Abstract
A series of pleuromutilin derivatives were synthesized by two synthetic procedures under mild reaction conditions and characterized by Nuclear Magnetic Resonance (NMR), Infrared Spectroscopy (IR), and High Resolution Mass Spectrometer (HRMS). Most of the derivatives with heterocyclic groups at the C-14 side of pleuromutilin exhibited excellent in vitro antibacterial activities against Staphylococcus aureus, methicillin-resistant Staphylococcus aureus (MRSA), methicillin-resistant Staphylococcus epidermidis (MRSE), and vancomycin-resistant Enterococcus (VRE) in vitro antibacterial activity. The synthesized derivatives which contained pyrimidine rings, 3a, 3b, and 3f, displayed modest antibacterial activities. Compound 3a, the most active antibacterial agent, displayed rapid bactericidal activity and affected bacterial growth in the same manner as that of tiamulin fumarate. Moreover, molecular docking studies of 3a and lefamulin provided similar information about the interactions between the compounds and 50S ribosomal subunit. The results of the study show that pyrimidine rings should be considered in the drug design of pleuromutilin derivatives.
1. Introduction
Antibiotics have been necessary life-saving drugs since the advent of penicillin in 1928 [1]. However, antibiotic-treatment faces total defeat as a result from drug resistance. Meanwhile, the discovery of antibiotics has gotten stuck in bottleneck. Although there have been more than 31 lead classes of antibiotics found and 160 kinds of antibiotics approved from the 1890s–1980s, few lead class antibiotics have been found in the past thirty years [1].
Many antibiotic treatment failures were associated with the spread of bacterial drug resistance, such as methicillin-resistant Staphylococcus aureus (MRSA), methicillin-resistant Staphylococcus epidermidis (MRSE), and vancomycin-resistant Enterococcus (VRE) [2]. Consequently, the study for clinically-available potent antibiotics that are effective against multidrug-resistant pathogens is becoming exceedingly urgent [3].
Pleuromutilin was first discovered and isolated from basidiomycetes, Pleurotus mutilus, and P. passeckerianus in 1951 [4]. Tiamulin (Figure 1) was the first derivative approved for veterinary use in 1979, following which valnemulin (Figure 1) was approved in 1999. It was not until 2007 that retapamulin (Figure 1) was used in human medicine [4]. The C-14 side of pleuromutilin is the modest modification position for designing a high antibacterial agent and thus improves biological activity and enhances water-solubility [5,6,7,8].
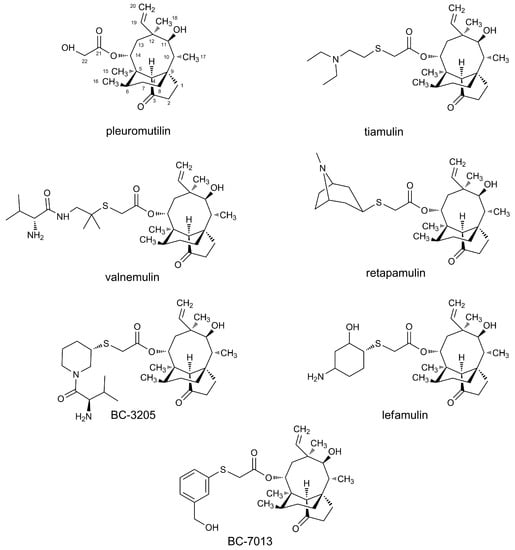
Figure 1.
Structural formulas of pleuromutilin and derivation thereof.
The antibacterial mechanisms of pleuromutilin derivatives are to inhibit protein synthesis by blocking the peptidyl transferase center (PTC). Their tricyclic mutilin core of pleuromutlin derivative intervenes the A-site and the C-14 side chain extends to the P-site, which disturbs the translation of mRNA [4,9,10,11,12].
Many authors realized that thioether’s presence in C-14 could improve its antibacterial activities [11,12]. Recently, three new potential drugs, BC-3205, BC-7013, and lefamulin, which contain heterocyclic groups, have been approved in clinical trials. Nabriva’s lead product, lefamulin, has entered phase III of clinical trials on community-acquired bacterial pneumonia [12] and has received U.S. Food and Drug Administrationfast-track status [13].
In the present project, we decided to screen potential derivatives on the pleuromutilin skeleton, as well as to increase our knowledge of the binding site by molecular docking.
2. Results and Discussion
2.1. Chemistry
Mutilin 14-tosyloxyacrtate (2) was prepared by pleuromutilin and p-toluene sulfonyl chloride under basic conditions in 78% yield. The pleuromutilin derivatives 3a–h were formed, as shown in Scheme 1. Mercaptan, used as an nucleophilic agent, easily attacked the C21-position of 14-tosyloxyacrtate under basic conditions. The synthetic route for compound 3i is depicted in Scheme 2. Compound 4 was prepared by reacting mutilin 14-tosyloxyacrtate (2) with sulfocarbamide under alkaline conditions. Compound 3i was obtained directly from compound 4 and 5 with a one pot reaction.
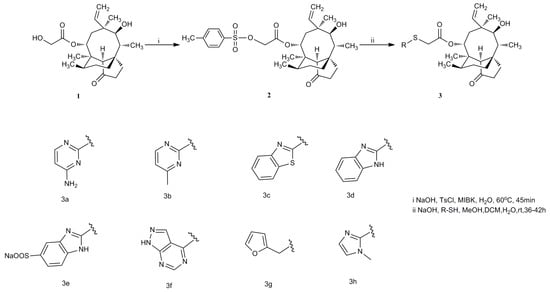
Scheme 1.
Synthesis of compounds 3a–3h.
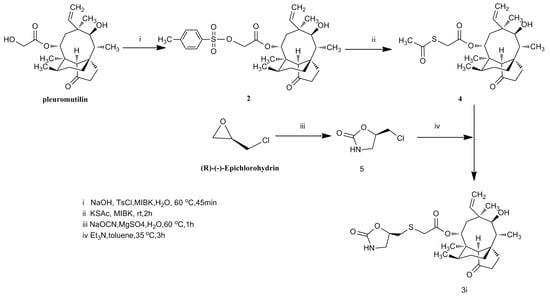
Scheme 2.
Synthesis of compound 3i.
All synthesis compounds were characterized by Nuclear Magnetic Resonance (NMR), Infrared Spectroscopy (IR) (Thermo Nicolet Corporation, Waltham, MA, USA.), and High Resolution Mass Spectrometer (HRMS) (Bruker Corporation, Billerica, MA, U.SA). A colorless crystal of compound 3a, block-like, was obtained by slow evaporation of a chloroform solution (Figure 2).
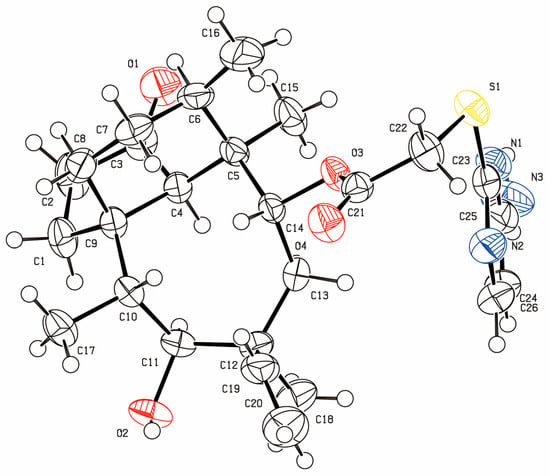
Figure 2.
Oak Ridge Thermal-Ellipsoid Plot Program (ORTEP) diagram for compound 3a with ellipsoids set at 75% probability.
2.2. Antibacterial Activity
The synthesized derivatives 3a–i were evaluated for their antibacterial activity against several representative Gram-positive strains, including Staphylococcus aureus, methicillin-resistant Staphylococcus aureus (MRSA), methicillin-resistant Staphylococcus epidermidis (MRSE), and vancomycin-resistant Enterococcus (VRE), and a Gram-negative bacterium, E. coli. Tiamulin fumarate served as a control. The results of minimum inhibitory concentrations (MICs) and lipophilicities (Clogp), which were predicted by ACD/Labs (Toronto, Ontario, ON, Canada), are shown in Table 1. The synthesized pleuromutilin derivatives exhibited modest to excellent antibacterial activity against S. aureus, MRSA, MRSE, and VRE (MIC, 0.0625–2 μg/mL), respectively, while they showed poor activity against E. coli. Compounds 3a, 3b, and 3f with a pyrimidine group showed higher antimicrobial activity than the other derivatives.

Table 1.
Antibacterial activity (Minimum Inhibitory Concentration) of the synthesized pleuromutilin derivatives.
The most potent compound, 3a, displayed promising antibacterial activity, the lowest MIC value in 3a–i and was therefore further evaluated for in vitro time-kill assay [14]. The bactericidal properties of 3a were compared at 1 × MIC and 6 × MIC against S. aureus (3a MIC = 0.0625 μg/mL, tiamulin MIC = 0.0625 μg/mL), and MRSA (3a MIC = 0.125 μg/mL, tiamulin MIC = 0.25 μg/mL) (Figure 2). As shown in Figure 3, 3a displayed a concentration-dependent effect, with faster killing kinetics at higher concentrations. Although 1 × MIC of compound 6a and tiamulin slowed bacterial propagation, their 6 × MIC achieved a 3-log10 reduction in 4–6 h. Compared to tiamulin, 3a showed more rapid bactericidal kinetics against S. aureus and MRSA with the same concentration (1 × MIC). Notably, 3a at 6 × MIC concentration reduced the viable count by approximately 6.8-log10 and achieved complete killing after 6 h of incubation.
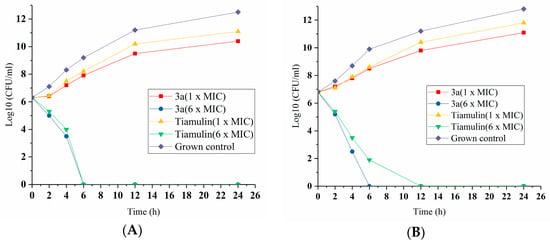
Figure 3.
Time-kill kinetics of compound 3a against S. aureus (ATCC 25923) (A) and MRSA (ATCC 43300) (B). Mean values of the CFU/mL (colony forming units per milliliter) were obtained from measurements taken in triplicate.
Further comparison of the physicochemical parameters of compounds 3a–i, having a relatively hydrophilic C-14 side chain, shows that at good solubility of all of these derivatives. The lipophilicity of compound 3a (ClogP = 3.23) is close to that of tiamulin, which contributes to its antibacterial activity comparable to that of tiamulin [15]. In summary, 3a exhibited signigicantly improved bactericidal activity compared to that of tiamulin.
2.3. Molecular Docking Studies
Molecular docking was performed with the aim of revealing the relations of the derivatives with their antibacterial activity at the atomic level. The suggestion that all pleuromutilin derivatives have been demonstrated to be effective might owe to their binding of the bacterial ribosome to PTC. To understand this phenomenon, we performed molecular docking to determine the reliability of this proposal. On applying Homdock software [16], the redocking of lefamulin into 5Hl7 [12] placed the compound in the PTC as that X-ray crystallography structure. The crystal structures was a typical complex of the 50 s ribosome about S. aureus and lefamulin. The docking results showed that the eight-membered ring can bind to the active site of the ribosome in the same manner as lefamulin with RMSD (Root-mean-square Deviation) at 0.88–1.12 Å (Figure 4, Table 2). Hydroxyl groups in all of the derivatives were located in a suitable position to form hydrogen bonds with G-2532, and some ester carbonyl groups were bound to C-2088 as hydrogen bonds. Although the tricyclic mutilin core did not form any hydrogen bonds with the PTC, it is stabilized by hydrophobic and Van der Waals interactions. Compounds 3b and 3d formed three hydrogen bonds with similar docking modes. Compound 3a, the most promising of the candidates (ΔGb = −8.855 kcal/mol), bonded to PTC as a typical characteristic based on the docking results. Interestingly, 3a, 3b, and 3f exhibited generally low energy and MIC values. The results were probably caused by its amine in pyrimidine, which plays an important role in the binding of the complex (Figure 4B).
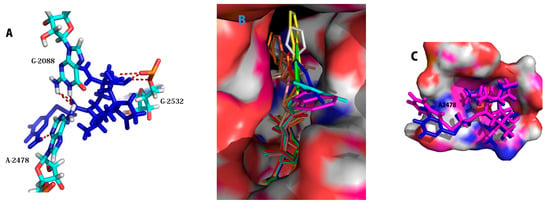
Figure 4.
(A) Docking modes of the synthesized compounds into 5HL7. Key amino acid residues and ligand-active site interactions are shown. 3a (blue) to 5HL7; Important residues are drawn in sticks and different color. Hydrogen bonds are showed as dashed red lines; (B) The best pose of the compounds obtained from the docking study in the active site of the peptidyl transferase center. (3a blue, 3b cyan, 3c yellow, 3d magentas, 3e oranges, 3f wheat, 3g gray, 3h green, 3i salmon, lefamulin red); (C) 3a (blue) and 3d (magentas) in 5HL7.
Table 2.
Binding RMSD (Root-mean-square Deviation), number of noncovalent molecular interactions and free energy.
Despite their similarities in H-binding, the activity of 3a was better than 3d. When we modeled the structural overlaps of compounds 3a and 3d, their side chain appeared markedly different. The pocket of PTC showed hydrophilicity (blue in Figure 4C) at the A-2478 position, which could make 3a more stable than 3d.
The binding sites of the pleuromutilin derivatives were different from that of other antibiotics, like macrocyclic and clindamycin. The macrocyclic ring was bound at aminoacyl-tRNAs at the bacterial ribosome (A-site) [17], which is the same side of the ribosomal tunnel as its side chain. In the crystal structure (PDB ID:1JZX), clindamycin also binds to the A-site of their PTC of bacterial ribosomes [18].
3. Experimental Section
3.1. Synthesis
General
All chemical reagents were purchased from J and K Chemical or Sigma-Aldrich Chemistry Co. (Darmstadt, Germany). Unless otherwise noted, all reactions were conducted under atmosphere. Thin-layer chromatography (TLC) analysis (Qingdao Haiyang Chemical Co., Ltd., Shandong, Qingdao, China) was used to monitor the reaction process. Column chromatography was carried out on silica gel (200–300 mesh). The products were eluted in an appropriate solvent mixture under air pressure. Concentration and evaporation of the solvent after reaction or extraction was carried out on a rotary evaporator. IR spectra were obtained on a Thermo Nicolet NEXUS-670 spectrometer (Thermo Nicolet Corporationcompany, Waltham, MA, USA) and recorded as KBr thin films and absorptions are reported in cm−1. HRMS were obtained with a Bruker Daltonics APEX II 47e mass spectrometer. NMR spectra (Bruker Corporation, Billerica, MA, USA) were recorded on a Bruker-400 MHz spectrometer (Bruker Corporation, Billerica, MA, USA) in appropriate solvents. Chemical shifts (δ) were expressed in parts per million (ppm) relative to the tetramethylsilane. Multiplicities of NMR signals are designated as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), br (broad), etc. 13C-NMR spectra were recorded on 100 MHz spectrometers. The single-crystal structure of the title compound was determined on a Bruker SMART APEX II X-diffractometer (Bruker Corporation, Billerica, MA, USA). All NMR, IR and HRMS datum were been added in Supplementary Materials.
14-O-(p-Toluene sulfonyloxyacetyl)mutilin (2). A 5 mL of NaOH aqueous solution (2 g, 50 mmol) was added dropwise to a mixture of pleuromutilin (7.57 g, 20 mmol) and p-toluenesulfonyl chloride (4.2 g, 22 mmol) in methyl isobutyl ketone (10 mL) and water (5 mL). The mixture was vigorously stirred for 45 min at 60 °C, then the reaction mixture was cooled to 10 °C and separated. The organic layer was washed with 5 mL water and 5 mL saturated sodium carbonate solution. The organic phase was dried overnight with anhydrous sodium sulfate. After filteration, the solvent was concentrated in vacuo to give 10.56 g of yellow oil. It was used in the next step without further purification. Yield: 93%. IR (KBr): 3446 (OH), 2924 (CH2), 2863 (CH2), 1732 (C=O), 1633 (C-C), 1597 (C=C), 1456 (C=C), 1371 (CH3), 1297 (C-O-C), 1233 (CH), 1117 (C-(C=O)-C), 1035 (C-O-C), 832 (CH), 664 (CH2=), 560 (CH2=) cm−1. 1H-NMR (400 MHz, CDCl3) δ 7.74 (d, J = 8.3 Hz, 2H), 7.26 (t, J = 13.3 Hz, 2H), 6.34 (dd, J = 17.4, 11.0 Hz, 1H), 5.70 (d, J = 8.5 Hz, 1H), 5.19 (dd, J = 55.1, 14.2 Hz, 2H), 4.47–4.33 (m, 2H), 3.28 (s, 1H), 2.38 (s, 3H), 2.25–2.09 (m, 3H), 2.01 (s, 1H), 1.99–1.88 (m, 1H), 1.71–1.62 (m, 1H), 1.61–1.51 (m, 2H), 1.46–1.38 (m, 2H), 1.35 (d, J = 7.8 Hz, 3H), 1.27 (d, J = 11.5 Hz, 1H), 1.18 (dd, J = 11.6, 4.5 Hz, 2H), 1.12–1.00 (m, 4H), 0.80 (d, J = 7.0 Hz, 3H), 0.55 (d, J = 7.0 Hz, 3H). 13C-NMR(100 MHz, CDCl3) δ215.71 (C=O), 163.87 (C=O), 144.29 (benzene-C), 137.70 (CH=), 131.63 (benzene-C), 128.91 (benzene-C), 127.09 (benzene-C), 116.38 (CH2=), 73.54 (CH), 69.29 (CH), 64.03 (CH), 57.02 (CH), 44.39 (C), 43.51 (CH2), 42.97 (C), 40.84 (C), 35.54 (CH), 35.03 (CH), 33.40 (CH2), 29.34 (CH3), 25.77(CH2), 25.39 (CH2), 23.81 (CH2), 20.68 (CH3), 15.53 (CH3), 13.76 (CH3), 10.47 (CH3). HRMS (ESI) calcd. [M + H]+ for C29H40O7S 533.250, found 533.2507.
14-O-(Acetic acidthioacetyl)mutilin (4). Compound 3 was prepared by stirring a mixing of compound 2 (10 mmol), potassium thioglycolate (20 mmol), and methyl isobutyl ketone (30 mL) in room temperature, and the mixture was stirred for 2 h. The mixture was extracted with water (10 mL). The organic phase were combined, dried over Na2SO4, and concentrated to give compound 3. Yield: 80%. IR (KBr): 3448 (OH), 2967 (CH2), 2924 (CH3), 2865 (CH2), 1731 (C=O), 1702 (C=O), 1453 (C-C), 1419, 1384 (CH3), 1295 (C-O-C), 1183 (CH), 1154 (C-O), 1115 (C-(C=O)-C), 1017 (C-O-C) cm−1. 1H-NMR (400 MHz, CDCl3) δ 6.58–6.30 (m, 1H), 5.72 (d, J = 8.4 Hz, 1H), 5.43–5.14 (m, 2H), 3.63 (s, 2H), 3.36 (d, J = 6.4 Hz, 1H), 2.36 (dd, J = 13.5, 3.5 Hz, 3H), 2.35–2.27 (m, 1H), 2.21 (dd, J = 17.4, 7.9 Hz, 1H), 2.13 (d, J = 14.9 Hz, 1H), 2.04 (dd, J = 26.1, 17.5 Hz, 1H), 1.73 (dd, J = 31.8, 10.6 Hz, 2H), 1.68–1.62 (m, 2H), 1.62–1.41 (m, 6H), 1.41–1.24 (m, 2H), 1.24–0.99 (m, 4H), 0.89 (t, J = 8.4 Hz, 3H), 0.78–0.61 (m, 3H). 13C-NMR (101 MHz, CDCl3) δ 216.90 (C=O), 193.45 (C=O), 167.32 (C=O), 138.92 (CH=), 117.19 (CH2=), 74.60 (CH), 70.09 (CH), 58.13 (CH), 45.45 (C), 44.71 (CH2), 44.01 (C), 41.89 (C), 36.74 (CH), 36.00 (CH3), 34.45 (CH2), 32.20 (CH2), 30.42 (CH2), 30.06 (CH2), 26.84 (CH2), 26.41 (CH3), 24.82 (CH2), 16.74 (CH3), 14.81(CH3), 11.43 (CH3). HRMS (ESI) calcd. [M + H]+ for C24H36O5S 437.2356, found 437.2339.
(R)-5-ChloroMethyl-2-oxazolidinone (5). The title compound was prepared by stirring a mixing of magnesium sulphate (10 mmol), sodium cyanate (10 mmol) and water (50 mL) in room temperature. (R)-(−)-Epichlorohydrin to the solution (5 mmol) was added dropwise to the mixture. The result mixture was stirred under 60 °C for 1 h. The reaction mixture was concentrated in vacuo and extracted by ethyl acetate. The two layers were separated, and the organic layer was dried over Na2SO4, filtered, and concentrated to dryness. Yield: 57%. IR (KBr): 3365 (NH), 1744 (C=O), 1429 (C-N), 1240 (C-C(=O)-O), 736 (C-Cl) cm−1. 1H-NMR (400 MHz, DMSO) δ 7.60 (s, 1H), 4.84 (dd, J = 9.6, 4.7 Hz, 1H), 4.04–3.73 (m, 2H), 3.59 (t, J = 9.0 Hz, 1H), 3.25 (dd, J = 9.0, 6.2 Hz, 1H). 13C-NMR (101 MHz, d6-DMSO) δ 158.68 (C=O), 74.35 (CH), 46.66 (CH2), 43.03(CH2). HRMS (ESI) calcd. [M + H]+ for C4H6ClNO 136.0159, found 136.0150.
General Procedure for Synthesis of Compounds 3a–3g. A mixing of thiols (1 mmol), sodium hydroxide (1.1 mmol), water (0.5 mL) and methanol (3 mL) were stirred in room temperature. After 30 min, compound 2 (1.1 mmol) in 5 mL CH2Cl2 was added dropwise to the mixture for 36 h–42 h. The mixture was concentrated in vacuo. The residue was dissolved by CH2Cl2. The solution was extracted three times with water. The organic phase was dried overnight with anhydrous sodium sulfate. The solvent was concentrated in vacuo to give crude products. The crude product was purified by silica gel column chromatography.
14-O-[(4-Amino-pyrimidinone-2-yl)thioacetyl]mutilin (3a). Compound 3a was prepared according to the general procedure from 14-O-(p-toluene sulfonyloxyacetyl) mutilinmutilin (2) and 4-amino-2-Pyrimidinone. The crude product was purified over silica gel column chromatography to give 4.09 g. Yield: 84%. IR (KBr): 3448 (OH), 2933 (CH2), 1730 (C=O), 1629 (C-C), 1583 (C=N), 1543 (C=C), 1467 (C=C), 1372 (CH3), 1249(C-C(=O)-O), 1153(C-O), 1117 (C-(C=O)-C), 1018 (C-O-C) cm−1. 1H-NMR (400 MHz, CDCl3) δ 7.89 (d, J = 5.1 Hz, 1H), 6.42 (dd, J = 17.1, 11.2 Hz, 1H), 6.04 (d, J = 5.2 Hz, 1H), 5.68 (d, J = 7.9 Hz, 1H), 5.17 (dd, J = 51.9, 14.0 Hz, 2H), 4.94 (s, 2H), 3.72 (dd, J = 32.8, 16.1 Hz, 2H), 3.28 (s, 1H), 2.24 (d, J = 6.5 Hz, 1H), 2.14 (dd, J = 14.8, 9.1 Hz, 2H), 2.02 (s, 1H), 1.94 (dd, J = 15.6, 8.5 Hz, 1H), 1.69 (d, J = 13.7 Hz, 1H), 1.61–1.52 (m, 2H), 1.48 (d, J = 12.0 Hz, 2H), 1.37 (s, 4H), 1.32–1.22 (m, 2H), 1.07 (s, 4H), 0.79 (d, J = 6.2 Hz, 3H), 0.68 (d, J = 6.2 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 216.11 (C=O), 168.72 (pyrimidine-C), 167.20 (C=O), 161.35 (pyrimidine-C), 154.88 (pyrimidine-C), 138.26 (CH=), 116.02 (CH2=), 100.18 (pyrimidine-C), 73.60 (CH), 68.51(CH), 57.36 (CH), 57.21(CH2), 44.46 (CH2), 43.47 (C), 42.93 (C), 40.87 (CH), 35.81 (CH), 35.02 (CH), 33.48 (CH2), 33.06 (CH2), 29.45(CH2), 25.89 (CH3), 23.84 (CH2), 15.74 (CH3), 13.92 (CH3), 10.44 (CH3). HRMS (ESI) calcd. [M + H]+ for C26H37N3O4S 488.2578, found 488.2570.
14-O-[(4-Methylpyrimidine-2-yl)thioacetyl]mutilin (3b). Compound 3b was prepared according to the general procedure from 14-O-(p-toluene sulfonyloxyacetyl) mutilin(2) and 4-methy-2-pyrimidinone. The crude product was purified over silica gel column chromatography to give 3.55 g. Yield: 73%. IR (KBr): 3439 (OH), 2935 (CH2), 1733 (C=O), 1658 (C-C), 1580 (C=N), 1535 (C=C), 1458 (C=C), 1396 (CH3), 1285 (C-O-C), 1118 (C-(C=O)-C) cm−1. 1H-NMR (400 MHz, CDCl3) δ 6.37 (dt, J = 33.7, 16.8 Hz, 1H), 5.99 (s, 1H), 5.69 (d, J = 8.4 Hz, 1H), 5.19 (dd, J = 56.5, 14.2 Hz, 2H), 3.88–3.73 (m, 2H), 3.29 (d, J = 5.6 Hz, 1H), 2.27–2.15 (m, 2H), 2.11 (d, J = 13.1 Hz, 3H), 2.02 (d, J = 6.4 Hz, 1H), 1.96 (d, J = 10.9 Hz, 1H), 1.69 (d, J = 14.2 Hz, 1H), 1.58 (dd, J = 21.0, 10.8 Hz, 2H), 1.49 (dd, J = 26.7, 13.3 Hz, 2H), 1.43–1.26 (m, 6H), 1.20 (dd, J = 17.5, 11.2 Hz, 2H), 1.05 (d, J = 20.9 Hz, 4H), 0.80 (d, J = 6.9 Hz, 3H), 0.67 (d, J = 6.9 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 215.94 (C=O), 165.80(C=O), 164.69 (pyrimidine-C), 164.11 (pyrimidine-C), 157.85 (pyrimidine-C), 137.90 (CH=), 116.27 (CH2=), 107.63 (pyrimidine-C), 73.55 (CH), 69.14 (CH), 57.08 (CH), 44.43 (C), 43.49 (CH2), 42.94 (C), 40.87 (C), 35.70 (CH), 35.00 (CH3), 33.44 (CH2), 32.23 (CH2), 29.39 (CH2), 25.84(CH3), 25.36(CH2), 23.82 (CH2), 23.09 (CH2), 15.84(CH3), 13.84 (CH3), 10.46 (CH3). HRMS (ESI) calcd. [M + H]+ for C27H38N2O4S 487.2625, found 487.2623.
14-O-[(Benzimidazole-2-yl)thioacetyl]mutilin (3c). Compound 3c was prepared according to the general procedure from 14-O-(p-toluene sulfonyloxyacetyl) mutilin (2) and 2-mercaptobenzothiazole. The crude product was purified over silica gel column chromatography to give 3.54 g. Yield: 67%. IR (KBr): 3442 (OH), 2929 (CH2), 1731 (C=O), 1458 (C=C), 1429 (C-C), 1274 (C-O-C), 1153 (C-O), 1117 (C-(C=O)-C) cm−1. 1H-NMR (400 MHz, CDCl3) δ 7.77 (dd, J = 19.9, 8.0 Hz, 2H), 7.39 (t, J = 7.7 Hz, 1H), 7.29 (t, J = 7.6 Hz, 1H), 6.42 (dd, J = 17.4, 11.0 Hz, 1H), 5.76 (d, J = 8.5 Hz, 1H), 5.19 (dd, J = 57.6, 14.2 Hz, 2H), 4.08 (dd, J = 41.4, 16.2 Hz, 2H), 3.31 (d, J = 6.4 Hz, 1H), 2.29 (dd, J = 14.1, 7.2 Hz, 1H), 2.20 (dd, J = 11.6, 6.3 Hz, 1H), 2.06 (s, 1H), 1.98 (dd, J = 16.0, 8.5 Hz, 1H), 1.82–1.64 (m, 2H), 1.64–1.51 (m, 2H), 1.50–1.33 (m, 6H), 1.24 (dd, J = 14.3, 7.4 Hz, 2H), 1.13–0.97 (m, 4H), 0.85 (d, J = 7.0 Hz, 3H), 0.77 (d, J = 6.9 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 215.94 (C=O), 165.84 (C=O), 163.54 (benzothiazole-C), 151.74 (benzothiazole-C), 137.76 (CH=), 134.49(benzothiazole-C), 125.02 (benzothiazole-C), 123.46 (benzothiazole-C), 120.68 (benzothiazole-C), 120.05 (benzothiazole-C), 116.21 (CH2=), 73.57 (CH), 69.20 (CH), 57.10 (CH), 44.42 (C), 43.41 (CH2), 42.90 (C), 40.86 (C), 35.74 (CH), 34.98 (CH), 34.67 (CH2), 33.43(CH2), 29.40 (CH2), 25.84 (CH2), 25.28 (CH3), 23.81 (CH2), 15.80(CH3), 13.81 (CH3), 10.44 (CH3). HRMS (ESI) calcd. [M + H]+ for C29H37NO4S 528.2237, found 528.2234.
14-O-[(Benzothiazole-2-yl)thioacetyl]mutilin (3d). Compound 3d was prepared according to the general procedure from 14-O-(p-toluene sulfonyloxyacetyl) mutilin (2) and 2-mercaptobenzimidazole. The crude product was purified over silica gel column chromatography to give 3.68 g. Yield 72%. IR (KBr): 3423 (OH), 2925 (CH2), 1726 (C=O), 1458 (C=C), 1439 (C-C), 1271 (C-O-C), 1152 (C-O), 1117 (C-(C=O)-C) cm−1. 1H-NMR (400 MHz, CDCl3) δ 7.50 (dd, J = 5.8, 3.1 Hz, 2H), 7.20 (dd, J = 6.0, 3.2 Hz, 2H), 6.41 (dd, J = 17.4, 11.0 Hz, 1H), 5.79 (d, J = 8.4 Hz, 1H), 5.18 (dd, J = 43.7, 14.2 Hz, 2H), 3.91 (s, 2H), 3.73 (q, J = 7.0 Hz, 1H), 3.35 (d, J = 6.4 Hz, 1H), 2.35–2.28 (m, 1H), 2.27–2.17 (m, 1H), 2.08 (s, 1H), 2.03 (dd, J = 16.1, 8.6 Hz, 1H), 1.82–1.65 (m, 2H), 1.64–1.52 (m, 2H), 1.49–1.32 (m, 6H), 1.24 (t, J = 7.0 Hz, 2H), 1.16–1.05 (m, 4H), 0.88 (d, J = 6.9 Hz, 3H), 0.72 (d, J = 7.0 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 215.89 (C=O), 167.78 (C=O), 147.20 (benzimidazole-C), 137.75 (CH=), 121.68 (benzimidazole-C), 116.28 (CH2=), 73.61 (CH), 69.82 (CH), 57.43 (CH), 57.09 (benzimidazole-C), 44.42 (C), 43.52 (CH2), 42.97 (C), 40.84 (C), 35.68 (CH), 35.04 (CH2), 34.16 (CH2), 33.42, 29.38 (CH2), 25.84 (CH2), 25.44 (CH3), 23.83 (CH2), 17.42 (CH2), 15.80 (CH3), 13.80 (CH3), 10.50 (CH3). HRMS (ESI) calcd. [M + H]+ for C29H38N2O4S 511.2526, found 511.2531.
14-O-[(5-Benzimidazolesulfonate-2-yl)thioacetyl]mutilin (3e). Compound 3e was prepared according to the general procedure from 14-O-(p-toluene sulfonyloxyacetyl) mutilin (2) and sodium 2-mercapto-5-benzimidazolesulfonate dihydrate. The crude product was purified over silica gel column chromatography to give 3.2 g. Yield: 54.2%. IR (KBr): 3448 (OH), 2940 (CH2), 1732 (C=O) 1298 (C-O-C), 1190 (S=O), 1178 (S=O) cm−1. 1H-NMR (400 MHz, DMSO) δ 7.80 (s, 1H), 7.69 (d, J = 8.5 Hz, 1H), 7.60 (d, J = 8.5 Hz, 1H), 6.04 (dd, J = 17.8, 11.2 Hz, 1H), 5.50 (d, J = 8.1 Hz, 1H), 4.96 (dd, J = 38.2, 14.5 Hz, 2H), 4.38 (d, J = 4.4 Hz, 1H), 3.36 (d, J = 5.5 Hz, 1H), 2.51 (s, 1H), 2.35 (s, 1H), 2.13 (dd, J = 21.0, 10.7 Hz, 1H), 2.08–1.96 (m, 2H), 1.90 (dd, J = 16.0, 8.1 Hz, 1H), 1.60 (s, 2H), 1.44 (d, J = 6.9 Hz, 1H), 1.33 (d, J = 7.6 Hz, 2H), 1.27–1.12 (m, 6H), 1.10–1.02 (m, 1H), 1.02–0.90 (m, 4H), 0.78 (d, J = 6.8 Hz, 3H), 0.58 (t, J = 9.3 Hz, 3H). 13C-NMR (101 MHz, DMSO) δ 217.48 (C=O), 166.34 (C=O), 150.52 (benzimidazole-C), 145.67 (benzimidazole-C), 141.16 (benzimidazole-C), 133.41 (CH=), 128.59 (benzimidazole-C), 125.97 (benzimidazole-C), 123.22 (benzimidazole-C), 115.68 (CH2=), 113.22 (CH2), 110.71 (benzimidazole-C), 72.92 (CH), 71.44 (CH), 57.42 (CH), 45.35 (C), 44.58 (CH2), 41.95 (C), 36.89 (CH), 36.64 (CH), 35.03 (CH2), 34.40 (CH2), 30.53 (CH2), 29.06 (CH2), 26.99 (CH2), 24.85 (CH2), 16.47 (CH3), 14.64 (CH3), 11.96 (CH3). HRMS (ESI) calcd. [M + H]+ for C29H38N2O7S2 591.2193, found 591.2192.
14-O-[(Pyryrazolo[3,4d]pyrimidine-4-yl)thioacetyl]mutilin (3f). Compound 3f was prepared according to the general procedure from 14-O-(p-toluene sulfonyloxyacetyl) mutilin (2) and 4-mercaptopyrazolo[3,4-d]pyrimidine. The crude product was purified over silica gel column chromatography to give 4.30 g. Yield: 84%. IR (KBr): 3431 (OH), 2934 (CH2), 1732 (C=O), 1567 (C=C), 1456 (C=C), 1406 (C-C), 1271 (C-O-C), 1152 (C-O), 1117 (C-(C=O)-C), 981 (CH) cm-1. 1H-NMR (400 MHz, CDCl3) δ 8.65 (d, J = 37.7 Hz, 1H), 8.29–8.08 (m, 1H), 6.43 (dt, J = 21.8, 10.9 Hz, 1H), 5.80 (d, J = 8.4 Hz, 1H), 5.38–5.29 (m, 1H), 5.24 (dd, J = 33.0, 14.5 Hz, 2H), 4.18–4.02 (m, 2H), 3.38 (d, J = 6.3 Hz, 1H), 2.31 (dd, J = 13.9, 7.1 Hz, 1H), 2.22 (dd, J = 13.0, 7.5 Hz, 1H), 2.11 (t, J = 8.4 Hz, 1H), 2.04 (d, J = 7.9 Hz, 1H), 1.73 (dd, J = 31.9, 9.2 Hz, 2H), 1.65 (dd, J = 17.1, 7.1 Hz, 2H), 1.58–1.37 (m, 6H), 1.36–1.22 (m, 2H), 1.19–1.06 (m, 4H), 0.88 (t, J = 11.3 Hz, 3H), 0.79 (t, J = 12.6 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 216.06 (C=O), 166.18 (C=O), 162.56 (pyrimidine-C), 153.05 (pyrimidine-C), 151.46 (pyrimidine-C), 137.88 (CH=), 131.82 (pyrazolo-C), 116.28 (CH2=), 110.65 (pyrimidine-C), 73.59 (CH), 69.22 (CH), 57.14 (CH), 44.46 (C), 43.57 (CH2), 42.92 (C), 40.88 (C), 35.75 (CH), 35.01 (CH2), 33.46 (CH2), 31.11 (CH2), 29.41 (CH2), 25.44 (CH3), 23.83 (CH2), 17.40(CH2), 15.74 (CH3), 13.86 (CH3), 10.47 (CH3). HRMS (ESI) calcd. [M + H]+ for C27H36N4O4S 513.2530, found 513.2527.
14-O-[(Furfuryl-2-yl)thioacetyl]mutilin (3g). Compound 3g was prepared according to the general procedure from 14-O-(p-toluene sulfonyloxyacetyl) mutilin (2) and furfuryl mercaptan. The crude product was purified over silica gel column chromatography to give 3.53 g. Yield: 74%. IR (KBr): 3547 (OH), 2933 (CH2), 2882 (CH2), 1731 (C=O), 1455 (C=C), 1281 (C-O-C), 1150 (C-O), 1115 (C-(C=O)-C) cm−1. 1H-NMR (400 MHz, CDCl3) δ 7.29 (s, 1H), 6.42 (dd, J = 17.4, 11.0 Hz, 1H), 6.31–6.06 (m, 2H), 5.71 (d, J = 8.4 Hz, 1H), 5.23 (dd, J = 57.0, 14.2 Hz, 2H), 3.75 (s, 2H), 3.30 (s, 1H), 3.02 (s, 2H), 2.29 (dd, J = 13.7, 6.8 Hz, 1H), 2.15 (dt, J = 19.6, 8.8 Hz, 2H), 2.09–1.98 (m, 2H), 1.75–1.66 (m, 1H), 1.64–1.55 (m, 2H), 1.53–1.35 (m, 6H), 1.30 (t, J = 14.9 Hz, 2H), 1.17–1.05 (m, 4H), 0.82 (d, J = 7.0 Hz, 3H), 0.68 (d, J = 6.8 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 215.99 (C=O), 167.67 (C=O), 149.27 (furan-C), 141.51(furan-C), 138.12 (CH=), 116.19 (CH2=), 109.37 (furan-C), 107.39 (furan-C), 73.65 (CH), 68.31 (CH), 57.21 (CH), 44.46 (C), 43.85 (CH2), 42.94 (C), 40.77 (C), 35.78 (CH), 35.04 (CH), 33.45 (CH2), 32.11 (CH2), 29.44 (CH2), 27.36 (CH2), 25.85 (CH3), 25.42 (CH2), 23.86 (CH2), 15.81 (CH3), 13.92 (CH3), 10.49 (CH3). HRMS (ESI) calcd. [M + H]+ for C27H38O5S 475.2513, found 475.2522.
14-O-[(1-Methylimidazole-2-yl)thioacetyl]mutilin (3h). Compound 3h was prepared according to the general procedure from 14-O-(p-toluene sulfonyloxyacetyl) mutilin (2) and 2-mercapto-1-methylimidazole. The crude product was purified over silica gel column chromatography to give 3.46 g. Yield: 73%. IR (KBr): 3423 (OH), 2924 (CH2), 2863 (CH2), 1717 (C=O), 1455 (C=C), 1410 (C-C), 1280 (C-O-C), 1145 (C-O), 1117 (C-(C=O)-C) cm−1. 1H-NMR (400 MHz, CDCl3) δ 6.88 (d, J = 49.1 Hz, 2H), 6.35 (dd, J = 17.4, 11.0 Hz, 1H), 5.63 (d, J = 8.5 Hz, 1H), 5.32–5.01 (m, 2H), 3.92–3.62 (m, 2H), 3.56 (d, J = 11.0 Hz, 3H), 3.27 (s, 1H), 2.22 (dt, J = 13.8, 7.0 Hz, 1H), 2.19–2.07 (m, 2H), 2.03 (d, J = 21.6 Hz, 1H), 1.93 (dd, J = 16.0, 8.6 Hz, 1H), 1.77–1.61 (m, 1H), 1.58–1.46 (m, 3H), 1.45–1.35 (m, 2H), 1.31 (d, J = 14.3 Hz, 3H), 1.26–1.13 (m, 2H), 1.12–0.97 (m, 4H), 0.80 (d, J = 7.0 Hz, 3H), 0.59 (d, J = 6.9 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 215.93 (C=O), 166.75 (C=O), 139.06 (imidazole-C), 138.01 (CH=), 128.55 (imidazole-C), 121.39 (imidazole-C), 116.07 (CH2=), 73.57 (CH), 68.77 (CH), 57.12 (CH), 44.42 (C), 43.48 (CH2), 42.94 (C), 40.77 (C), 36.08 (CH), 35.70 (CH2), 35.00 (CH3), 33.44 (CH2), 32.35 (CH2), 29.40 (CH2), 25.83 (CH2), 25.41(CH3), 23.82 (CH2), 15.61 (CH3), 13.79 (CH3), 10.44 (CH3). HRMS (ESI) calcd. [M + H]+ for C26H38N2O4S 475.2625, found 475.2630.
14-O-(2-oxazolidinone,5-(methyl)-)(thioacetyl)mutilin (3i). A mixing of (R)-5-chloroMethyl-2-oxazolidinone (5) (1 mmol), sodiumiodide (0.1 mmol), and acetone (10 mL) were stirred in room temperature. After 30 min, the reaction solution was filtered and concentrated. Compound 4 (1.1 mmol) and triethylamine (20 mL) was added under N2. The solvent was stirred in 40 °C for 10 h. The mixture was extracted with water (10ml) and HCl (2N, 10 mL). The organic layers were concentrated in vacuo to give crude products. The crude product was purified by silica gel column chromatography. Yield: 62%. IR (KBr): 3422 (O), 2933 (CH2), 1735 (C=O), 1686 (C=O), 1458 (C=C), 1420 (C-C), 1284 (C-O-C), 1151 (C-O), 1117 (C-(C=O)-C) cm−1. 1H-NMR (400 MHz, CDCl3) δ 6.63–6.35 (m, 1H), 6.17 (d, J = 8.2 Hz, 1H), 5.75 (d, J = 8.2 Hz, 1H), 5.27 (dd, J = 51.9, 14.2 Hz, 2H), 4.92–4.67 (m, 1H), 3.82–3.62 (m, 1H), 3.45–3.32 (m, 2H), 3.30–3.07 (m, 2H), 2.99 (ddd, J = 9.9, 8.9, 4.4 Hz, 1H), 2.87 (dt, J = 12.8, 5.2 Hz, 1H), 2.34 (s, 1H), 2.28–2.17 (m, 2H), 2.11 (s, 1H), 2.08 (d, J = 8.6 Hz, 1H), 1.77 (d, J = 14.3 Hz, 1H), 1.65 (d, J = 10.4 Hz, 2H), 1.52 (dd, J = 25.3, 6.7 Hz, 2H), 1.44 (d, J = 1.0 Hz, 4H), 1.39 (s, 1H), 1.35–1.26 (m, 1H), 1.16 (d, J = 14.9 Hz, 4H), 0.89 (d, J = 6.8 Hz, 3H), 0.73 (d, J = 6.8 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 216.99 (C=O), 168.63 (C=O), 159.41 (C=O), 139.16 (CH=), 117.14 (CH2=), 75.66 (CH), 74.61 (C), 69.70 (CH), 58.17 (CH), 45.46 (C), 45.06 (CH2), 44.88 (CH2), 43.95 (C), 41.78 (C), 36.75 (CH), 36.03 (CH), 35.99, 34.81 (CH), 34.45 (CH2), 30.42 (CH2), 26.86 (CH3), 26.41 (CH2), 24.84 (CH2), 16.82 (CH3), 14.88 (CH3), 11.48 (CH3). HRMS (ESI) calcd. [M + Na]+ for C26H39NO6S 516.2395, found 516.2394.
3.2. Biological Evaluation
3.2.1. MIC Testing
The MIC values of 3a–i and tiamulin fumarate against bacteria were determined using the broth dilution method [13]. Stock solutions of compounds were prepared in DMSO. The compounds were added to the test tube and serially diluted in Mueller-Hinton broth (the final concentration is 0.0625 μg/mL). Five bacteria, including S. aureus, MRSA, MRSE, VRE, and one Gram-negative bacterium, E. coli, were cultivated and added to the tube. The initial concentration of bacteria cannot be lower than 105 CFU/mL. The broth was incubated at 36.7 °C for 18–24 h. MICs were read when the change of clarity in the broth was observed in the control test tube.
3.2.2. Bactericidal time-kill kinetics
The two bacterias were prepared in Muellere Hinton broth at 37 °C for 6 h with shaking. The solution of compound 3a and tiamulin fumarate, 1 × MIC and 6 × MIC, were added to the bacterial suspension so that the final concentrations were 106~107 CFU/mL, respectively. After specified time intervals (0, 1, 2, 4, 6, 12, and 24 h), 20 mL aliquots were serially diluted in 0.9% saline, plated on sterile Muellere Hinton agar plates, and incubated at 37 °C for 24 h. The viable colonies were counted and represented as log10 (CFU/mL). The same procedure was repeated in triplicate.
3.3. Molecular Modeling Studies
The 50 s ribosomal of S. aureus in complex with lefamulin (PDB ID: 5HL7) [12] was simulated useHomdock software in the Chil2 package (University of Pittsburgh, Pittsburgh, PA, U.S., version 0.99). The package contains a Graph-based molecular alignment (GMA) tool and a Monte-Carlo/Simulated Annealing (MC/SA) algorithm-based docking (GlamDock) (University of Pittsburghcompany, Pittsburgh, PA, USA, version 0.99) tool. Lefamulin was the template for flexible molecular alignment, and the interaction was optimized by GlamDock according to the ChillScore scoring function based on ChemScore with a smooth, improved potential. All the compounds were prepared with Avogadro software [19], including a 5000 steps Steepest Descent and 1000 steps Conjugate Gradients geometry optimization based on the MMFF94 force field. The docking site was set to base lefamulin. All compounds have compared to original conformation of 5HL7, which was kept for binding affinity comparison. Compounds and receptors were estimated by ChillScore. Hydrogen bonds and other interactions were detected and generated by PyMol 1.5.03 [20,21].
4. Conclusions
In summary, a series of novel pleuromutilin derivatives bearing heterocyclic ring at the C-14 side chain were synthesized. Our results show that a heterocyclic substituent bearing a amine group leads to excellent in vitro antibacterial activity against S. aureus, MRSA, MRSE, and VRE. Compounds that contain pyrimidine rings prepared in this thesis showed moderate to excellent biological activities, and the hydrophilicity of the side chain showed some correlations to its activity. Compound 3a, the most effective compound, showed rapid bactericidal activity against S. aureus and MRSA in time-kill assay. Molecular docking studies also revealed that 3a displayed lower Gibbs free energy. Thus, compound 3a has been selected for further evaluation as a promising candidate for treating bacterial infection.
Supplementary Materials
Samples of Compounds 2, 4, 5, 3a, 3b, 3c, 3d, 3e, 3f, 3g, 3h and 3i are available from Supplementary Materials.
Acknowledgments
This work was financed by Central Agricultural Scientific Research Institutions (No.1610322016007), National Key Technology Support Program (No. 2015BAD11B02) and Agricultural Science and Technology Innovation Program (ASTIP, No. CAASASTIP-2014-LIHPS-04).
Author Contributions
Supervision of the whole work was conducted by Yunpeng Yi; Study conception and experiments were designed by Pengchegn Dong, Yunpeng Yi, Yunxing Fu and Ruofeng Shang; Experiments were conducted by Wenwen Qin; Experimental data were analyzed and interpreted by Yu Liu and Jiangping Liang; Manuscript was prepared by Yunpeng Yi and Ruofeng Shang.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Singh, S.B.; Young, K.; Silver, L.L. What is an “Ideal” Antibiotic? Discovery Challenges and Path forward. Biochem. Pharmacol. 2017, 133, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Ai, X.; Pu, X.; Yi, Y.; Liu, Y.; Xu, S.; Liang, J.; Shang, R. Synthesis and Pharmacological Evaluation of Novel Pleuromutilin Derivatives with Substituted Benzimidazole Moieties. Molecules 2016, 21, 1488. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, F.; Hervey, A.; Robbins, W.J. Antibiotic Substances from Basidiomycetes VIII. Pleurotus Multilus (Fr.) Sacc. and Pleurotus Passeckerianus Pilat. Proc. Natl. Acad. Sci. USA 1951, 37, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Eyal, Z.; Matzov, D.; Krupkin, M.; Paukner, S.; Riedl, R.; Rozenberg, H.; Zimmerman, E.; Bashan, A.; Yonath, A. A novel pleuromutilin antibacterial compound, its binding mode and selectivity mechanism. Sci. Rep. 2016, 6, 39004. [Google Scholar] [CrossRef] [PubMed]
- Novak, R.; Shlaes, D.M. The pleuromutilin antibiotics: A new class for human use. Curr. Opin. Investig. Drugs 2010, 11, 182–191. [Google Scholar] [PubMed]
- Riedl, K. Studies on pleuromutilin and some of its derivatives. J. Antibiot. 1976, 29, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Bacque, E.; Pautrat, F.; Zard, S.Z. A Flexible Strategy for the Divergent Modification of Pleuromutilin. Chem. Commun. 2002, 34, 2312–2313. [Google Scholar] [CrossRef]
- Egger, H.; Reinshagen, H. New pleuromutilin derivatives with enhanced antimicrobial activity. I. Synthesis. Jpn. J. Antibiot. 1976, 29, 915–922. [Google Scholar] [CrossRef]
- Poulsen, S.M.; Karlsson, M.; Johansson, L.B.; Vester, B. The pleuromutilin drugs tiamulin and valnemulin bind to the RNA at the peptidyl transferase centre on the ribosome. Mol. Microbiol. 2001, 41, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Schlünzen, F.; Pyetan, E.; Fucini, P.; Yonath, A.; Harms, J.M. Inhibition of peptide bond formation by pleuromutilins: The structure of the 50S ribosomal subunit from Deinococcus radiodurans in complex with tiamulin. Mol. Microbiol. 2004, 54, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Bashan, A.; Auerbach-Nevo, T.; Yaggie, R.D.; Gontarek, R.R.; Yonath, A. Induced-Fit Tightens Pleuromutilins Binding to Ribosomes and Remote Interactions Enable Their Selectivity. Proc. Natl. Acad. Sci. USA 2007, 104, 4291–4296. [Google Scholar]
- Eyal, Z.; Matzov, D.; Krupkin, M.; Wekselman, I.; Paukner, S.; Zimmerman, E.; Rozenberg, H.; Bashan, A.; Yonath, A. Structural insights into species-specific features of the ribosome from the pathogen Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2015, 112, 5805–5814. [Google Scholar] [CrossRef] [PubMed]
- Thakare, R.; Dasgupta, A.; Chopra, S. LEFAMULIN Pleuromutilin antibacterial, Treatment of pneumonia and ABSSI. Drugs Future 2016, 157–167. [Google Scholar]
- Barry, A.L.; Craig, W.A.; Nadler, H.; Reller, L.B.; Sanders, C.C.; Swenson, J.M. Methods for determining bactericidal activity of antimicrobial agents: Approved guideline. NCCLS Document M26-A 1999, 19. [Google Scholar]
- Klich, K.; Pyta, K.; Kubicka, M.M.; Ruszkowski, P.; Celewicz, L.; Gajecka, M.; Przybylski, P. Synthesis, Antibacterial, and Anticancer Evaluation of Novel Spiramycin-Like Conjugates Containing C(5) Triazole Arm. J. Med. Chem. 2016, 59, 7963–7973. [Google Scholar] [CrossRef] [PubMed]
- Marialke, J.; Tietze, S.; Apostolakis, J. Similarity Based Docking. J. Chem. Inf. Model. 2008, 48, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Domagalska, J.; Janas, A.; Pyta, K.; Pecyna, P.; Ruszkowski, P.; Celewicz, L.; Gajecka, M.; Bartl, F.; Przybylski, P. 16-Membered Macrolide Lactone Derivatives Bearing a Triazole-Functionalized Arm at the Aglycone C13 Position as Antibacterial and Anticancer Agents. ChemMedChem 2016, 11, 1886–1891. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.N. On the specificity of antibiotics targeting the large ribosomal subunit. Ann. N. Y. Acad. Sci. 2011, 1241, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL User’s Manual. Available online: https://www.scienceopen.com/document?vid=602cf44e-e5ab-49bd-bb13-5f4fef98741d (accessed on 24 January 2015).
- The PyMOL Molecular Graphics System. Available online: http://www.mendeley.com/catalog/pymol-molecular-graphics-system-version-11/ (accessed on 24 January 2015).
Sample Availability: Samples of the compounds are available from the authors. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).