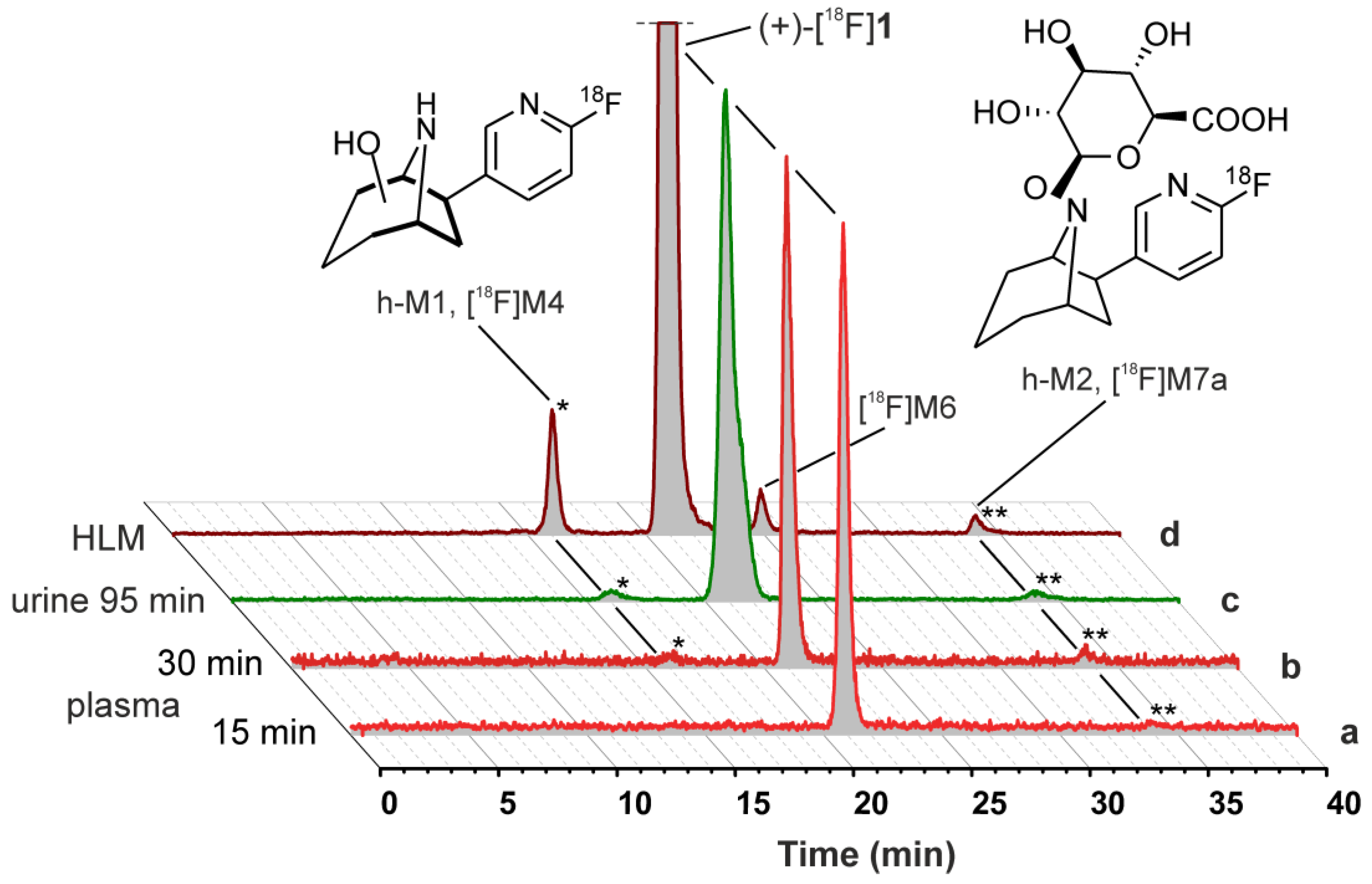
2.5. Metabolism Studies of (+)-[18F]1 in Humans
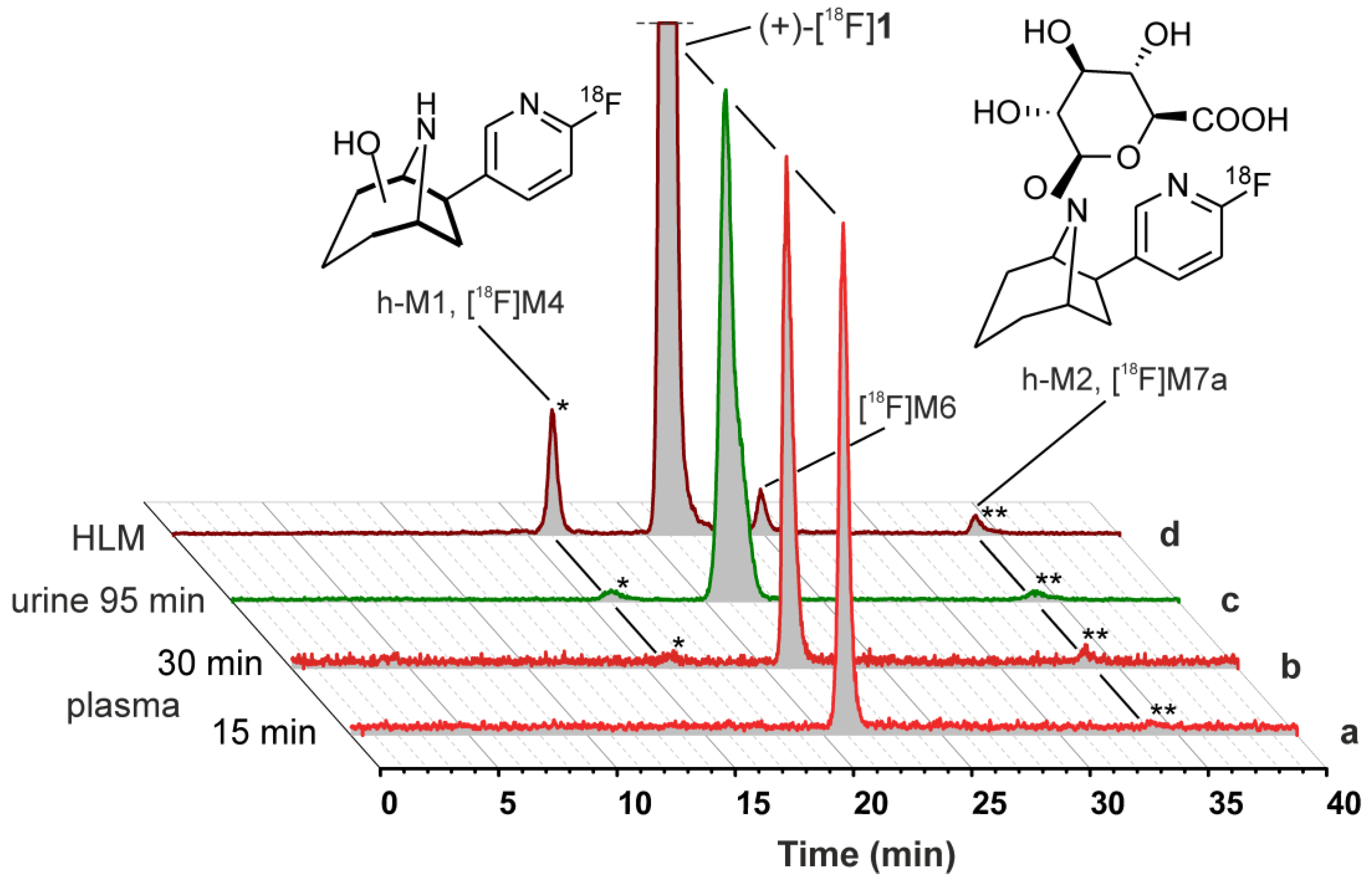
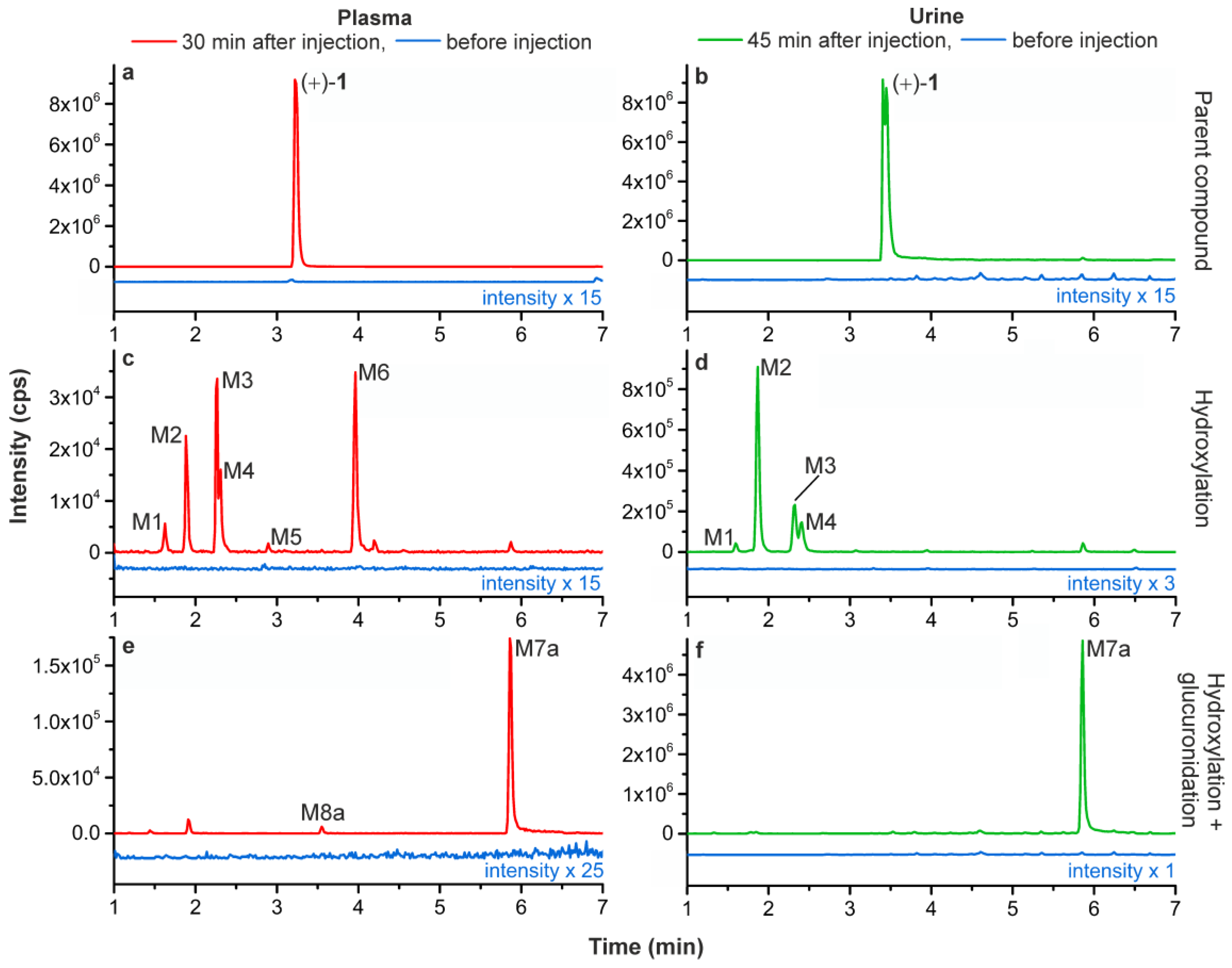
2.5.1. Metabolic Stability of (+)-[18F]1 and Detection of Radiometabolites
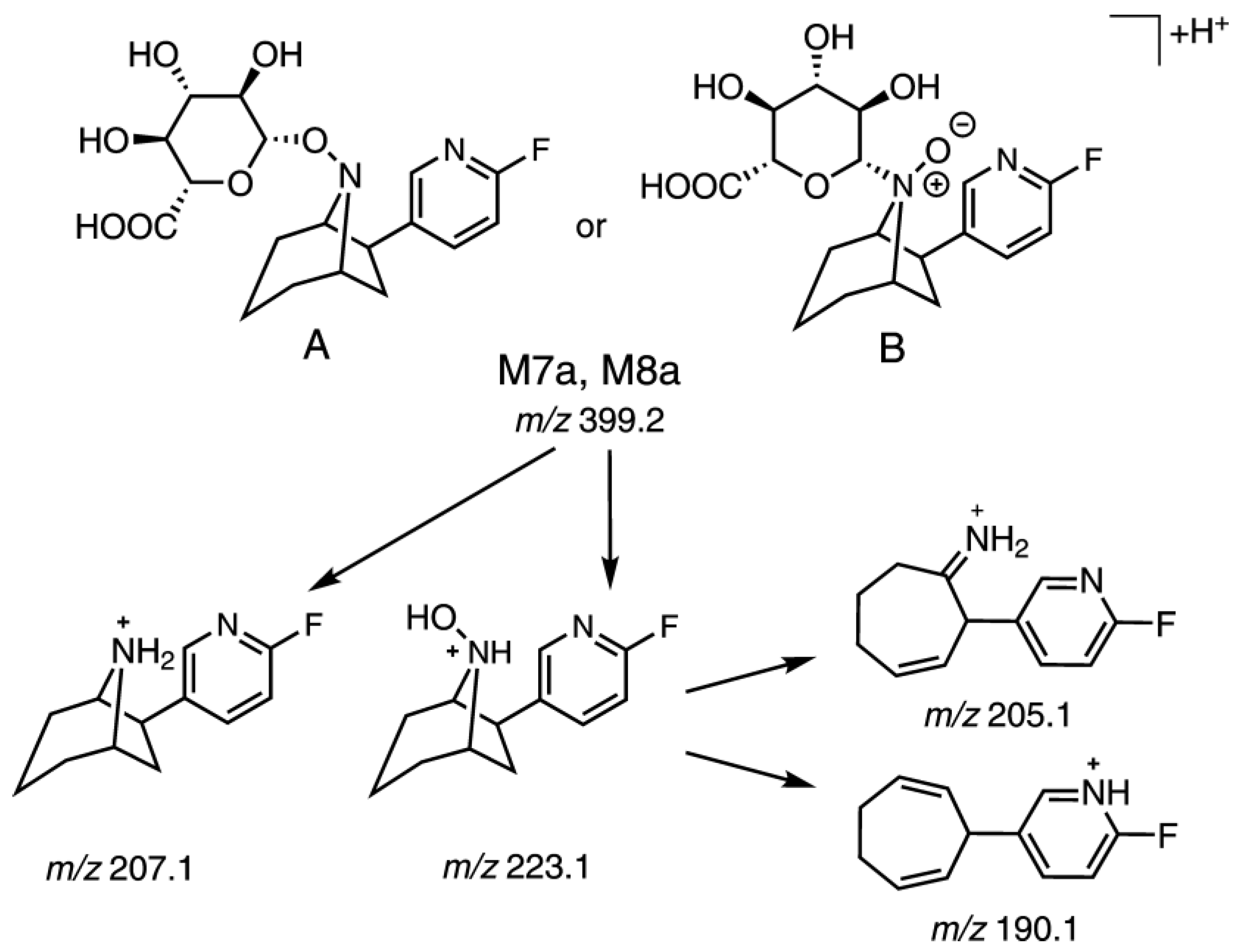
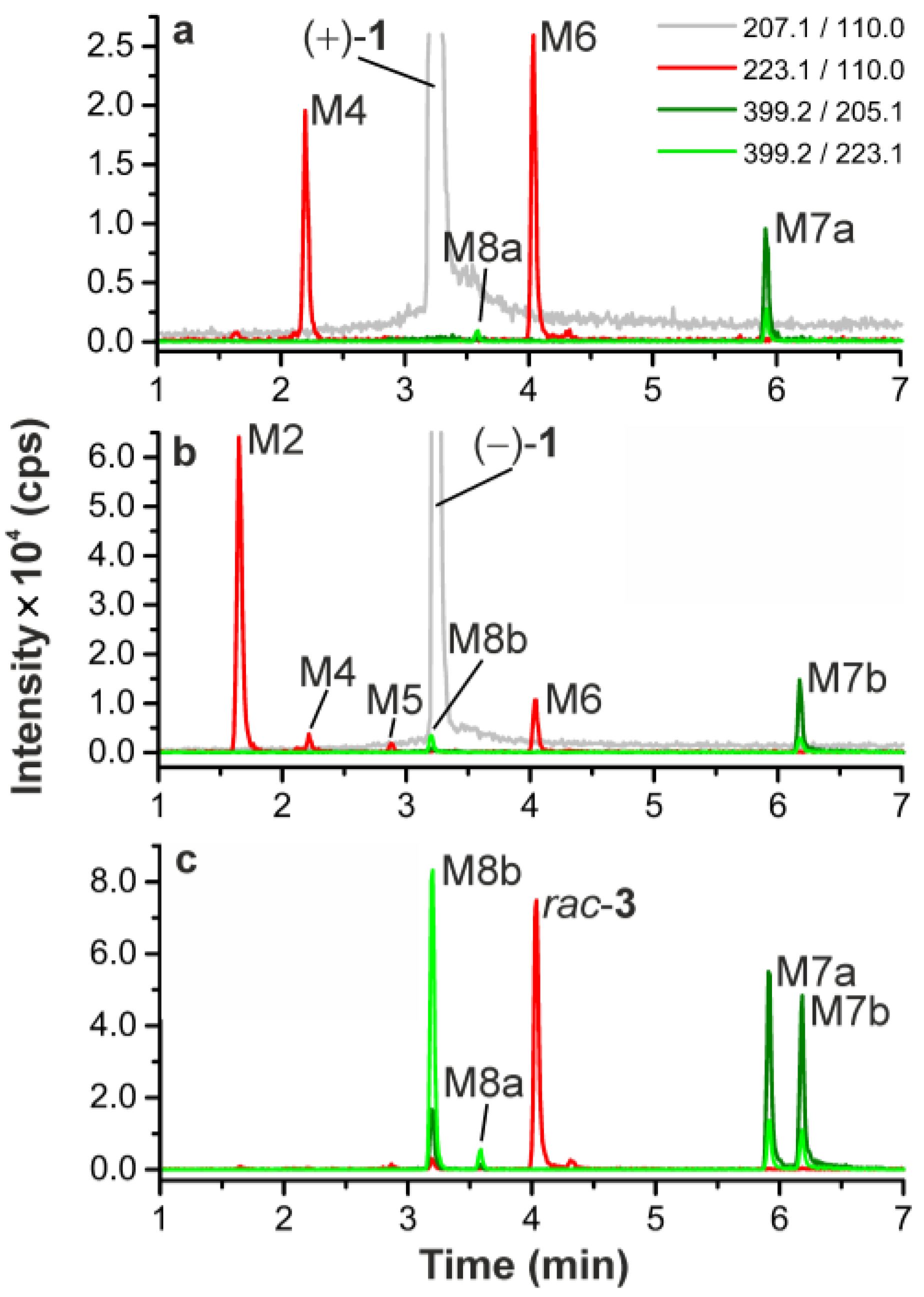
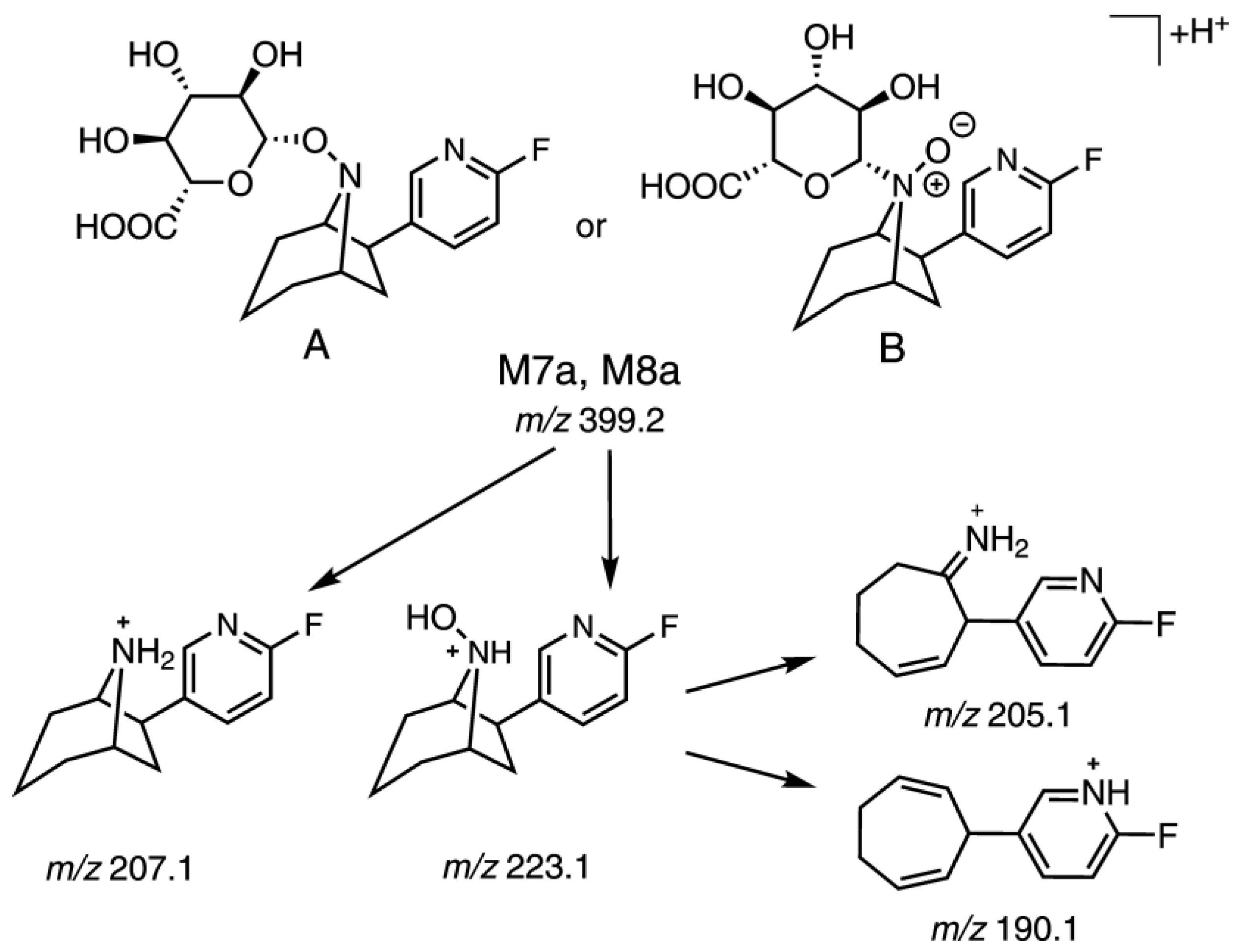
2.5.2. Identification of Radiometabolites of (+)-[18F]1 formed in Humans
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Radiosynthesis of (+)-[18F]1
4.3. Synthesis of Reference Compound rac-3
4.3.1. (+/−)-exo-6-(6-Fluoro-pyridin-3-yl)-8-aza-bicyclo[3.2.1]octan-8-ol ((rac)-8-hydroxy-flubatine, rac-3)
4.4. In Vivo Metabolism Study of (+)-1 in Pigs
4.5. Microsomal Incubations
4.6. Investigation of (+)-[18F]1 in Humans
4.7. LC-MS/MS Analyses
4.8. Radio-HPLC
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Badio, B.; Garraffo, H.M.; Spande, T.F.; Daly, J.W. Epibatidine: Discovery and definition as a potent analgesic and nicotinic agonist. Med. Chem. Res. 1994, 4, 440–448. [Google Scholar]
- Smits, R.; Fischer, S.; Hiller, A.; Deuther-Conrad, W.; Wenzel, B.; Patt, M.; Cumming, P.; Steinbach, J.; Sabri, O.; Brust, P.; et al. Synthesis and biological evaluation of both enantiomers of [18F]flubatine, promising radiotracers with fast kinetics for the imaging of α4β2-nicotinic acetylcholine receptors. Bioorg. Med. Chem. 2014, 22, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Sabri, O.; Becker, G.-A.; Meyer, P.M.; Hesse, S.; Wilke, S.; Graef, S.; Patt, M.; Luthardt, J.; Wagenknecht, G.; Hoepping, A.; et al. First-in-human PET quantification study of cerebral α4β2* nicotinic acetylcholine receptors using the novel specific radioligand (−)-[18F]Flubatine. NeuroImage 2015, 118, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, F.-A.; Smits, R.; Fischer, S.; Donat, C.K.; Hoepping, A.; Brust, P.; Steinbach, J. LC-MS Supported Studies on the in Vitro Metabolism of both Enantiomers of Flubatine and the in vivo Metabolism of (+)-[18F]Flubatine—A Positron Emission Tomography Radioligand for Imaging α4β2 Nicotinic Acetylcholine Receptors. Molecules 2016, 21, 1200. [Google Scholar] [CrossRef] [PubMed]
- Brust, P.; Patt, J.T.; Deuther-Conrad, W.; Becker, G.; Patt, M.; Schildan, A.; Sorger, D.; Kendziorra, K.; Meyer, P.; Steinbach, J.; et al. In vivo measurement of nicotinic acetylcholine receptors with [18F]norchloro-fluoro-homoepibatidine. Synapse 2008, 62, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Hockley, B.G.; Stewart, M.N.; Sherman, P.; Quesada, C.; Kilbourn, M.R.; Albin, R.L.; Scott, P.J.H. (-)-[18F]Flubatine: Evaluation in rhesus monkeys and a report of the first fully automated radiosynthesis validated for clinical use. J. Labelled Comp. Radiopharm. 2013, 56, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Gallezot, J.-D.; Esterlis, I.; Bois, F.; Zheng, M.-Q.; Lin, S.-F.; Kloczynski, T.; Krystal, J.H.; Huang, Y.; Sabri, O.; Carson, R.E.; et al. Evaluation of the sensitivity of the novel α4β2* nicotinic acetylcholine receptor PET radioligand 18F-(−)-NCFHEB to increases in synaptic acetylcholine levels in rhesus monkeys. Synapse 2014, 68, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Bois, F.; Gallezot, J.-D.; Zheng, M.-Q.; Lin, S.-F.; Esterlis, I.; Cosgrove, K.P.; Carson, R.E.; Huang, Y. Evaluation of [18F]-(–)-norchlorofluorohomoepibatidine ([18F]-(–)-NCFHEB) as a PET radioligand to image the nicotinic acetylcholine receptors in non-human primates. Nucl. Med. Biol. 2015, 42, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Patt, M.; Becker, G.A.; Grossmann, U.; Habermann, B.; Schildan, A.; Wilke, S.; Deuther-Conrad, W.; Graef, S.; Fischer, S.; Smits, R.; et al. Evaluation of metabolism, plasma protein binding and other biological parameters after administration of (–)-[18F]Flubatine in humans. Nucl. Med. Biol. 2014, 41, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Hillmer, A.T.; Esterlis, I.; Gallezot, J.D.; Bois, F.; Zheng, M.Q.; Nabulsi, N.; Lin, S.F.; Papke, R.L.; Huang, Y.; Sabri, O.; et al. Imaging of cerebral α4β2* nicotinic acetylcholine receptors with (–)-[18F]Flubatine PET: Implementation of bolus plus constant infusion and sensitivity to acetylcholine in human brain. NeuroImage 2016, 141, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Hillmer, A.T.; Nabulsi, N.; Matuskey, D.; Lim, K.; Lin, S.-F.; Esterlis, I.; Carson, R.E.; Huang, Y.; Cosgrove, K.P. Evaluation of (–)-[18F]Flubatine-specific binding: Implications for reference region approaches. Synapse 2017, 72. [Google Scholar] [CrossRef]
- Ma, Y.; Kiesewetter, D.O.; Lang, L.; Gu, D.; Chen, X. Applications of LC-MS in PET Radioligand Development and Metabolic Elucidation. Curr. Drug Metab. 2010, 11, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Kerns, E.H. LC/MS applications in drug development. Mass. Spectrom. Rev. 1999, 18, 187–279. [Google Scholar] [CrossRef]
- Lin, J.H.; Lu, A. Y. H. Role of Pharmacokinetics and Metabolism in Drug Discovery and Development. Pharmacol. Rev. 1997, 49, 403. [Google Scholar] [PubMed]
- Martignoni, M.; Groothuis, G.M.M.; de Kanter, R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin. Drug Metab. Toxicol. 2006, 2, 875–894. [Google Scholar] [CrossRef] [PubMed]
- Testa, B.; Krämer, S.D. The Biochemistry of Drug Metabolism—An Introduction: Part 1. Principles and Overview. Chem. Biodivers. 2006, 3, 1053–1101. [Google Scholar] [CrossRef] [PubMed]
- Trufelli, H.; Palma, P.; Famiglini, G.; Cappiello, A. An overview of matrix effects in liquid chromatography-mass spectrometry. Mass. Spectrom. Rev. 2011, 30, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Liu, X. The Conduct of Drug Metabolism Studies Considered Good Practice (II): In vitro Experiments. Curr. Drug Metab. 2007, 8, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Asha, S.; Vidyavathi, M. Role of Human Liver Microsomes in In Vitro Metabolism of Drugs-A Review. Appl. Biochem. Biotechnol. 2010, 160, 1699–1722. [Google Scholar] [CrossRef] [PubMed]
- Lever, S.Z.; Fan, K.-H.; Lever, J.R. Tactics for preclinical validation of receptor-binding radiotracers. Nucl. Med. Biol. 2016, 44, 4–30. [Google Scholar] [CrossRef] [PubMed]
- German Clinical Trials Register/Deutsches Register Klinischer Studien (DRKS): DRKS00005819. Available online: https://www.drks.de/drks_web/navigate.do?navigationId=trial.HTML&TRIAL_ID=DRKS00005819 (accessed on 14 February 2018).
- Fischer, S.; Hiller, A.; Smits, R.; Hoepping, A.; Funke, U.; Wenzel, B.; Cumming, P.; Sabri, O.; Steinbach, J.; Brust, P. Radiosynthesis of racemic and enantiomerically pure (−)-[18F]flubatine—A promising PET radiotracer for neuroimaging of α4β2 nicotinic acetylcholine receptors. Appl. Radiat. Isot. 2013, 74, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Patt, M.; Schildan, A.; Habermann, B.; Fischer, S.; Hiller, A.; Deuther-Conrad, W.; Wilke, S.; Smits, R.; Hoepping, A.; Wagenknecht, G.; et al. Fully automated radiosynthesis of both enantiomers of [18F]Flubatine under GMP conditions for human application. Appl. Radiat. Isot. 2013, 80, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.N.; Hockley, B.G.; Scott, P.J.H. Synthesis of (−)-[18F]Flubatine ([18F]FLBT). In Radiochemical Syntheses: Further Radiopharmaceuticals for Positron Emission Tomography and New Strategies for Their Production; Scott, P.J.H., Ed.; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2015; pp. 1–11. [Google Scholar]
- Miller, R.R.; Doss, G.A.; Stearns, R. Identification of a Hydroxylamine Glucuronide Metabolite of an Oral Hypoglycemic Agent. Drug Metab. Disp. 2004, 32, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Trontelj, T. Quantification of Glucuronide Metabolites in Biological Matrices by LC-MS/MS. In Tandem Mass Spectrometry-Applications and Principles; Prasain, J., Ed.; INTECHOPEN: London, UK; Available online: https://www.intechopen.com/books/tandem-mass-spectrometry-applications-and-principles/quantification-of-glucuronide-metabolites-in-biological-matrices-by-lc-ms-ms (accessed on 14 February 2018). [CrossRef]
- Kaivosaari, S.; Finel, M.; Koskinen, M. N-glucuronidation of drugs and other xenobiotics by human and animal UDP-glucuronosyltransferases. Xenobiotica 2011, 41, 652–669. [Google Scholar] [CrossRef] [PubMed]
- Bolleddula, J.; DeMent, K.; Driscoll, J.P.; Worboys, P.; Brassil, P.J.; Bourdet, D.L. Biotransformation and bioactivation reactions of alicyclic amines in drug molecules. Drug Metab. Rev. 2014, 46, 379–419. [Google Scholar] [CrossRef] [PubMed]
- Holčapek, M.; Kolářová, L.; Nobilis, M. High-performance liquid chromatography–tandem mass spectrometry in the identification and determination of phase I and phase II drug metabolites. Anal. Bioanal. Chem. 2008, 391, 59–78. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | tR (min) a | MRM Transition | EPI Fragmentation b (% Intensity in Brackets) | Identification | |
|---|---|---|---|---|---|
| Plasma | Urine | ||||
| M1 | 1.62 | 1.60 | 223.1/110.0 | 81.9 (100), 205.0 (71), 223.0 (32), 163.1 (26), 162.1 (23), 110.0 (17), 136.0 (15), 131.0 (15), 103.9 (14), 188.1 (9) | Hydroxylation |
| M2 | 1.89 | 1.87 | 223.1/110.0 | 223.1 (100), 136.0 (40), 110.0 (39), 162.1 (37), 180.1 (36), 188.0 (28), 179.1 (25), 114.0 (13), 124.0 (13), 206.0 (11), 160.0 (9), 138.0 (8), 142.0 (4), 205.1 (4) | |
| M3 | 2.26 | 2.32 | 223.1/110.0 | 223.1 (100), 150.0 (63), 164.1 (44), 110.0 (42), 188.1 (41), 82.0 (39), 135.1 (31), 206.1 (18), 176.1 (16), 120.1 (14), 162.1 (12), 124.0 (12), 205.1 (12), 134.0 (12), 136.1 (12), 163.1 (10) | |
| M4 | 2.31 | 2.40 | 223.1/110.0 | 81.9 (100), 205.1 (60), 188.0 (39), 177.1 (31), 223.1 (23), 110.0 (22), 176.1 (9), 160.0 (7) | |
| M5 | 2.89 | - c | 223.1/110.0 | no EPI spectrum due to low intensity | |
| M6 | 3.96 | - c | 223.1/110.0 | no EPI spectrum due to low intensity | |
| M7a | 5.87 | 5.86 | 399.2/223.1 | 205.2 (100), 223.2 (44), 136.1 (34), 203.2 (26), 110.0 (13), 162.1 (11), 177.2 (9), 83.0 (9), 207.2 (8), 189.1 (6), 109.1 (6), 190.2 (6) | Hydroxylation + glucuronidation |
| Metabolite a | tR (min) b | Substrate | MRM Transitions | EPI Fragmentation c (% Intensity in Brackets) | Identification |
|---|---|---|---|---|---|
| M7b | 6.18 | (−)-1 or rac-3 | 399.2/223.1 399.2/205.1 | 205.1241 (100), 223.1 (60), 136.0 (37), 203.1 (20), 110.0 (18), 177.1 (16), 83.0 (11), 207.1 (11), 162.0 (11), 82.0 (10), 116.0 (8), 163.1 (8), 190.1 (7), 124.0 (7) | Hydroxylation + Glucuronidation d |
| M8a | 3.60 | (+)-1 or rac-3 | 399.2/223.1 399.2/205.1 | 223.1 (100), 205.1 (21), 124.1 (6), 110.2 (6), 136.0 (6), 84.9 (4), 113.0 (4) | |
| M8b | 3.22 | (−)-1 or rac-3 | 399.2/223.1 399.2/205.1 | 223.1 (100), 205.1 (27), 136.1 (16), 84.9 (9), 110.0 (8), 150.0 (6), 203.2 (6), 82.9 (6), 67.9 (4), 190.1 (4), 113.0 (3) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ludwig, F.-A.; Fischer, S.; Smits, R.; Deuther-Conrad, W.; Hoepping, A.; Tiepolt, S.; Patt, M.; Sabri, O.; Brust, P.
Exploring the Metabolism of (+)-[18F]Flubatine In Vitro and In Vivo: LC-MS/MS Aided Identification of Radiometabolites in a Clinical PET Study
Ludwig F-A, Fischer S, Smits R, Deuther-Conrad W, Hoepping A, Tiepolt S, Patt M, Sabri O, Brust P.
Exploring the Metabolism of (+)-[18F]Flubatine In Vitro and In Vivo: LC-MS/MS Aided Identification of Radiometabolites in a Clinical PET Study
Ludwig, Friedrich-Alexander, Steffen Fischer, René Smits, Winnie Deuther-Conrad, Alexander Hoepping, Solveig Tiepolt, Marianne Patt, Osama Sabri, and Peter Brust.
2018. "Exploring the Metabolism of (+)-[18F]Flubatine In Vitro and In Vivo: LC-MS/MS Aided Identification of Radiometabolites in a Clinical PET Study
Ludwig, F.-A., Fischer, S., Smits, R., Deuther-Conrad, W., Hoepping, A., Tiepolt, S., Patt, M., Sabri, O., & Brust, P.
(2018). Exploring the Metabolism of (+)-[18F]Flubatine In Vitro and In Vivo: LC-MS/MS Aided Identification of Radiometabolites in a Clinical PET Study