Abstract
A series of optically active poly(diphenylacetylene) derivatives bearing a chiral substituent (poly-2S) or chiral and achiral substituents (poly-(2Sx-co-31−x)) on all of their pendant phenyl rings were synthesized by the reaction of poly(bis(4-carboxyphenyl)acetylene) with (S)-1-phenylethylamine ((S)-2) or benzylamine (3) in the presence of a condensing reagent. Their chiroptical properties and chiral recognition abilities as chiral stationary phases (CSPs) for high-performance liquid chromatography (HPLC) were investigated. Poly-2S and poly-(2Sx-co-31−x) (0.06 < x < 0.71) formed a preferred-handed helical conformation with opposite helical senses after thermal annealing despite possessing the same chiral pendant (h-poly-2S and h-poly-(2Sx-co-31−x)). Furthermore, h-poly-2S and h-poly-(2S0.36-co-30.64) emitted circularly polarized luminescence with opposite signs. h-Poly-2S showed higher chiral recognition abilities toward a larger number of racemates than poly-2S without a preferred-handed helicity and the previously reported preferred-handed poly(diphenylacetylene) derivative bearing the same chiral substituent on half of its pendant phenyl rings. h-Poly-(2S0.36-co-30.64) also exhibited good chiral recognition abilities toward several racemates, though the elution order of some enantiomers was reversed compared with h-poly-2S.
1. Introduction
It is well known that a pair of enantiomers often show significant differences in their physiological activities. The development of efficient techniques for the separation of enantiomers is therefore important in several fields, especially in the pharmaceutical industry. The direct separation of enantiomers by high-performance liquid chromatography (HPLC) using a chiral stationary phase (CSP) is currently one of the most popular and effective methods for both analyzing the composition of enantiomeric mixtures and purifying such mixtures to give pure enantiomers [1,2,3,4,5,6]. However, the success of this method is entirely dependent on the development of effective CSPs with excellent resolving abilities. A large number of optically active small molecules [7,8,9,10,11] and polymers [1,2,3,12,13,14,15] have been evaluated to date as CSPs for HPLC. However, the development of novel CSPs with excellent resolving abilities is still highly desired because there are still several chiral compounds that cannot be resolved using existing commercially available CSPs.
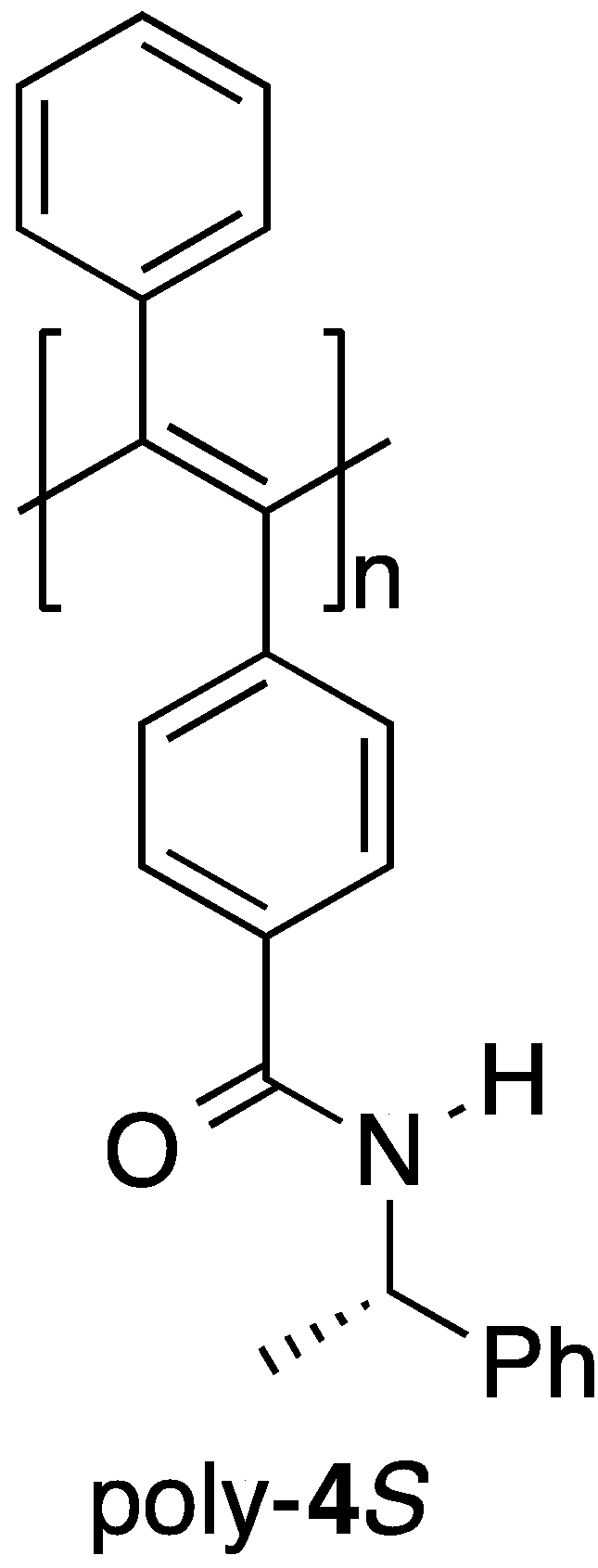
Optically active poly(phenylacetylene)s with a preferred-handed helicity have been prepared [16] and some of these systems have been reported to exhibit good chiral recognition abilities as CSPs for HPLC because of their preferred-handed helical conformation [17,18,19,20,21,22,23,24,25,26]. However, very few optically active poly(diphenylacetylene)s have been prepared to date, which has limited research towards evaluating the scope and efficiency of the chiral recognition abilities of these materials [27,28,29,30,31,32,33,34,35]. We recently reported the first example of an optically active poly(diphenylacetylene)-based CSP for HPLC [36]. In this particular case, we synthesized an optically active poly(diphenylacetylene) bearing a chiral pendant amide moiety (poly-4S) (Figure 1) by the reaction of an optically inactive precursor polymer bearing a pendant carboxyl group with the optically active amine (S)-1-phenylethylamine (S)-2. The results revealed that this polymer showed good chiral recognition ability towards various racemates when it was used as a CSP for HPLC and that its chiral recognition ability was significantly dependent on its preferred-handed helical conformation. Notably, the preferred-handed helical conformation of the polymer was induced by the thermal annealing process (h-poly-4S), which was applied after the introduction of the optically active pendants via a polymer reaction. The chiral recognition abilities of h-poly-4S with a preferred-handed helicity were greater than those of poly-4S without a preferred-handed helicity. This result indicated that the macromolecular helicity induced in the polymer backbone by thermal annealing as a consequence of the effect of the chiral pendant groups was playing an important role in the high chiral recognition ability of this material. This finding therefore inspired us to synthesize several other optically active poly(diphenylacetylene)s with the aim of developing more practically useful poly(diphenylacetylene)-based CSPs for HPLC. Although poly(diphenylacetylene)s have pendant phenyl rings on all of the carbons in their main chain, only half of the pendent phenyl rings in poly-4S feature an amide functional group, which could act as effective chiral recognition sites. In this study, we designed and synthesized several novel optically active poly(diphenylacetylene)s bearing chiral or achiral substituents, which were connected to the pendant phenyl rings of the polymer through an amide bond (poly-2S and poly-(2Sx-co-31–x)). We also investigated the chiroptical properties and chiral recognition abilities of these materials as CSPs for HPLC.
Figure 1.
The structure of poly-4S.
2. Results and Discussion
The synthetic route used to prepare poly-2S and poly-(2Sx-co-31–x) is shown in Scheme 1. The direct polymerization of diphenylacetylene monomers bearing polar functional groups, such as carboxyl and amide groups, hardly proceeds because the group 5 transition metals used for polymerization of disubstituted acetylenes are intolerant to such polar functional groups [37,38]. With this in mind, the carboxyl groups of the diphenylacetylene monomer (1) were protected as the corresponding n-heptyl esters prior to being polymerized in toluene with WCl6–Ph4Sn at 100 °C. Notably, these conditions have been reported to be effective for the polymerization of diphenylacetylenes bearing ester groups [32,39]. It was also envisaged that the long alkyl (n-heptyl) chains of the ester groups would enhance the solubility of the resulting polymer in toluene. The polymerization reaction of 1 proceeded smoothly to give poly-1 with a moderate molecular weight (Mn = 1.3 × 104, Mw/Mn = 1.6) in high yield (77%). The ester groups in poly-1 were then hydrolyzed under alkaline conditions to give poly-1-H. The complete removal of the n-heptyl esters was confirmed by 1H-NMR and IR spectroscopy (Figures S1 and S2). Poly-2S was prepared in a similar manner to that previously reported for poly-4S [36], by the polymer reaction of poly-1-H with (S)-2 using 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMT-MM) as a condensing reagent. The near complete introduction of the (S)-2 residue was confirmed by 1H NMR, IR and elemental analyses (Figures S1 and S3). Poly-(2Sx-co-31–x)s were synthesized by the two-step polymer reaction of poly-1-H with (S)-2, followed by benzylamine (3) using DMT-MM as a condensing reagent. Poly-1-H was initially reacted with (S)-2 in the presence of a controlled amount of DMT-MM to allow for the introduction of a small number of (S)-2 units as the pendant groups. The resulting material was then reacted with an excess of 3 in the presence of an excess of DMT-MM. The (S)-2 unit contents (x) of the resulting poly-(2Sx-co-31–x)s were determined by 1H-NMR analysis (see Figure S3). Poly-(2Sx-co-31–x)s containing different amounts of the (S)-2 units were obtained by changing the feed ratio of DMT-MM relative to poly-1-H whilst maintaining that of (S)-2 relative to DMT-MM ([(S)-2]/[DMT-MM] = 2) (Table S1). The (S)-2 unit contents of the resulting polymers were found to correspond to approximately one-third of an equivalent of the DMT-MM feed in the first step.
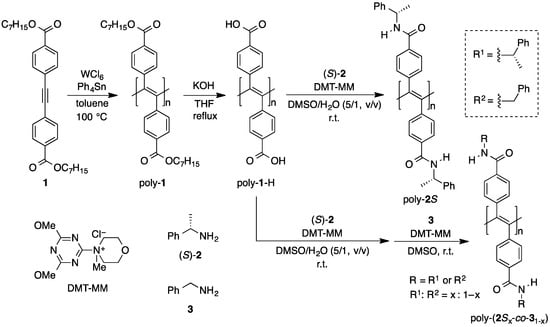
Scheme 1.
Synthesis of poly-2S and poly-(2Sx-co-31−x).
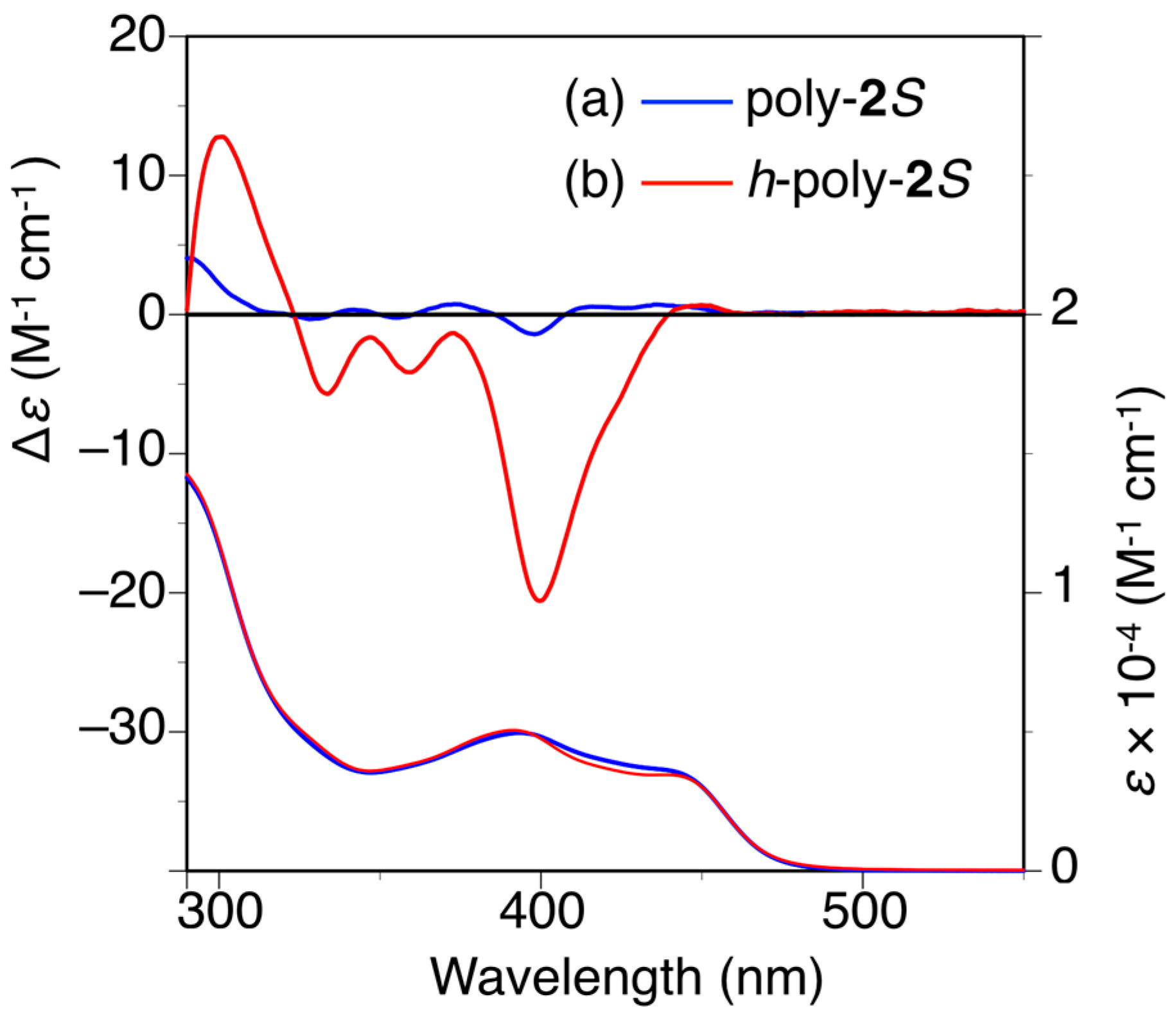
The circular dichroism (CD) spectrum of poly-2S showed almost no circular dichroism (CD) in the wavelength region longer than 300 nm ascribed to the absorption of the polymer backbone in N,N-dimethylformamide (DMF) immediately after the preparation of the sample (Figure 2a) [27,32,33,34,35]. However, an apparent Cotton effect was observed around 400 nm after one day. Furthermore, the CD intensity very slowly increased with time in DMF at 25 °C, but it did not reach a constant value even after 8 days (Figure S4a). When the solution was annealed at higher temperatures, the CD intensity rapidly increased to reach a plateau (Δε400 = ca. −21) after 5 h at 90 °C or 2 h at 120 °C, which was accompanied by a negligible change in the absorption spectra (Figure 2b and Figures S4b and S5). These results indicated that a predominantly one-handed helical conformation was induced in the polymer backbone by thermal annealing owing to the effect of the optically active pendants (h-poly-2S). A similar thermal annealing was necessary for poly-4S to form the preferred-handed helical conformation (h-poly-4S) [27,36]. This result was attributed to steric hindrance between the neighboring pendant phenyl rings, which was large enough to prevent poly-2S and poly-4S from being transformed into their preferred-handed helical conformations after the introduction of the optically active pendants.
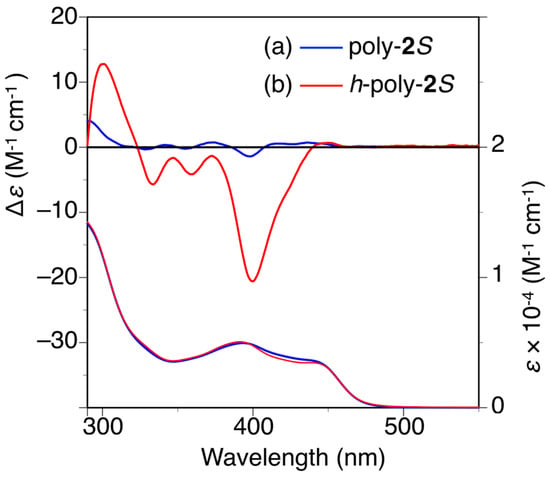
Figure 2.
CD and absorption spectra of poly-2S (a) and h-poly-2S (b) in N,N-dimethylformamide (DMF) at 25 °C. h-Poly-2S was obtained by annealing poly-2S in DMF at 120 °C for 2 h.
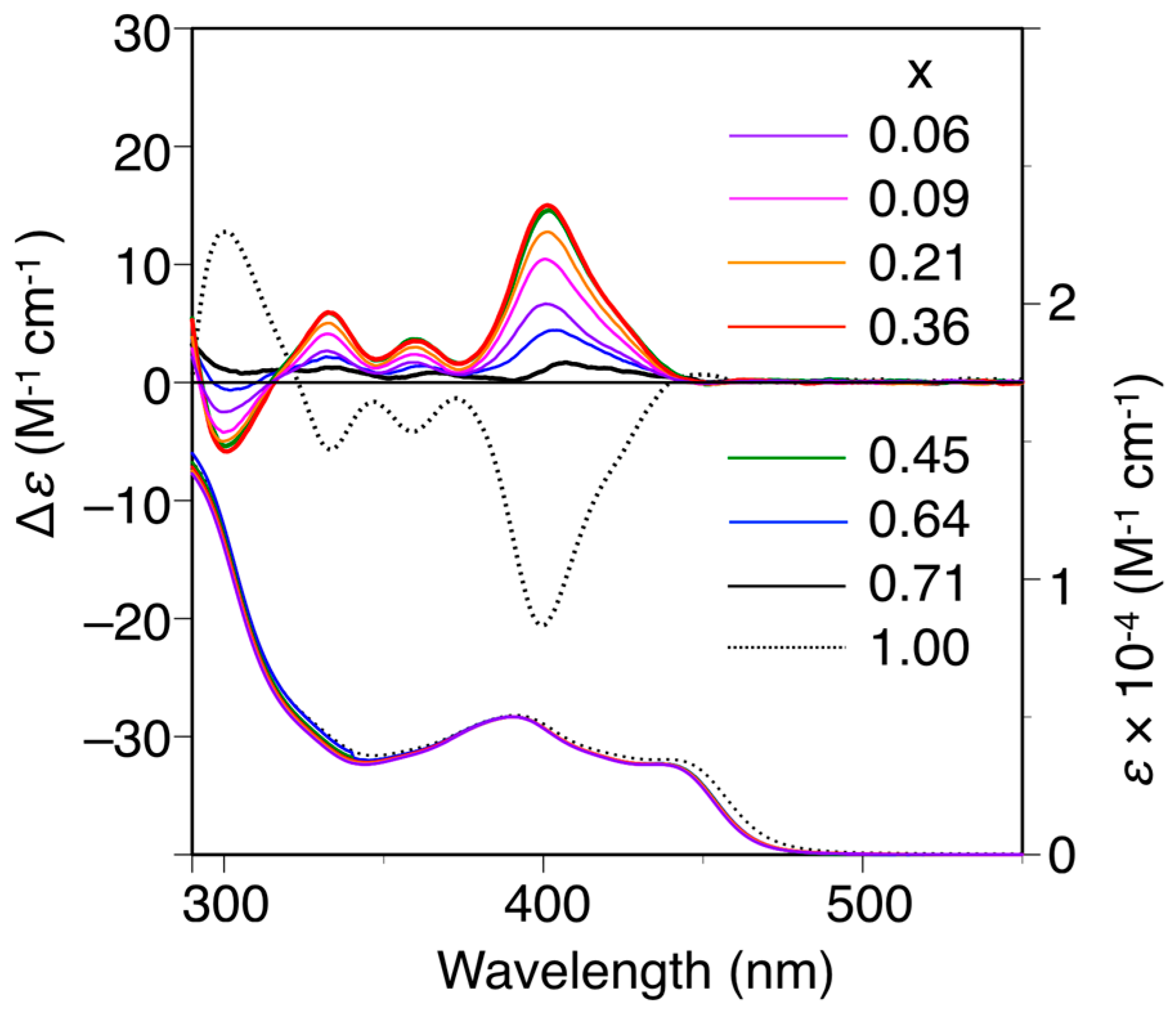
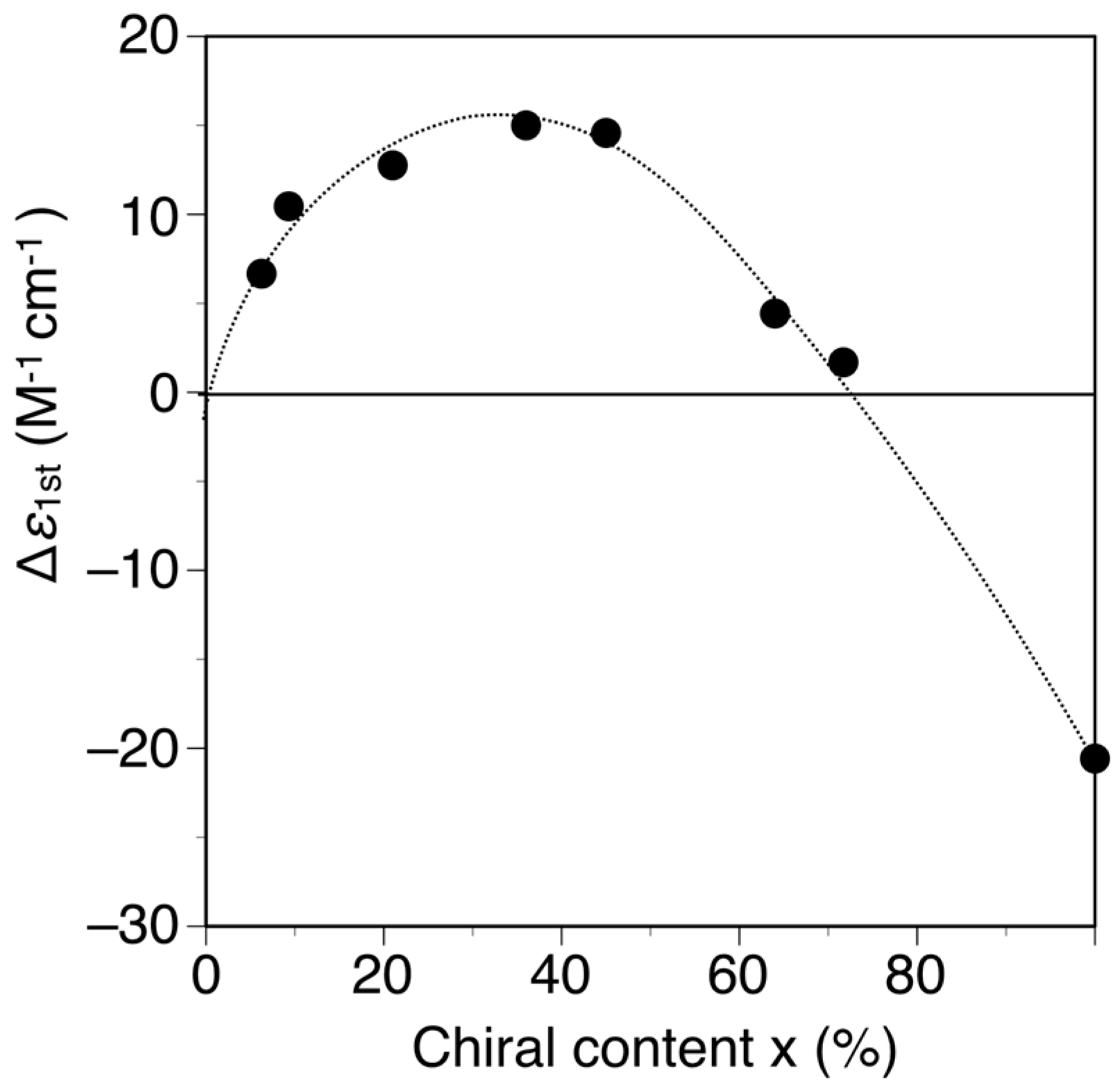
The poly-(2Sx-co-31−x)s were converted to the corresponding h-poly-(2Sx-co-31−x)s with predominantly one-handed helical conformations in the same way by annealing the corresponding DMF solutions at 120 °C for 2 h. Figure 3 shows the CD and absorption spectra of the h-poly-(2Sx-co-31–x)s in DMF at 25 °C together with those of h-poly-2S. The CD intensities of these polymers (Δε of the first Cotton effect) were plotted against their chiral (S)-2 unit contents, as shown in Figure 4. Interestingly, the Cotton effects of the h-poly-(2Sx-co-31–x)s gave the opposite sign to that of h-poly-2S when the (S)-2 content was below ca. 71 mol %. Furthermore, the CD spectrum of h-poly-(2S0.36-co-30.64) was almost a mirror image of that of h-poly-2S accompanied with a negligible change in the absorption. These results indicated that the poly-(2Sx-co-31–x)s (x < 0.71) existed in a predominantly one-handed helical conformation with an opposite helical-sense to that of h-poly-2S. This system therefore allowed for the selective formation of right- and left-handed helical structures using a single enantiomer of 2 as the chiral unit, with the actual outcome being dependent on the mole fraction of 2. Similar helical sense inversion behaviors in the copolymers consisting of chiral and achiral units depending on the contents of the chiral units have been also reported in polyisocyanates [40], polysilanes [41], polyacetylenes [42,43,44], polyisocyanides [45] and poly(quinoxaline-2,3-diyl)s [46]. It has also been reported that these phenomena can be explained using a modified version of the Ising model proposed by Sato et al., where the helix-sense is determined by the interactions between the neighboring pendant groups, chiral–chiral units and chiral–achiral units, which can stabilize the opposite helix-sense to each other [47].
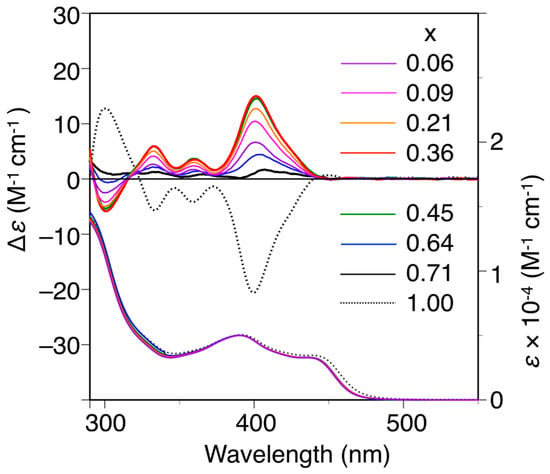
Figure 3.
CD and absorption spectra of poly-(2Sx-co-31–x) and h-poly-2S in DMF at 25 °C. h-poly-(2Sx-co-31–x) and h-poly-2 were obtained by annealing poly-(2Sx-co-31–x) and poly-2S, respectively, in DMF at 120 °C for 2 h.
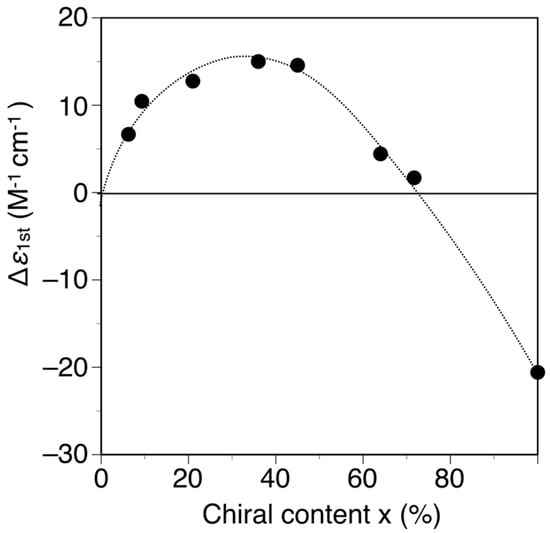
Figure 4.
Plots of the Δε1st values of h-poly-(2Sx-co-31–x) and h-poly-2S in DMF at 25 °C vs. their chiral (S)-2 unit contents (x).
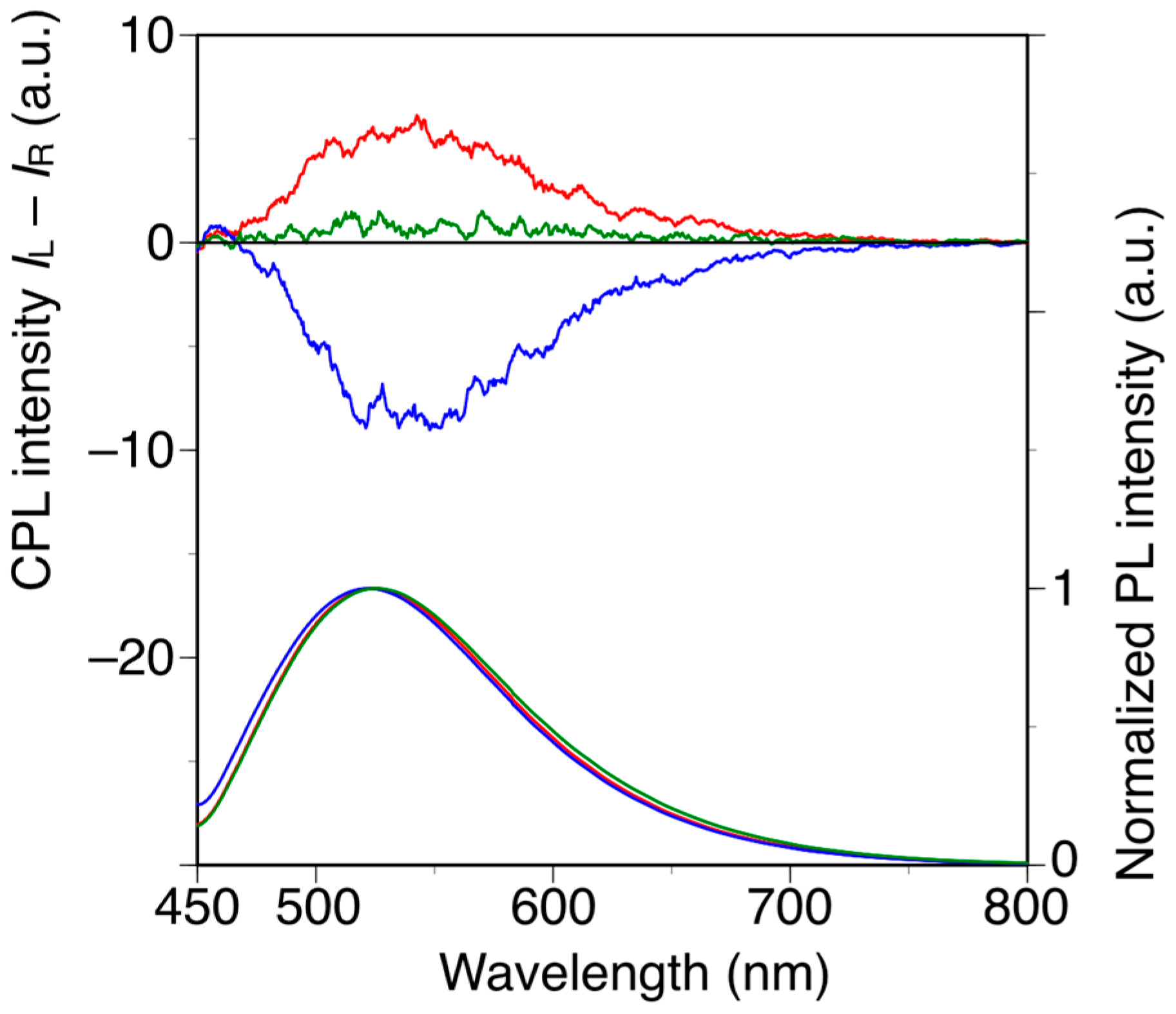
Poly(diphenylacetylene)s are highly emissive without the introduction of any other fluorescent substituents, whereas poly(phenylacetylene)s are generally non-emissive materials [38,48]. With this in mind, we investigated the relationship between the main-chain conformation and the circularly polarized luminescence (CPL) properties of h-poly-2S and h-poly-(2S0.36-co-30.64). The CPL and photoluminescence (PL) spectra of h-poly-2S, h-poly-(2S0.36-co-30.64) and poly-(2S0.36-co-30.64) in DMF are shown in Figure 5. Photographic images of these solutions under irradiation at 365 nm are shown in Figure S6. h-Poly-2S and h-poly-(2S0.36-co-30.64) showed equal and opposite CPL bands around 520 nm, which were consistent with the corresponding PL bands. In contrast, no apparent CPL signal was observed for poly-(2S0.36-co-30.64). The signs of the CPL signals at 520 nm were identical to those of the CD band around 400 nm and the values of the emission dissymmetry factor (glum), which is defined as glum = 2(IL − IR)/(IL + IR), where IL and IR are the intensities of the left- and right-handed circularly polarized light, respectively, were estimated to be 1.2 × 10−3 for h-poly-2S and 0.8 × 10−3 for h-poly-(2S0.36-co-30.64). These results indicated that h-poly-2S and h-poly-(2S0.36-co-30.64) can emit opposite CPL originating from the preferred-handed helical conformation with opposite helical sense to each other despite having the same central chirality in their pendants.
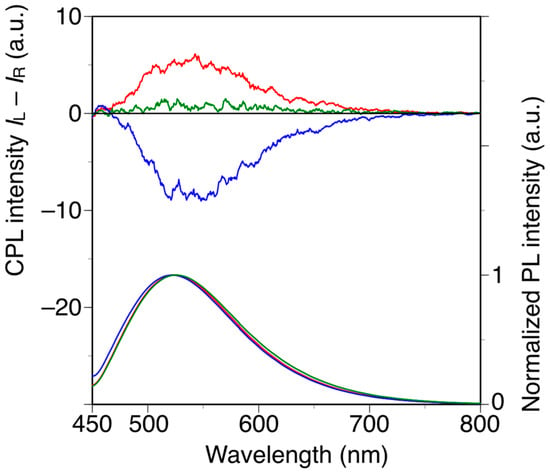
Figure 5.
CPL and PL spectra of h-poly-2 (blue line), h-poly-(2S0.36-co-30.64) (red line) and poly-(2S0.36-co-30.64) (green line) in DMF at 25 °C with excitation at 388 nm. [polymer] = 1.0 × 10−5 M.
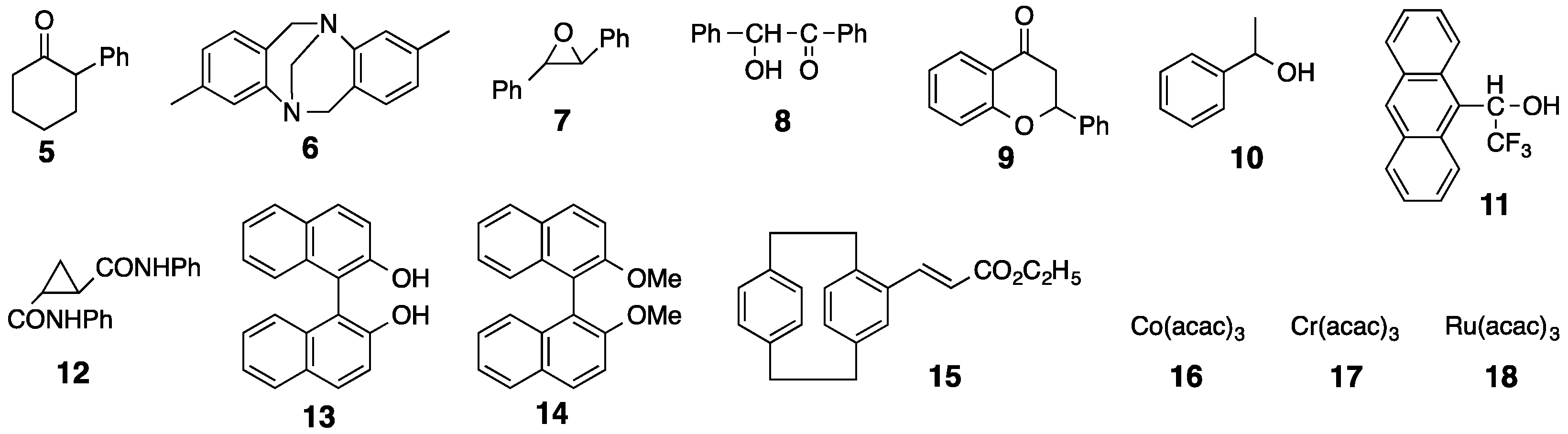
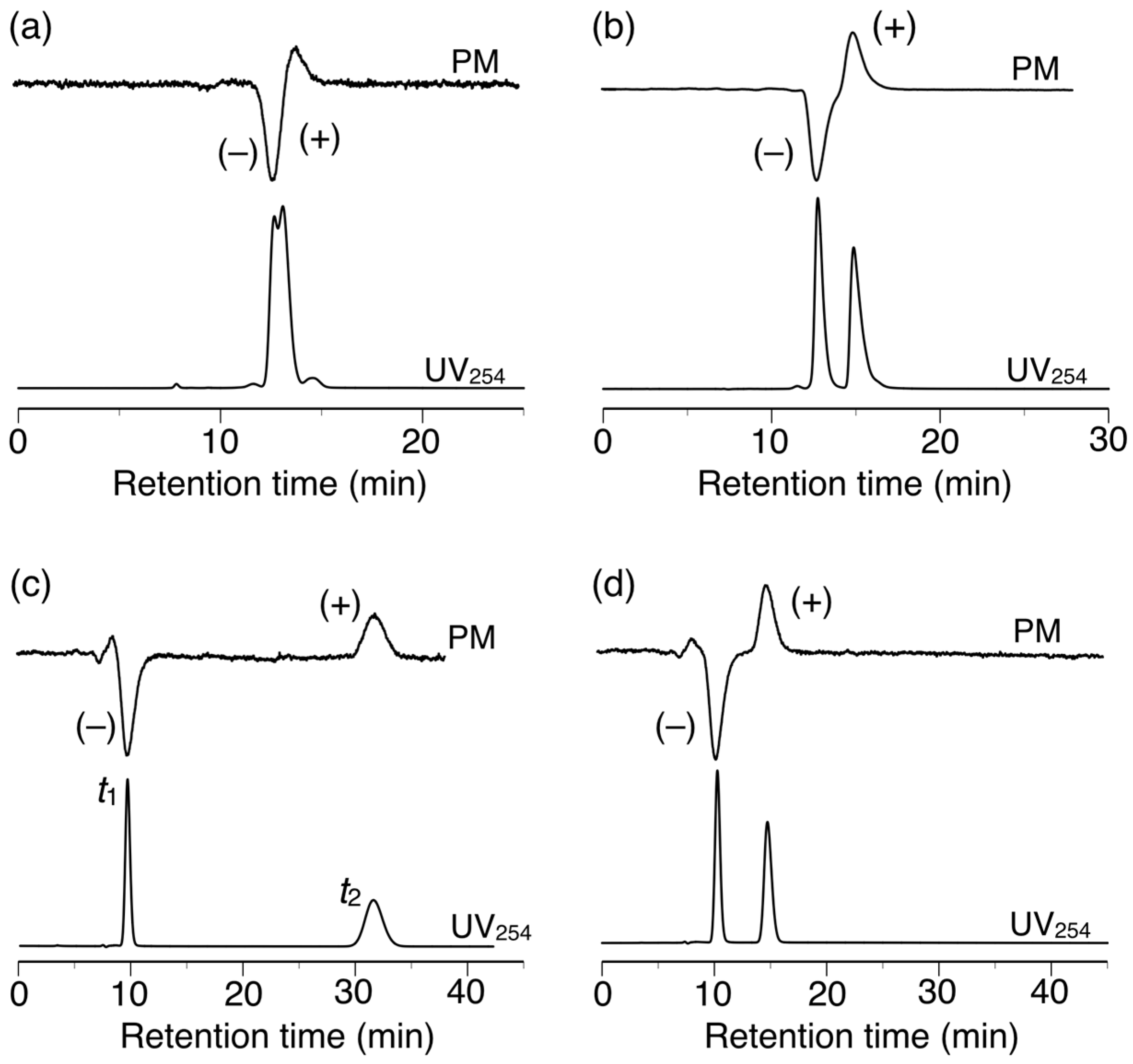
The chiral recognition abilities of poly-2S, h-poly-2S and h-poly-(2S0.36-co-30.64) as CSPs for HPLC were evaluated using the various racemic compounds with different functional groups (5–12) including axial (13, 14) or planar (15) chiral compounds as well as chiral metal complexes (16–18) (Figure 6). The packing materials were prepared by coating macroporous silica gel (particle size 7 μm, pore size 100 nm) with DMF solutions of the corresponding polymers [49]. The obtained packing materials were subsequently packed into a stainless steel column (25 × 0.20 cm (i.d.)) using a conventional slurry method [50]. The chromatographic resolution results are summarized in Table 1 and the chromatograms obtained for the resolutions of racemic 15 and 16 on poly-2S- and h-poly-2S-based CSPs are shown in Figure 7. Ultraviolet (UV) and polarimetric (PM) detectors were used for the detection and identification of the peaks, respectively. As shown in Figure 7c, the different enantiomers of 16 were eluted with retention times of t1 ((–)-enantiomer) and t2 ((+)-enantiomer), which showed a complete baseline separation on the poly-2S-based CSP. The retention factor k1 (k1 = (t1 − t0)/t0) for the first eluted enantiomer, the separation factor α (α = (t2 − t0)/(t1 − t0)) and the resolution factor RS (RS = 2(t2 − t1)/(w1 + w2)), where t0 is the hold-up time and w1 and w2 are the band widths of the first- and second-eluted enantiomers on the base line, were determined to be 0.14, 19.5 and 10.2, respectively.
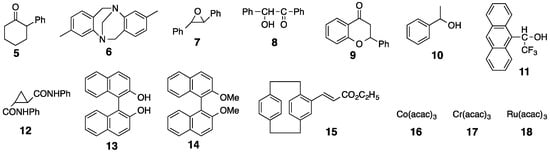
Figure 6.
Structures of racemates 5–18.

Table 1.
Resolution results for racemates 5–18 on poly-2S, h-poly-2S, h-poly-(2S0.36-co-30.64) and h-poly-4S a.
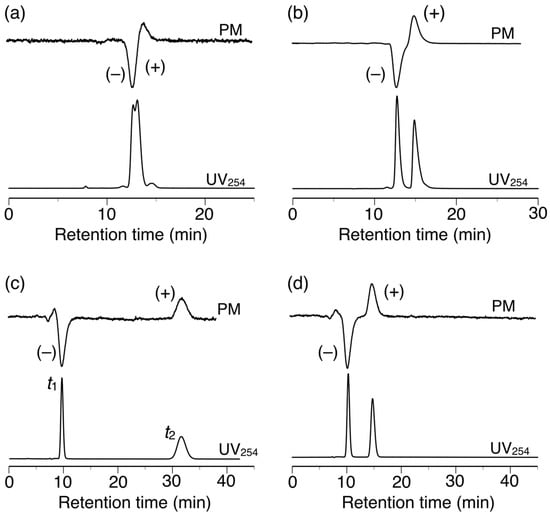
Figure 7.
HPLC chromatograms for the resolution of 15 (a,b) and 16 (c,d) on poly-2S (a,c) and h-poly-2S (b,d). Eluent: hexane–2-propanol (99:1, v/v) (a,b), MeOH–H2O (90:10, v/v).
Poly-2S partially or completely resolved nine of the 14 racemates tested in the current study. Notably, poly-2S exhibited excellent chiral recognition abilities toward chiral metal acetylacetonate complexes 16–18, showing complete baseline separations in all three cases, together with very high α values (α > 15) (Figure 7c). Methanol/H2O (90:10, v/v) was used as the eluent because the enantiomers of 16–18 were not eluted with hexane-2-propanol.
h-Poly-2S showed higher chiral recognition ability than poly-2S toward seven racemates. For example, h-poly-2S resolved 15 with complete baseline separation (Figure 7b), whereas poly-2S only achieved a partial separation of the same compound (Figure 7a). Moreover, h-poly-2S resolved racemates 8 and 9, which were barely separated on poly-2S. These results therefore indicated that the preferred-handed helical conformation induced in h-poly-2S was playing an important role in its excellent chiral recognition ability, which was consistent with the results of our previous report for h-poly-4S [36]. As expected, poly-2S and h-poly-2S both exhibited higher enantioselectivities toward nine of the racemates than our previously reported poly-4S and h-poly-4S systems, respectively, bearing the same (S)-2 unit on half of their phenyl pendants [36]. This result could be attributed to an increase in the number of the amide pendants acting as effective chiral recognition sites compared with the previous systems. In our previous study, we also observed an interesting inversion in the order of the enantioseparation of the chiral metal complexes 16–18 on the poly-4S- and h-poly-4S-based CSPs, despite the fact that they had the same optically active groups of their pendant moieties [36]. This phenomenon can be explained by the opposite enantioselectivity between the central chirality in the optically active pendants and the induced macromolecular helicity of the polymer backbone toward these racemates, which would represent a negative synergetic effect. In contrast, h-poly-2S and poly-2S completely resolved racemates 16–18 with good enantioselectivities (α = 2.9–3.6), but the elution order was the same on both CSPs (Figure 7c,d). The effect of the induced macromolecular helicity of h-poly-2S on the separation of these racemates therefore appeared to have been cancelled out by the central chirality of the pendants, because h-poly-2S had twice as many chiral pendants as h-poly-4S.
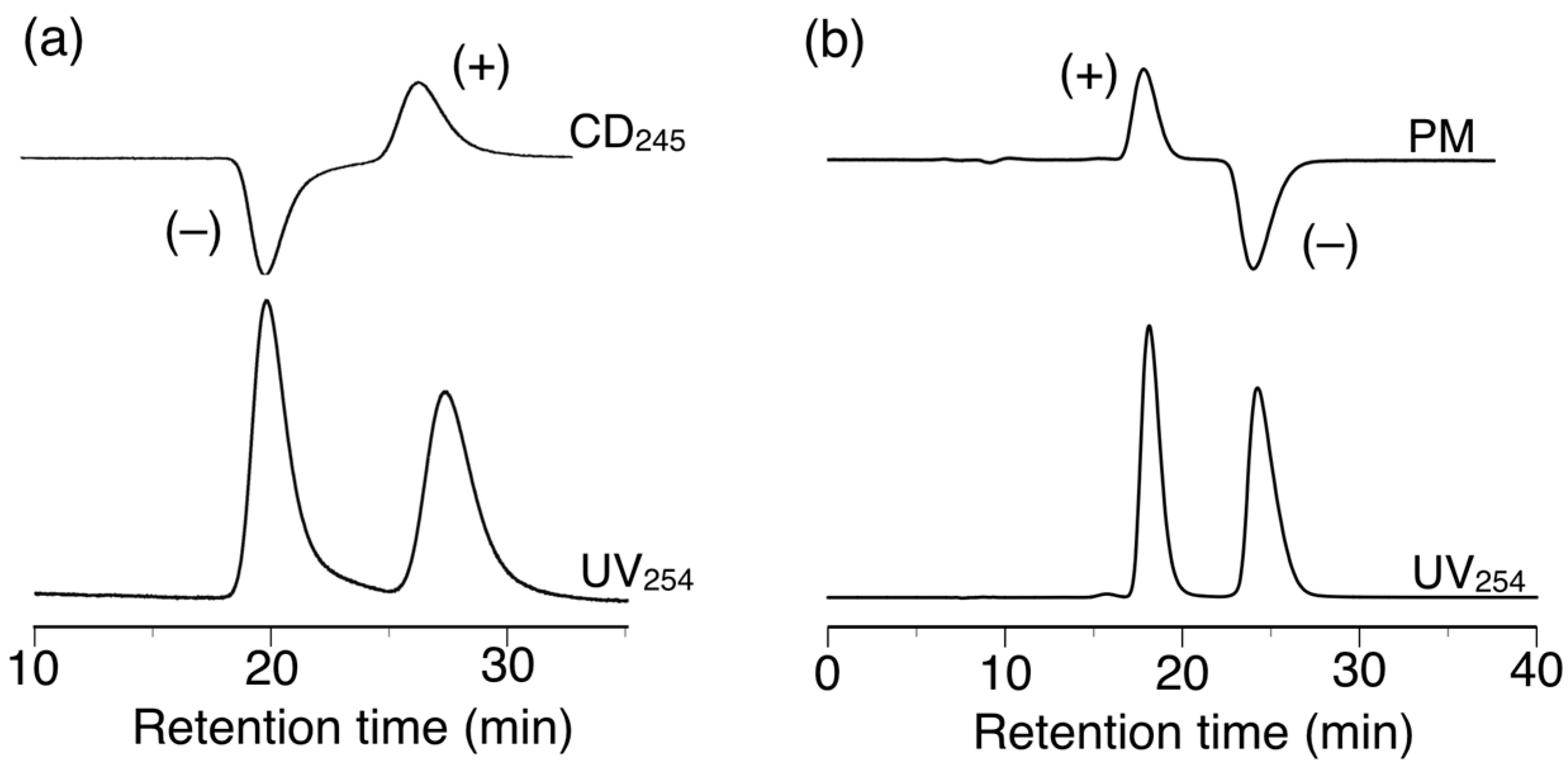
h-Poly-(2S0.36-co-30.64) exhibited different chiral recognition properties to h-poly-2S and successfully resolved 11 of the 14 racemates evaluated in the current study. It is noteworthy that h-poly-(2S0.36-co-30.64) resolved racemates 6 and 14, which were not resolved by poly-2S or h-poly-2S (Table 1 and Figure 8a). Among the enantiomers separated using h-poly-(2S0.36-co-30.64), the elution orders of six of the racemates (5–7, 9, 13 and 15) were the opposite of those achieved on h-poly-2S. In particular, the complete baseline separation of the racemic cyclophane derivative 15 with a planar chirality was achieved on h-poly-2S and h-poly-(2S0.36-co-30.64) with a completely reversed elution order; with the (–)-enantiomer eluting prior to the (+)-enantiomer on h-poly-2S (α = 1.38) (Figure 7b) and vice versa on h-poly-(2S0.36-co-30.64) (α = 1.56) (Figure 8b). These results clearly suggested that the macromolecular helicity induced in these polymers was making a major contribution to the resolution of these racemates.
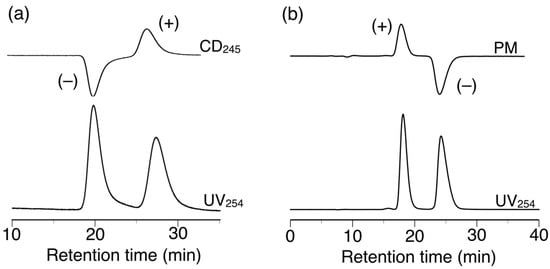
Figure 8.
HPLC chromatograms for the resolution of 14 (a) and 15 (b) on h-poly-(2S0.36-co-30.64). Eluent: hexane–2-propanol (99:1, v/v).
3. Materials and Methods
3.1. Reagents and Materials
Copper(I) iodide (CuI) and tungsten(VI) chloride (WCl6) were purchased from Kanto Kagaku (Tokyo, Japan) and Aldrich (Milwaukee, WI, USA), respectively. Bis(triphenylphosphine)palladium(II) dichloride (Pd(PPh3)2Cl2) and tetraphenyltin (Ph4Sn) were purchased from Tokyo Kasei (TCI, Tokyo, Japan). Triphenylphosphine (PPh3) and 3 were purchased from Wako Chemicals (Osaka, Japan). Porous spherical silica gel with a mean particle size of 7 μm and a mean pore diameter of 100 nm (Daiso gel SP-1000-7), trimethylsilylacetylene and (S)-2 were kindly provided by Daiso Chemical (Osaka, Japan), Shin-Etsu Chemical (Tokyo, Japan) and Yamakawa Chemical (Tokyo, Japan), respectively. Anhydrous tetrahydrofuran (THF), toluene, dichloromethane, N,N-dimethylformamide (DMF) and pyridine were obtained from Kanto Kagaku. Anhydrous dimethyl sulfoxide (DMSO) was purchased from Aldrich. Triethylamine (TEA) was dried over KOH pellets and distilled onto KOH under nitrogen. These solvents were subsequently stored under nitrogen before being used. Heptyl 4-iodobenzoate [36] and 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMT-MM) [51] were prepared according to the previously reported methods.
3.2. Instruments
Melting points were measured on a Yanako melting point apparatus (Yanako, Kyoto, Japan) and reported as the uncorrected values. NMR spectra were recorded on a JNM-ECA 500 (JEOL, Tokyo, Japan) (500 MHz for 1H, 125 MHz for 13C) spectrometer using CDCl3, DMSO-d6 or DMSO-d6-D2O as the solvent. TMS was used as an internal reference for the samples run in CDCl3 (1H and 13C), whereas the residual solvent peaks were used as internal reference peaks for the samples run in DMSO-d6 and DMSO-d6-D2O (1H and 13C). IR spectra were recorded with a JASCO Fourier Transform IR-460 spectrophotometer (JASCO, Hachioji, Japan). Absorption and CD spectra were measured in a 1.0 mm quartz cell on a JASCO V-650 spectrophotometer and a JASCO J-725 spectropolarimeter, respectively. The temperature was controlled with a JASCO PTC-348WI apparatus. The polymer concentration was calculated based on the monomer units. The CPL and PL spectra were recorded on a JASCO CPL-300 spectrometer and a JASCO FP-6300 spectrofluorometer, respectively, at room temperature. Size exclusion chromatography (SEC) measurements were performed on a JASCO PU-2080 liquid chromatograph equipped with a UV-vis (JASCO UV-970) detector at 40 °C using a Shodex (Tokyo, Japan) KF-805F SEC column. The temperature was controlled with a JASCO CO-1560 column oven. THF was used as the eluent at a flow rate of 1.0 mL/min. Molecular weight calibration curves were generated using polystyrene standards (Tosoh, Tokyo, Japan). The enantiomers were separated by chromatography using a JASCO PU-2080 liquid chromatograph equipped with a multiwavelength detector (JASCO MD-2018) and a polarimetric detector (JASCO OR-990, Hg without filter) or a CD detector (JASCO CD-2095) at ambient temperature. Elemental analyses were performed at the Research Institute for Instrumental Analysis of Advanced Science Research Center, Kanazawa University, Kanazawa, Japan.
3.3. Synthesis of Monomer
Bis(4-heptyloxycarbonylphenyl)acetylene (1) was prepared according to the route shown in Scheme S1.
Synthesis of heptyl 4-((trimethylsilyl)ethynyl)benzoate: To a solution of heptyl 4-iodobenzoate (10.1 g, 29.3 mmol) in anhydrous TEA (50 mL) was added trimethylsilylacetylene (4.30 mL, 31.7 mmol), Ph3P (126 mg, 0.480 mmol), CuI (135 mg, 0.192 mmol) and Pd(PPh3)2Cl2 (81.8 mg, 0.117 mmol), and the resulting solution was stirred at room temperature for 20 h. The reaction mixture was filtered through Celite, and the filtrate was evaporated to dryness to give a residue, which was purified by column chromatography over silica gel eluting with a 1:30 (v/v) mixture of ethyl acetate and hexane to give the desired product as a yellow oil (9.23 g, 98% yield). 1H-NMR (500 MHz, CDCl3, r.t.): δ 7.96 (d, J = 8.0 Hz, 2H, Ar–H), 7.51 (d, J = 8.0 Hz, 2H, Ar–H), 4.30 (t, J = 6.6 Hz, 2H, OCH2CH2), 1.76 (quint, J = 6.6 Hz, 2H, OCH2CH2), 1.25–1.46 (m, 8H, 4CH2), 0.89 (t, J = 6.6 Hz, 3H, CH3), 0.26 (s, 9H, TMS).
Synthesis of heptyl 4-ethynylbenzoate: To a solution of heptyl 4-((trimethylsilyl)ethynyl)benzoate (5.96 g, 18.8 mmol) in a 3:1 (v/v) mixture of THF and methanol (360 mL) was added K2CO3 (12.9 g, 93.6 mmol), and the resulting mixture was stirred at room temperature for 1 h. The suspension was then filtered and the filtrate was concentrated under reduced pressure. The concentrated solution was diluted with ethyl acetate and washed sequentially with aqueous 1 N HCl and saturated NaHCO3 solution, before being dried over Na2SO4. The solvent was removed by evaporation to give the crude product as a residue, which was purified by column chromatography over silica gel eluting with a 1:30 (v/v) mixture of ethyl acetate and hexane to give the desired product as a colorless oil (3.56 g, 77% yield). 1H-NMR (500 MHz, CDCl3, r.t.): δ 7.99 (d, J = 8.6 Hz, 2H, Ar–H), 7.55 (d, J = 8.0 Hz, 2H, Ar–H), 4.31 (t, J = 6.6 Hz, 2H, OCH2CH2), 3.22 (s, 1H, CH), 1.76 (quint, J = 6.6 Hz, 2H, OCH2CH2), 1.26–1.46 (m, 8H, 4CH2), 0.89 (t, J = 6.9 Hz, 3H, CH3).
Synthesis of bis(4-heptyloxycarbonylphenyl)acetylene (1): To a solution of heptyl 4-iodobenzoate (5.03 g, 14.5 mmol) in anhydrous TEA (25 mL) was added Ph3P (57.1 mg, 0.218 mmol), CuI (63.6 mg, 0.334 mmol), Pd(PPh3)2Cl2 (40.8 mg, 0.0581 mmol) and heptyl 4-ethynylbenzoate (3.55 g, 14.5 mmol), and the resulting mixture was stirred at room temperature for 20 h. The reaction mixture was then filtered through Celite and the filtrate was concentrated under reduced pressure to give a residue, which was diluted with ethyl acetate and washed sequentially with water and brine, before being dried over Na2SO4. The solvent was removed under reduced pressure to give a residue, which was purified by column chromatography over silica gel eluting with a 1:25 (v/v) mixture of ethyl acetate and hexane to give the crude product. The crude product was subsequently purified by recrystallization from a mixture of toluene and methanol to give the desired product as a white solid (5.11 g, 77% yield). M.p.: 61.3–62.4 °C. IR (KBr, cm−1): 1943 (νC≡C), 1707 (νC=O). 1H-NMR (500 MHz, CDCl3, r.t.): δ 8.04 (d, J = 8.6 Hz, 4H, Ar–H), 7.60 (d, J = 8.0 Hz, 4H, Ar–H), 4.33 (t, J = 6.6 Hz, 4H, 2OCH2CH2), 1.76 (quint, J = 6.6 Hz, 4H, 2OCH2CH2), 1.25–1.47 (m, 16H, 8CH2), 0.90 (t, J = 6.9 Hz, 6H, 2CH3). 13C-NMR (125 MHz, CDCl3, r.t.): δ 166.19, 131.76, 130.46, 129.68, 127.38, 91.49, 65.56, 31.88, 29.11, 28.85, 26.15, 22.75, 14.23. Elemental analysis: Calcd. for C30H38O4: C, 77.89; H, 8.28. Found: C, 77.60; H, 8.37.
3.4. Polymerization
The polymerization reaction was carried out in a Schlenk flask under argon using a WCl6-Ph4Sn catalyst system according to the literature method [32,36]. Monomer 1 (1.05 g, 2.27 mmol), WCl6 (90 mg, 0.23 mmol) and Ph4Sn (98 mg, 0.23 mmol) were placed in a Schlenk flask under argon, followed by anhydrous toluene (4.5 mL), which was added using a syringe. The resulting mixture was heated at 100 °C under stirring for 24 h. The reaction mixture was then cooled to room temperature and treated with a large amount of methanol to precipitate the polymer, which was collected by centrifugation and then dried in vacuo at room temperature overnight. The dried polymer was purified by reprecipitation from toluene using a 3:1 (v/v) mixture of methanol and THF as an antisolvent system, and the resulting solid was dried in vacuo at room temperature overnight to give poly-1-Hep (0.81 g, 77% yield). The molecular weight (Mn) of poly-1-Hep was estimated to be 1.3 × 104 (Mw/Mn = 1.6) by SEC using polystyrene standards with THF as the eluent. IR (KBr, cm−1): 1721 (νC=O of ester). 1H-NMR (500 MHz, CDCl3, 25 °C): δ 7.16–7.28 (br, 4H, Ar–H), 6.41–6.71 (br, 2H, Ar–H), 5.92–6.15 (br, 2H, Ar–H), 4.03–4.48 (br, 4H, 2OCH2CH2), 1.60–1.93 (br, 4H, 2OCH2CH2), 1.25–1.47 (br, 16H, 8CH2), 0.79–1.04 (br, 6H, 2CH3). 13C-NMR (125 MHz, CDCl3, 25 °C): δ 165.60, 146.48, 146.33, 130.63, 128.88, 128.34, 126.99, 65.44, 31.90, 29.17, 28.67, 25.98, 22.74, 14.18. Elemental analysis: Calcd. for C30H38O4: C, 77.89; H, 8.28. Found: C, 77.40; H, 8.42.
Poly-1-Hep was converted to poly-1-H by the hydrolysis of its ester groups as follows. A solution of poly-1-Hep (0.71 g) in THF (30 mL) was treated with a 4 N solution of aqueous KOH (25 mL), and the resulting mixture was stirred at 80 °C for 3 h. The mixture was cooled to ambient temperature and the THF was removed under reduced pressure to give a concentrated mixture, which was treated with a 4 N aqueous solution of KOH (25 mL). The resulting mixture was then stirred at 80 °C for 72 h to complete the hydrolysis. The solution was subsequently cooled to ambient temperature and acidified with a 6 N aqueous HCl solution, resulting in the precipitation of poly-1-H, which was collected by centrifugation, washed with water and dried in vacuo at room temperature overnight to give the desired product (243 mg, 60% yield). IR (KBr, cm−1): 1700 (νC=O of carboxylic acid). 1H-NMR (500 MHz, D2O/DMSO-d6 (1/1, v/v), 25 °C): δ 5.30–8.00 (br, 8H, Ar–H). 13C-NMR (125 MHz, D2O/DMSO-d6 (1/2, v/v), 25 °C): δ 126.99, 128.06, 129.61, 131.18, 147.10, 167.65. Elemental analysis: Calcd. for C16H10O4: C, 72.18; H, 3.79. Found: C, 72.36; H, 3.98.
3.5. Synthesis of Poly-2S by Using a Polymer Reaction
A solution of poly-1-H (150 mg, 0.56 mmol) and (S)-2 (278 μL, 2.2 mmol) in a 5:1 (v/v) mixture of DMSO and H2O (26 mL) was treated with DMT-MM (620 mg, 2.2 mmol), and the resulting mixture was stirred at room temperature for 24 h. The mixture was then added to a large volume of methanol, resulting in the precipitation of the poly-2S product, which was collected by centrifugation and then dried in vacuo at room temperature. The resulting poly-2S product was found to contain approximately 100 mol % of (S)-2 as the pendant group to each monomer unit, as determined by 1H-NMR and elemental analyses (Figure S3a).
Spectroscopic data for poly-2S: IR (KBr, cm−1): 1635 (νC=O of amide), 1533 (νN–H of amide). 1H-NMR (500 MHz, CDCl3, 25 °C): δ 8.40–7.60 (br, 2H, NH), 7.60–4.70 (br, 20H, aromatic, CHCH3), 2.00–1.10 (br, 6H, CHCH3). Elemental analysis: Calcd. for C32H28N2O2·0.85H2O: C, 78.78; H, 6.14; N, 5.74. Found: C, 78.85; H, 5.88; N, 5.84.
3.6. Synthesis of Poly-(2Sx-co-31−x) by Using Polymer Reactions
Typical procedure for synthesis of poly-(2Sx-co-31−x) by polymer reaction of poly-1-H with (S)-2 followed by 3 using DMT-MM as a condensing reagent is described below. To a solution of poly-1-H (100 mg, 0.37 mmol) and (S)-2 (67 μL, 0.54 mmol) in DMSO (6 mL) was added DMT-MM (75 mg, 0.27 mmol), and the resulting mixture was stirred at room temperature for 12 h. The reaction mixture was then poured into a 5:1 (v/v) mixture of hexane and ethanol, and the resulting precipitated polymer was collected by centrifugation, washed sequentially with a 1 N aqueous solution of HCl, water and methanol, and then dried under vacuum at room temperature overnight. The dried polymer was dissolved in DMSO (6 mL) and treated with an excess of 3 (200 μL, 1.87 mmol) in the presence of DMT-MM (258 mg, 0.93 mmol) for 12 h at room temperature. The reaction mixture was then poured into a large volume of methanol, and the resulting precipitated polymer collected by centrifugation and dried under vacuum at room temperature overnight to yield poly-(2S0.36-co-30.64) (125 mg, 74%). The (S)-2 content of the polymer was determined to be 36 mol % by 1H-NMR analysis (Figure S3b).
Spectroscopic data for poly-(2S0.36-co-30.64): IR (KBr, cm−1): 1638 (νC=O of amide), 1523 (νN–H of amide). 1H-NMR (500 MHz, CDCl3, 25 °C): δ 8.75–8.02 (br, 2H, NH), 7.66–5.75 (br, 18H, aromatic), 5.27–4.80 (br, CHCH3), 4.58–3.96 (br, CH2), 1.57–0.85 (br, CHCH3). Calcd. for 0.36C32H28N2O2·0.64C30H24N2O2·0.6H2O: C, 79.27; H, 5.77; N, 6.02. Elemental analysis: Found: C, 79.13; H, 5.53; N, 5.98.
The other poly-(2Sx-co-31−x)s were synthesized in the same way by varying the feed ratio of DMT-MM relative to poly-1-H whilst maintaining that of (S)-2 against DMT-MM ([(S)-2]/[DMT-MM] = 2) (see Table S1). The (S)-2 unit contents (x) of the poly-(2Sx-co-31−x)s were determined by 1H-NMR analysis.
3.7. Preparation of High-Performance Liquid Chromatography (HPLC) Columns
A solution of poly-2, h-poly-2S or h-poly-(2S0.36-co-30.64) (100 mg) in DMF (1 mL) was used to coat some macroporous silica gel (400 mg) according to a previously reported method [49], and the solvent was then evaporated under reduced pressure. The poly-2, h-poly-2S and h-poly-(2S0.36-co-30.64) contents on the silica gel were estimated to be 18.3, 17.6 and 19.9 wt %, respectively, by thermogravimetric analysis. After fractionating the packing materials with sieves, the resulting materials were packed into a stainless steel tube (25 × 0.20 cm (i.d.)) using a conventional high-pressure slurry packing technique with an ECONO-PACKER MODEL CPP-085 (Chemco, Osaka, Japan) [50]. The plate numbers for the columns were 3700 (poly-2S), 3500 (h-poly-2S) and 3300 (h-poly-(2S0.36-co-30.64)), respectively, for the elution of benzene with hexane/2-propanol (90:10, v/v) at a flow rate of 0.1 mL/min. The hold-up times (t0) were estimated using 1,3,5-tri-tert-butylbenzene and acetone as the non-retained compounds for the normal and reverse phases, respectively [52].
4. Conclusions
In conclusion, we have synthesized a series of optically active poly(diphenylacetylene)s bearing amide functional groups on all of their pendant phenyl rings. These systems consist of a chiral unit (poly-2S) or both chiral and achiral units (poly-(2Sx-co-31–x)) and were prepared by the reaction of an optically inactive poly(diphenylacetylene) bearing carboxyl groups with chiral or achiral amines. The resulting polymers were evaluated in terms of their chiroptical properties and chiral recognition abilities toward various racemates such as chiral stationary phases (CSPs) for high-performance liquid chromatography (HPLC). We found that poly-2S and poly-(2Sx-co-31–x) folded into a preferred-handed helical conformation (h-poly-2S and h-poly-(2Sx-co-31–x)) by thermal annealing. Notably, the helical senses of these materials varied considerably depending on their chiral unit contents. Furthermore, the circular dichroism (CD) and circularly polarized luminescence (CPL) spectra of h-poly-2S and h-poly-(2S0.36-co-30.64) were almost mirror images of each other. We have demonstrated that these polymers can efficiently resolve various racemates when used as CSPs for HPLC and that the enantioselectivities and elution orders of the enantiomers were significantly influenced by the helical structures induced in these polymers. The elution orders of some enantiomers were reversed on the h-poly-2S- and h-poly-(2S0.36-co-30.64)-based CSPs because of the opposite helicities induced in these polymers. Moreover, poly-2S and h-poly-2S showed higher chiral recognition abilities toward many racemates than the previously reported poly(diphenylacetylene) system bearing the same chiral substituent on only half of its pendant phenyl rings. This result was attributed to an increase in the number of the amide pendants acting as effective chiral recognition sites [36]. Poly(diphenylacetylene)s are thermally and chemically more stable than poly(phenylacetylene)s and are highly emissive without introducing any other fluorescent substituents. We therefore believe that these findings will not only allow for the design of useful poly(diphenylacetylene)-based CSPs for HPLC with much higher chiral recognition abilities toward diverse racemates, but that they will also assist in the development of new circularly polarized luminescence materials.
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/21/11/1487/s1.
Acknowledgments
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grants-in-Aid for Scientific Research (B), Grant No. 16H04154.
Author Contributions
K.M. conceived the project, designed the experiments and wrote the manuscript. M.M. principally performed the experiments. Y.S. performed the enantioseparation experiments. S.K. and T.I. performed the data analysis. All of the authors discussed the results and edited the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Shen, J.; Okamoto, Y. Efficient separation of enantiomers using stereoregular chiral polymers. Chem. Rev. 2016, 116, 1094–1138. [Google Scholar] [CrossRef] [PubMed]
- Ikai, T.; Okamoto, Y. Structure control of polysaccharide derivatives for efficient separation of enantiomers by chromatography. Chem. Rev. 2009, 109, 6077–6101. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Ikai, T. Chiral HPLC for efficient resolution of enantiomers. Chem. Soc. Rev. 2008, 37, 2593–2608. [Google Scholar] [CrossRef] [PubMed]
- Francotte, E.; Lindner, W. Chirality in Drug Research; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Subramanian, G. Chiral Separation Techniques: A practical Approach, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Andersson, S.; Allenmark, S.G. Preparative chiral chromatographic resolution of enantiomers in drug discovery. J. Biochem. Biophys. Methods 2002, 54, 11–23. [Google Scholar] [CrossRef]
- Gasparrini, F.; Misiti, D.; Villani, C. High-performance liquid chromatography chiral stationary phases based on low-molecular-mass selectors. J. Chromatogr. A 2001, 906, 35–50. [Google Scholar] [CrossRef]
- Ward, T.J.; Farris Iii, A.B. Chiral separations using the macrocyclic antibiotics: A review. J. Chromatogr. A 2001, 906, 73–89. [Google Scholar] [CrossRef]
- Davankov, V.A. Enantioselective ligand exchange in modern separation techniques. J. Chromatogr. A 2003, 1000, 891–915. [Google Scholar] [CrossRef]
- Lämmerhofer, M.; Lindner, W. Liquid chromatographic enantiomer separation and chiral recognition by cinchona alkaloid-derived enantioselective separation materials. In Advances in Chromatography; CRC Press: Boca Raton, FL, USA, 2007; Volume 46, pp. 1–107. [Google Scholar]
- D’Acquarica, I.; Gasparrini, F.; Misiti, D.; Pierini, M.; Villani, C. HPLC chiral stationary phases containing macrocyclic antibiotics: Practical aspects and recognition mechanism. In Advances in Chromatography; CRC Press: Boca Raton, FL, USA, 2007; Volume 46, pp. 109–173. [Google Scholar]
- Yamamoto, C.; Okamoto, Y. Optically active polymers for chiral separation. Bull. Chem. Soc. Jpn. 2004, 77, 227–257. [Google Scholar] [CrossRef]
- Yashima, E. Polysaccharide-based chiral stationary phases for high-performance liquid chromatographic enantioseparation. J. Chromatogr. A 2001, 906, 105–125. [Google Scholar] [CrossRef]
- Nakano, T. Optically active synthetic polymers as chiral stationary phases in HPLC. J. Chromatogr. A 2001, 906, 205–225. [Google Scholar] [CrossRef]
- Okamoto, Y.; Yashima, E. Polysaccharide derivatives for chromatographic separation of enantiomers. Angew. Chem. Int. Ed. 1998, 37, 1020–1043. [Google Scholar] [CrossRef]
- Yashima, E.; Maeda, K.; Iida, H.; Furusho, Y.; Nagai, K. Helical polymers: Synthesis, structures, and functions. Chem. Rev. 2009, 109, 6102–6211. [Google Scholar] [CrossRef] [PubMed]
- Ishidate, R.; Shimomura, K.; Ikai, T.; Kanoh, S.; Maeda, K. Macromolecular helicity induction and memory in a poly(biphenylylacetylene) bearing an ester group and its application to a chiral stationary phase for high-performance liquid chromatography. Chem. Lett. 2015, 44, 946–948. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, H.; Yang, T.; Ma, R.; Liu, L.; Sakai, R.; Satoh, T.; Kakuchi, T.; Okamoto, Y. Synthesis and chiral recognition of helical poly(phenylacetylene)s bearing l-phenylglycinol and its phenylcarbamates as pendants. J. Polym. Sci. A Polym. Chem. 2015, 53, 809–821. [Google Scholar] [CrossRef]
- Zhang, C.; Ma, R.; Wang, H.; Sakai, R.; Satoh, T.; Kakuchi, T.; Liu, L.; Okamoto, Y. Influence of helical structure on chiral recognition of poly(phenylacetylene)s bearing phenylcarbamate residues of l-phenylglycinol and amide linage as pendants. Chirality 2015, 27, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Anger, E.; Iida, H.; Yamaguchi, T.; Hayashi, K.; Kumano, D.; Crassous, J.; Vanthuyne, N.; Roussel, C.; Yashima, E. Synthesis and chiral recognition ability of helical polyacetylenes bearing helicene pendants. Polym. Chem. 2014, 5, 4909–4914. [Google Scholar] [CrossRef]
- Shimomura, K.; Ikai, T.; Kanoh, S.; Yashima, E.; Maeda, K. Switchable enantioseparation based on macromolecular memory of a helical polyacetylene in the solid state. Nat. Chem. 2014, 6, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, H.; Geng, Q.; Yang, T.; Liu, L.; Sakai, R.; Satoh, T.; Kakuchi, T.; Okamoto, Y. Synthesis of helical poly(phenylacetylene)s with amide linkage bearing l-phenylalanine and l-phenylglycine ethyl ester pendants and their applications as chiral stationary phases for HPLC. Macromolecules 2013, 46, 8406–8415. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, F.; Li, Y.; Shen, X.; Xu, X.; Sakai, R.; Satoh, T.; Kakuchi, T.; Okamoto, Y. Influence of stereoregularity and linkage groups on chiral recognition of poly(phenylacetylene) derivatives bearing l-leucine ethyl ester pendants as chiral stationary phases for HPLC. J. Polym. Sci. A Polym. Chem. 2013, 51, 2271–2278. [Google Scholar] [CrossRef]
- Naito, Y.; Tang, Z.; Iida, H.; Miyabe, T.; Yashima, E. Enantioseparation on helical poly(phenylacetylene)s bearing cinchona alkaloid pendants as chiral stationary phases for HPLC. Chem. Lett. 2012, 41, 809–811. [Google Scholar] [CrossRef]
- Yashima, E.; Matsushima, T.; Nimura, T.; Okamoto, Y. Enantioseparation on optically active stereoregular polyphenylacetylene derivatives as chiral stationary phases for HPLC. Korea Polym. J. 1996, 4, 139–146. [Google Scholar]
- Yashima, E.; Huang, S.; Okamoto, Y. An optically active stereoregular polyphenylacetylene derivative as a novel chiral stationary phase for HPLC. J. Chem. Soc. Chem. Commun. 1994, 1811–1812. [Google Scholar] [CrossRef]
- Kim, H.; Seo, K.-U.; Jin, Y.-J.; Lee, C.-L.; Teraguchi, M.; Kaneko, T.; Aoki, T.; Kwak, G. Highly emissive, optically active poly(diphenylacetylene) having a bulky chiral side group. ACS Macro Lett. 2016, 5, 622–625. [Google Scholar] [CrossRef]
- Kim, H.; Lee, D.; Lee, S.; Suzuki, N.; Fujiki, M.; Lee, C.-L.; Kwak, G. Optically active conjugated polymer from solvent chirality transfer polymerization in monoterpenes. Macromol. Rapid Commun. 2013, 34, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- San Jose, B.A.; Matsushita, S.; Akagi, K. Lyotropic chiral nematic liquid crystalline aliphatic conjugated polymers based on disubstituted polyacetylene derivatives that exhibit high dissymmetry factors in circularly polarized luminescence. J. Am. Chem. Soc. 2012, 134, 19795–19807. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.A.; Qin, A.; Tong, L.; Zhao, H.; Zhao, Q.; Sun, J.Z.; Tang, B.Z. Synthesis of functional disubstituted polyacetylenes bearing highly polar functionalities via activated ester strategy. ACS Macro Lett. 2012, 1, 75–79. [Google Scholar] [CrossRef]
- Lee, D.; Jin, Y.-J.; Kim, H.; Suzuki, N.; Fujiki, M.; Sakaguchi, T.; Kim, S.K.; Lee, W.-E.; Kwak, G. Solvent-to-polymer chirality transfer in intramolecular stack structure. Macromolecules 2012, 45, 5379–5386. [Google Scholar] [CrossRef]
- Jim, C.K.W.; Lam, J.W.Y.; Leung, C.W.T.; Qin, A.; Mahtab, F.; Tang, B.Z. Helical and luminescent disubstituted polyacetylenes: Synthesis, helicity, and light emission of poly(diphenylacetylene)s bearing chiral menthyl pendant groups. Macromolecules 2011, 44, 2427–2437. [Google Scholar] [CrossRef]
- Fukushima, T.; Tsuchihara, K. Syntheses and chirality control of optically active poly(diphenylacetylene) derivatives. Macromolecules 2009, 42, 5453–5460. [Google Scholar] [CrossRef]
- Teraguchi, M.; Suzuki, J.; Kaneko, T.; Aoki, T.; Masuda, T. Enantioselective permeation through membranes of chiral helical polymers prepared by depinanylsilylation of poly(diphenylacetylene) with a high content of the pinanylsilyl group. Macromolecules 2003, 36, 9694–9697. [Google Scholar] [CrossRef]
- Aoki, T.; Kobayashi, Y.; Kaneko, T.; Oikawa, E.; Yamamura, Y.; Fujita, Y.; Teraguchi, M.; Nomura, R.; Masuda, T. Synthesis and Properties of Polymers from Disubstituted Acetylenes with Chiral Pinanyl Groups. Macromolecules 1998, 32, 79–85. [Google Scholar] [CrossRef]
- Maeda, K.; Maruta, M.; Shimomura, K.; Ikai, T.; Kanoh, S. Chiral recognition ability of an optically active poly(diphenylacetylene) as a chiral stationary phase for HPLC. Chem. Lett. 2016, 45, 1063–1065. [Google Scholar] [CrossRef]
- Shiotsuki, M.; Sanda, F.; Masuda, T. Polymerization of substituted acetylenes and features of the formed polymers. Polym. Chem. 2011, 2, 1044–1058. [Google Scholar] [CrossRef]
- Liu, J.; Lam, J.W.Y.; Tang, B.Z. Acetylenic polymers: Syntheses, structures, and functions. Chem. Rev. 2009, 109, 5799–5867. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.Z.; Qin, A.; Lam, J.W.Y.; Sun, J.Z.; Dong, Y.; Häussler, M.; Liu, J.; Xu, H.P.; Zheng, Q.; Tang, B.Z. Disubstituted polyacetylenes containing photopolymerizable vinyl groups and polar ester functionality: Polymer synthesis, aggregation-enhanced emission, and fluorescent pattern formation. Macromolecules 2007, 40, 3159–3166. [Google Scholar] [CrossRef]
- Maeda, K.; Okamoto, Y. Synthesis and conformational characteristics of poly(phenyl isocyanate)s bearing an optically active ester group. Macromolecules 1999, 32, 974–980. [Google Scholar] [CrossRef]
- Koe, J.R.; Fujiki, M.; Motonaga, M.; Nakashima, H. Cooperative helical order in optically active poly(diarylsilylenes). Macromolecules 2001, 34, 1082–1089. [Google Scholar] [CrossRef]
- Morino, K.; Maeda, K.; Okamoto, Y.; Yashima, E.; Sato, T. Temperature dependence of helical structures of poly(phenylacetylene) derivatives bearing an optically active substituent. Chem. Eur. J. 2002, 8, 5112–5120. [Google Scholar] [CrossRef]
- Deng, J.P.; Tabei, J.; Shiotsuki, M.; Sanda, F.; Masuda, T. Variation of helical pitches driven by the composition of N-propargylamide copolymers. Macromolecules 2004, 37, 9715–9721. [Google Scholar] [CrossRef]
- Tabei, J.; Shiotsuki, M.; Sato, T.; Sanda, F.; Masuda, T. Control of helix sense by composition of chiral-achiral copolymers of N-propargylbenzamides. Chem. Eur. J. 2005, 11, 3591–3598. [Google Scholar] [CrossRef] [PubMed]
- Takei, F.; Onitsuka, K.; Takahashi, S.; Terao, K.; Sato, T. Control of helical structure in random copolymers of chiral and achiral aryl isocyanides prepared with palladium-platinum mu-ethynediyl complexes. Macromolecules 2007, 40, 5245–5254. [Google Scholar] [CrossRef]
- Nagata, Y.; Nishikawa, T.; Suginome, M. Exerting control over the helical chirality in the main chain of sergeants-and-soldiers-type poly(quinoxaline-2,3-diyl)s by changing from random to block copolymerization protocols. J. Am. Chem. Soc. 2015, 137, 4070–4073. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Terao, K.; Teramoto, A.; Fujiki, M. On the composition-driven helical screw-sense inversion of chiral-achiral random copolymers. Macromolecules 2002, 35, 5355–5357. [Google Scholar] [CrossRef]
- San Jose, B.A.; Matsushita, S.; Moroishi, Y.; Akagi, K. Disubstituted liquid crystalline polyacetylene derivatives that exhibit linearly polarized blue and green emissions. Macromolecules 2011, 44, 6288–6302. [Google Scholar] [CrossRef]
- Okamoto, Y.; Aburatani, R.; Hatada, K. Chromatographic chiral resolution: XIV. Cellulose tribenzoate derivatives as chiral stationary phases for high-performance liquid chromatography. J. Chromatogr. A 1987, 389, 95–102. [Google Scholar] [CrossRef]
- Okamoto, Y.; Kawashima, M.; Hatada, K. Chromatographic resolution: XI. Controlled chiral recognition of cellulose triphenylcarbamate derivatives supported on silica gel. J. Chromatogr. A 1986, 363, 173–186. [Google Scholar] [CrossRef]
- Kunishima, M.; Kawachi, C.; Monta, J.; Terao, K.; Iwasaki, F.; Tani, S. 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride: An efficient condensing agent leading to the formation of amides and esters. Tetrahedron 1999, 55, 13159–13170. [Google Scholar] [CrossRef]
- Koller, H.; Rimböck, K.-H.; Mannschreck, A. High-pressure liquid chromatography on triacetylcellulose. J. Chromatogr. A 1983, 282, 89–94. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).