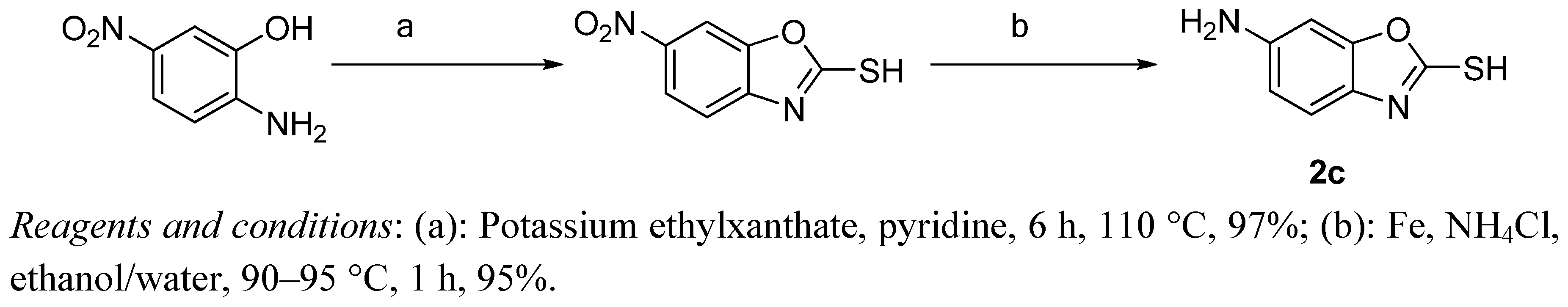3. Experimental
3.1. General
The human cancer cell lines, HepG2, HCT116, PC-9, Hela, SKOV-3, A549, A375, H460 and A431 were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA). Dulbecco’s modified Eagle medium (DMEM) and RPMI 1640 were purchased from Gibco (Grand Island, NY, USA). Fetal bovine serum (FBS) was obtained from Hyclone (Logan, UT, USA). The purity of compound screened in biological assays was determined to be >98% by HPLC analysis. An Symmetry C18 (75 mm × 4.6 mm, i.d. 3.5μm) (Waters, Milford, MA, USA) was used with a t elution of acetonitrile and HPLC-grade water as mobile phase. 1H-NMR were recorded at 400 MHz on a Varian spectrometer (Varian, Palo Alto, CA, USA) model Gemini 400. Mass Spectra (MS) were measured by Q-TOF Priemier mass spectrometer utilizing electrospray ionization (ESI) (Micromass, Manchester, UK).
3.2. Preparation of 6-Aminobenzo[d]oxazole-2-thiol (2c)
3.2.1. 6-Nitrobenzo[d]oxazole-2-thiol
6-Nitrobenzo[d]oxazole-2-thiol was synthesized according to a modified literature method [
7]. A suspension of 2-amino-5-nitrophenol (3.08 g, 20 mmol) and potassium ethylxanthate (3.36 g, 21 mmol) in dry pyridine (40 mL) was stirred at 120 °C for 6 h, and then at room temperature for another 16 h. 2 M HCl solution was added to the solution to adjust the pH value to 6. The resulting precipitate was collected by filtration, washed with petroleum ether and then dried in vacuum to afford 3.22 g (97%) of the title compound.
1H-NMR (DMSO-
d6) δ: 7.39 (d, 1H,
J = 8 Hz), 7.86 (t, 1H,
J = 8 Hz), 8.22 (dd, 1H,
J1 = 4 Hz,
J2 = 8 Hz)), 8.41 (s, 1H). ESI-MS (
m/z, %): 197.1 (M+H)
+.
3.2.2. 6-Aminobenzo[d]oxazole-2-thiol (2c)
A mixture of 6-nitrobenzo[d]oxazole-2-thiol (5 g, 25 mmol), iron powder (3.36 g, 60 mmol), and ammonium chloride (2.67 g, 50 mmol) was refluxed in a mixed solvent of ethanol (25 mL) and H2O (8 mL). After completion of the reaction, the mixture was filtered while hot and washed with petroleum ether. Standing at room temperature gave compound 2c as a bright yellow solid. Yield: 3.7 g, 90%. 1H-NMR (DMSO-d6) δ: 6.51 (dd, 2H, J1 = 4 Hz, J2 = 8 Hz), 6.64 (d, 2H, J = 4 Hz), 6.90 (d, 2H, J = 8 Hz). ESI-MS (m/z, %): 167.1 (M+H)+.
3.3. 2-Chloro-N-(4-chlorobenzyl)acetamide (3a)
2-Chloroacetyl chloride (4.79 g, 42 mmol) was added dropwise with stirring to a mixture of (4-chlorophenyl) methanamine (4.96 g, 35 mmol) in dichloromethane (50 mL) cooled to 0 °C. The resulting mixture was allowed to warm to ambient temperature and stirred for 1 h. After filtering, the solid was washed with petroleum ether and dried under vacuum for 12 h at 25–30 °C. The title compound 3a was obtained by recrystalization from ethanol. Yield: 7.25 g, 95%.1H-NMR (400 MHz, DMSO-d6): δ 4.13 (s, 2H), 4.29 (d, J = 6.0 Hz, 2H), 7.28 (d, J = 4.4 Hz, 2H), 7.39 (d, J = 8.0 Hz, 2H), 8.77 (s, 1H). ESI-MS (m/z, %): 218.0 (M−H)+.
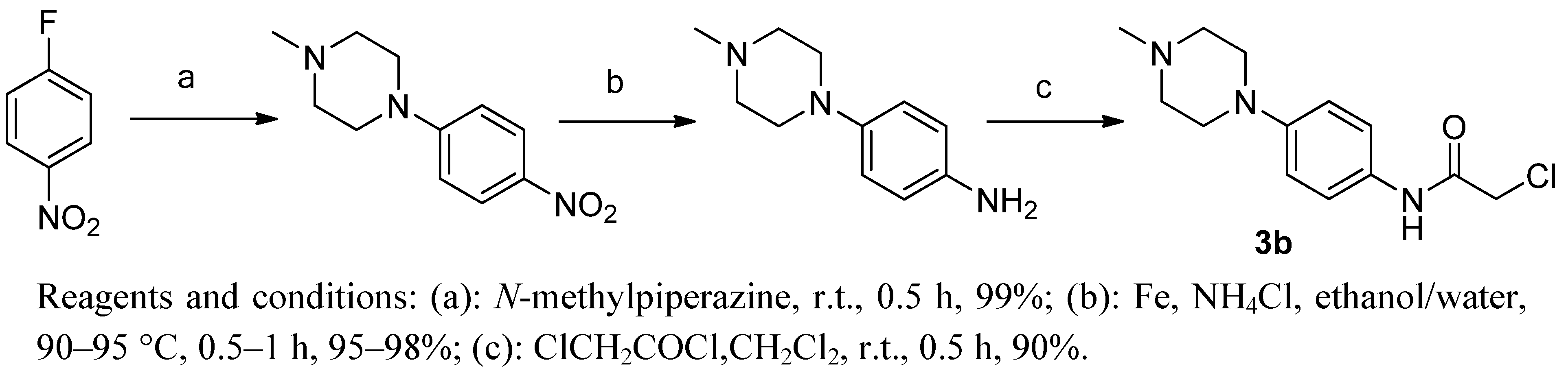
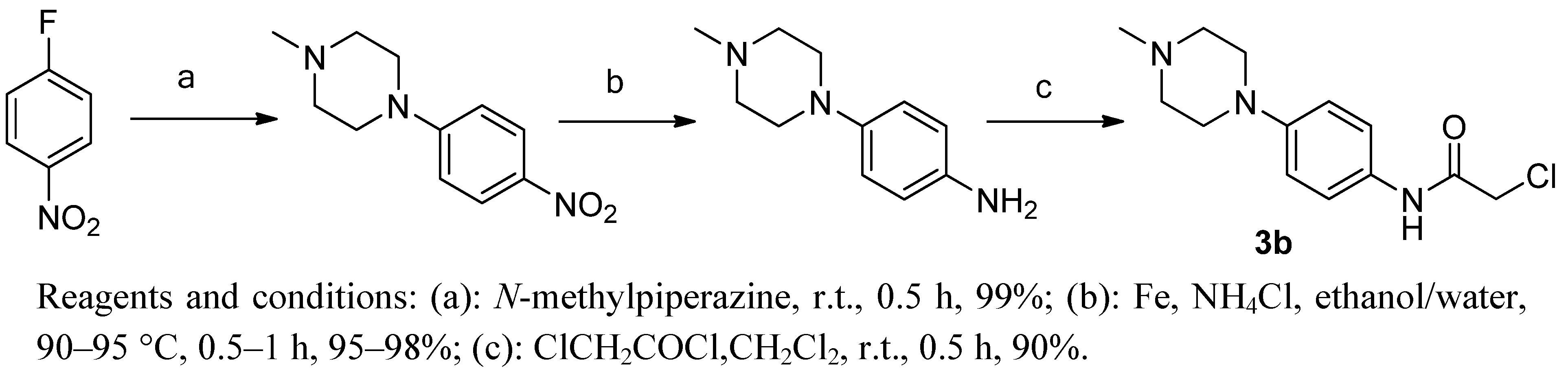
3.4. 2-Chloro-N-(4-(4-methylpiperazin-1-yl)phenyl)acetamide (3b)
3.4.1. 1-Methyl-4-(4-nitrophenyl)piperazine
Briefly, 1-methylpiperazine (15.6 g, 156 mmol) was added slowly while stirring to a solution of 4-fluoronitrobenzene (20 g, 142 mmol) at room temperature. The mixture was stirred for 1 h, then filtered and washed with petroleum ether to give product as an orange solid. Yield: 30.3 g, 96.43%.1H-NMR (DMSO-d6) δ: 2.22 (s, 3H), 2.43 (t, 4H, J = 4 Hz), 3.45 (t, 4H, J = 4 Hz), 7.03 (d, 2H, J = 8 Hz), 8.05 (d, 2H, J = 8 Hz). ESI-MS (m/z, %): 222.0 (M+H)+.
3.4.2. 4-(4-Methylpiperazin-1-yl)aniline
A mixture of 1-methyl-4-(4-nitrophenyl) piperazine (14.8 g, 67 mmol), iron powder (15 g, 268 mmol), ammonium chloride (7.2 g, 134 mmol) was refluxed in a mixed solvent of ethanol (60 mL) and H2O (20 mL). After completion of the reaction, the mixture was filtered while hot and washed with petroleum ether. After standing at room temperature an off-white acicular solid was obtained. Yield: 12.2 g, 95.31%. 1H-NMR (DMSO-d6) δ: 2.22 (s, 3H), 2.5 (t, 4H, J = 4 Hz), 2.75 (t, 4H, J = 4 Hz), 3.22 (s, 2H), 6.55 (d, 2H, J = 8 Hz), 6.74 (d, 2H, J = 8 Hz). ESI-MS (m/z, %): 192.0 (M+H)+.
3.4.3. 2-Chloro-N-(4-(4-methylpiperazin-1-yl)phenyl)acetamide (3b)
2-Chloroacetyl chloride (4.79 g, 42 mmol) was added dropwise with stirring to a mixture of 1-methyl-4-(4-nitrophenyl) piperazine (6.69 g, 35 mmol) in dichloromethane (50 mL) cooled at 0 °C. The resulting mixture was allowed to warm to ambient temperature and stirred for 1 h. After filtering, the solid was washed with petroleum ether and dried under vacuum for 12 h at 25–30 °C. The title compound 3b was obtained by recrystalization from a solution of ethanol. Yield: 8.48 g, 90.51%.1H-NMR (DMSO-d6) δ: 2.21 (s, 3H), 2.43 (t, 4H, J = 4.8 Hz), 3.07 (t, 4H, J = 4.8 Hz), 4.2 (s, 2H), 6.90 (d, 2H, J = 8 Hz), 7.42 (d, 2H, J = 8 Hz), 10.10 (s, 1H). ESI-MS (m/z, %): 268.1 (M+H)+.
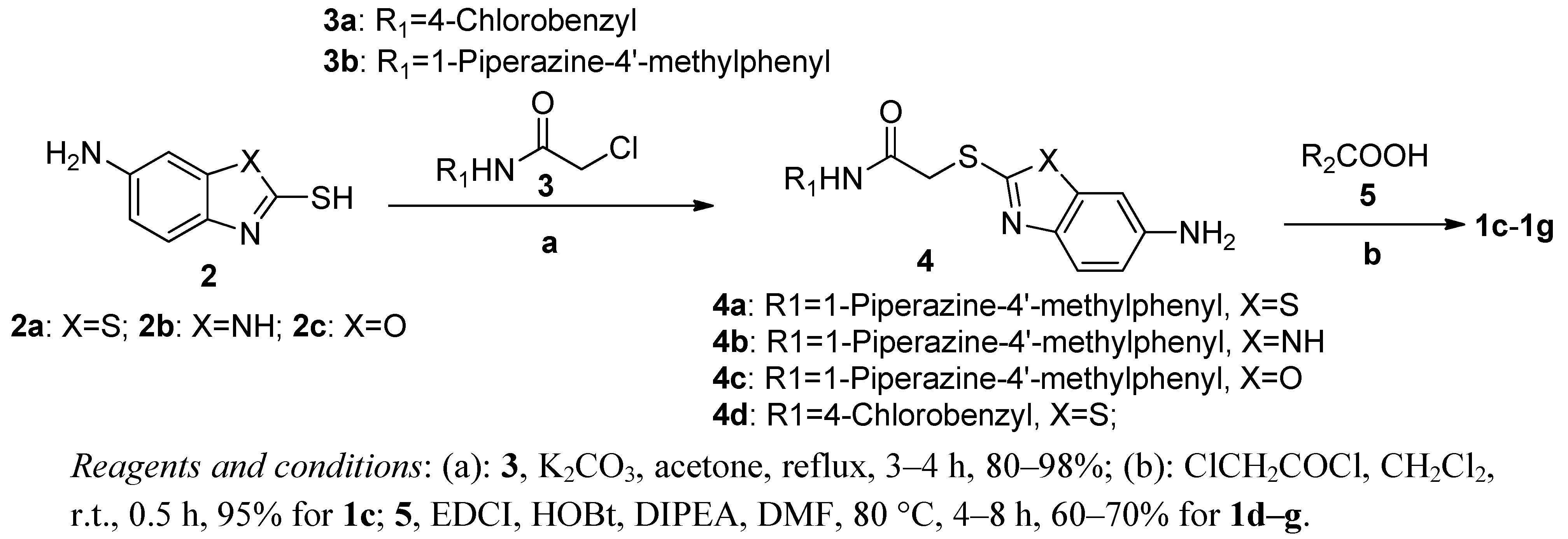
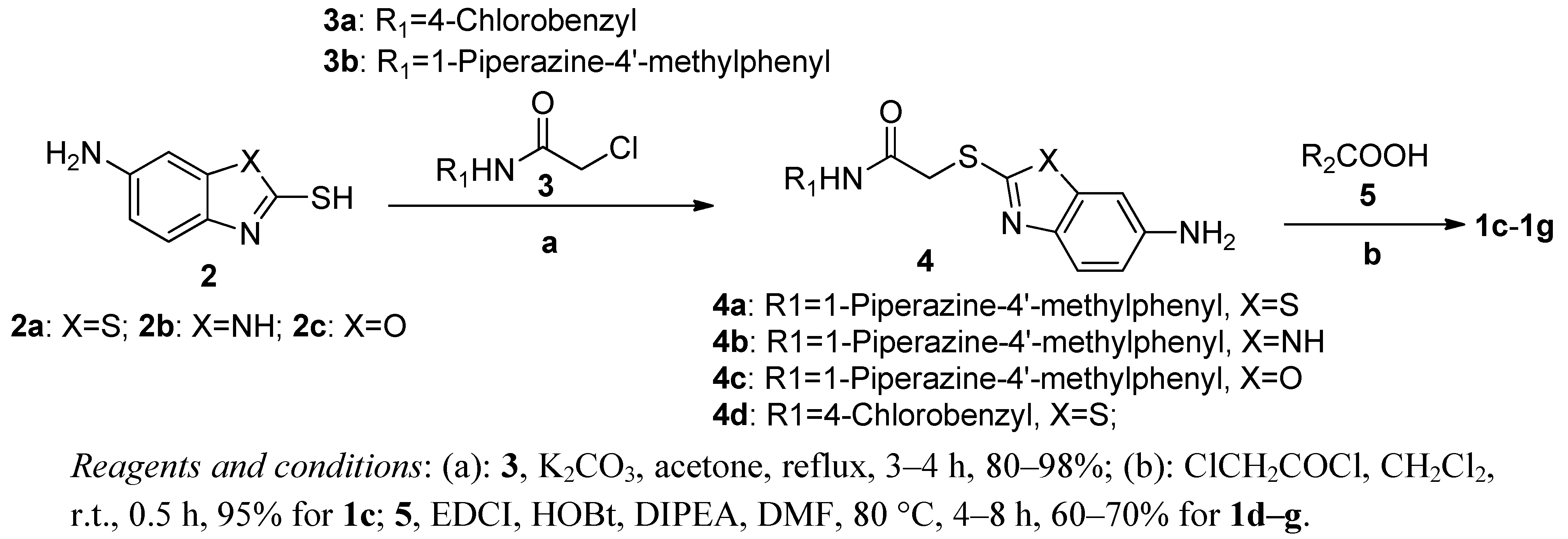
3.5. General Procedure for Preparing Compounds 4a–d
Compound 3a–b (13.76 mmol) was added to a mixture of 2a–c (16.51 mmol) and potassium carbonate (20.64 mmol) in acetone (25 mL) while refluxing with stirring. The resulting mixture was stirred under reflux for 4 h, then cooled to ambient temperature. After removal of acetone on a rotary evaporator, the solid was dissolved in ethyl acetate (100 mL) and washed with water (2 × 10 mL), saturated solution of sodium bicarbonate (2 × 20 mL) and sodium chloride (2 × 20 mL). Then, the organic phase was dried over Na2SO4 and concentrated in vacuo to give a solid product. The title compounds 4a–d were thus obtained, and could be used directly for the next step without further purification. The yields of 4a, 4b, 4c and 4d, were 92%, 90%, 90% and 93%, respectively.
2-(6-aminobenzo[d]thiazol-2-ylthio)-N-(4-(4-methylpiperazin-1-yl)phenyl)acetamide (4a): 1H-NMR (DMSO-d6) δ: 2.24 (s, 3H), 2.44 (t, 4H, J = 4 Hz), 3.06 (t, 4H, J = 4 Hz), 4.14 (s, 2H), 5.38 (s, 2H), 6.70 (dd, 1H, J1 = 2 Hz, J2 = 8.8 Hz), 6.88 (d, 2H, J = 9.2 Hz), 7.00 (d, 1H, J = 2.4 Hz), 7.41 (d, 2H, J = 8.8 Hz), 7.49 (d, 1H, J = 8.8 Hz), 10.16 (s, 1H). ESI-MS (m/z, %): 414.2 (M+H)+.
2-(6-amino-1H-benzo[d]imidazol-2-ylthio)-N-(4-(4-methylpiperazin-1-yl)phenyl)acetamide (4b): 1H-NMR (DMSO-d6) δ: 2.23 (s, 3H), 2.47 (s, 4H), 3.07 (s, 4H), 4.21 (s, 2H), 5.31 (s, 2H), 6.46 (d, 1H, J = 4 Hz), 6.56 (s, 1H), 6.88 (d, 2H, J = 8.8 Hz), 7.16 (s, 1H), 7.42 (d, 2H, J = 8.8 Hz), 10.40 (s, 1H), 12.13 (s, 1H). ESI-MS (m/z, %): 397.3 (M+H)+.
2-(6-aminobenzo[d]oxazol-2-ylthio)-N-(4-(4-methylpiperazin-1-yl)phenyl)acetamide (4c): 1H-NMR (DMSO-d6) δ: 2.23 (s, 3H), 2.46 (t, 4H, J = 4 Hz), 3.07 (t, 4H, J = 4 Hz), 4.23 (s, 2H), 5.31 (s, 2H), 6.56 (dd, 1H, J1 = 2 Hz, J2 = 8.4 Hz), 6.71 (d, 1H, J = 1.6 Hz), 6.89 (d, 2H, J = 8 Hz), 7.22 (t, 1H, J = 8 Hz), 7.41 (d, 2H, J = 8 Hz), 10.16 (s, 1H). ESI-MS (m/z, %): 398.2 (M+H)+.
2-(6-aminobenzo[d]thiazol-2-ylthio)-N-(4-chlorobenzyl)acetamide (4d): 1H-NMR (DMSO-d6) δ: 4.09 (s, 2H), 4.30 (d, 2H, J = 5.6 Hz), 5.40 (s, 2H), 6.73 (dd, 1H, J1 = 1.6 Hz, J2 = 8.8 Hz), 7.01 (d, 1H, J = 1.6 Hz), 7.28 (q, 4H, J = 8.4 Hz), 7.49 (d, 1H, J = 8.4 Hz), 8.81 (t, 1H, J = 5.6 Hz). ESI-MS (m/z, %): 364.0 (M+H)+.
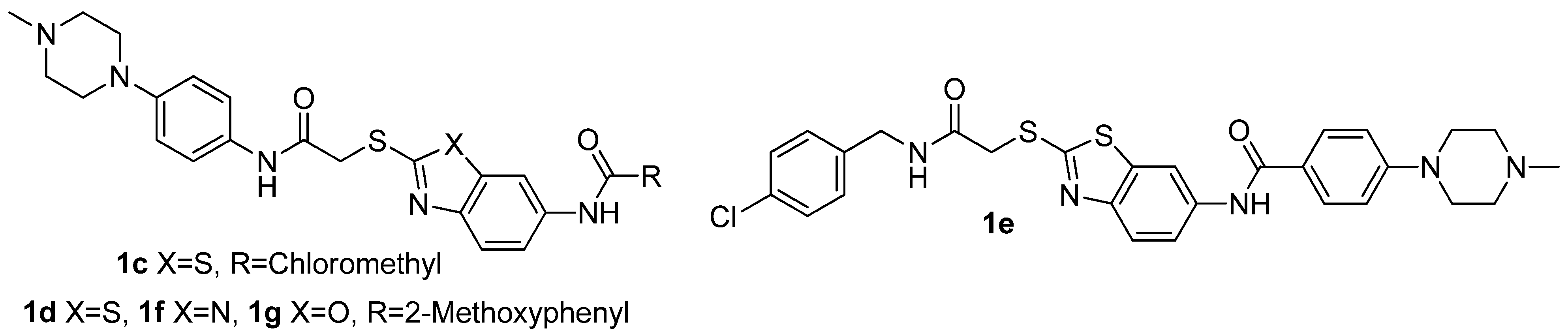
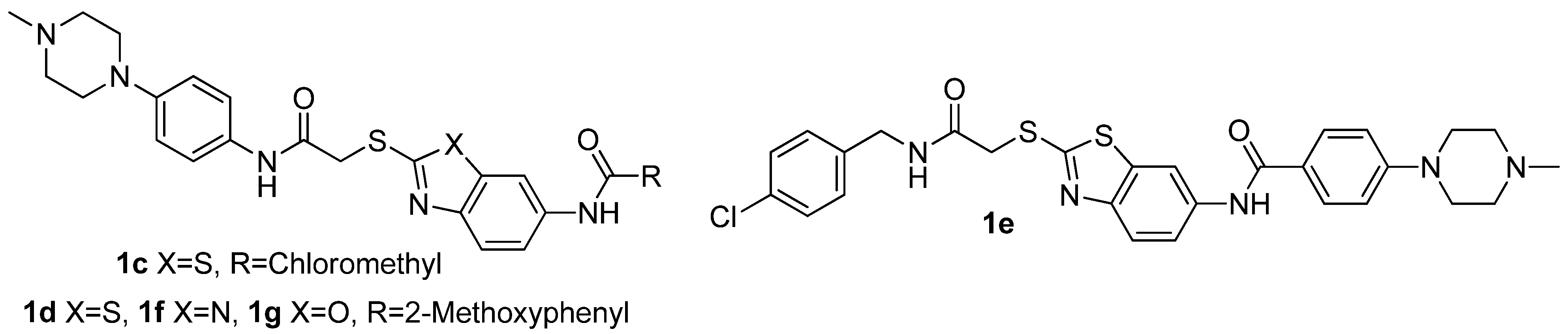
3.6. 2-Chloro-N-(2-(2-(4-(4-methylpiperazin-1-yl)phenylamino)-2-oxoethylthio)benzothiazol-6-yl)-acetamide (1c)
2-Chloroacetyl chloride (43 mg, 0.38 mmol) was added dropwise with stirring to a solution of 4a (80 mg, 0.19 mmol) in dichloromethane (4 mL) originally at 0 °C. The resulting mixture was allowed to warm to ambient temperature and stirred for 1 h. After filtering, the solid was washed with petroleum ether and dried under vacuum for 12 h at 25–30 °C. The title compound 1c (an off-white solid) were obtained by recrystallization from ethanol. Yield: 72 mg, 77.42%. m.p. 245.8–246.1 °C. 1H-NMR (DMSO-d6) δ: 2.21 (s, 3H), 2.43 (t, J = 4.4 Hz, 4H), 3.07 (t, 4H, J = 4.8 Hz), 4.30 (s, 2H), 4.35 (s, 2H), 6.93 (d, 2H, J = 8.8Hz), 7.49 (d, 2H, J = 8.8 Hz), 7.55 (dd, 1H, J = 2 Hz, 8.8 Hz), 7.79 (d, 1H, J = 8.8 Hz), 8.39 (d, 1H, J = 2 Hz), 10.32 (s, 1H), 10.62 (s, 1H); ESI-MS (m/z, %): 490.3 (M+H)+.
3.7. General Procedure for Preparing Compounds 1d–g
A mixture of compound 4a–d (0.5 mmol), 2-methoxybenzoic acid hydrate or 4-(4-methylpiperazin-1-yl)benzoic acid (0.75 mmol), EDCI (0.75 mmol), HOBt (0.75 mmol), DIPEA (0.75 mmol) was warmed up to 80 °C in DMF (5 mL) with stirring. After completion of reaction, the mixture was diluted with ethyl acetate (100 mL). The resulting mixture was washed with washed with water (2 × 20 mL), saturated solution of sodium bicarbonate (2 × 20 mL) and sodium chloride (2 × 20 mL). Then, the organic phase was dried over Na2SO4 and concentrated in vacuo to give a solid product. The title compounds were obtained by recrystallization from ethanol or ethyl acetate.
2-Methoxy-N-(2-(2-(4-(4-methylpiperazin-1-yl)phenylamino)-2-oxoethylthio)benzo[d]thiazol-6-yl)benzamide (1d). Off-white solid. Yield: 76 mg, 75.47%. m.p. 172.1–172.4 °C. 1H-NMR (DMSO-d6) δ: 2.10 (s, 3H), 2.22 (t, 4H, J = 4.4 Hz), 3.08 (t, 4H, J = 4.8 Hz), 3.91 (s, 3H), 4.35 (s, 3H), 6.91 (d, 2H, J = 8.8 Hz), 7.09 (t, 1H, J = 7.2 Hz), 7.20 (d, 1H, J = 8.4 Hz), 7.48 (d, 2H, J = 8.4 Hz), 7.53 (t, 1H, J = 8 Hz), 7.67 (q, 2H, J = 8.8 Hz), 7.80 (d, 1H, J = 8.4 Hz), 8.55 (s, 1H), 10.22 (s,1H), 10.36 (s,1H); ESI-MS (m/z, %): 548.0 (M+H)+.
N-(2-(2-(4-Chlorobenzylamino)-2-oxoethylthio)benzo[d]thiazol-6-yl)-4-(4-methylpiperazin-1-yl)-benzamide (1e). Yellow solid. Yield: 184 mg, 65.36%. m.p. 177.8–179.2 °C. 1H-NMR (DMSO-d6) δ: 2.09 (s, 3H), 2.45 (t, 4H, J = 4.4 Hz), 3.30 (t, 4H, J = 4.8 Hz), 4.19 (s, 2H), 4.31 (d, 2H, J = 6 Hz), 7.03 (d, 2H, J = 8.8 Hz), 7.28 (d, 2H, J = 8.8 Hz), 7.32 (d, 2H, J = 8.4 Hz), 7.76 (s, 2H), 7.89 (d, 2H, J = 8.8 Hz), 8.52 (s, 1H), 8.85 (t, 1H, J = 6Hz), 10.17 (s, 1H); ESI-MS (m/z, %): 566.2 (M+H)+.
2-Methoxy-N-(2-(2-(4-(4-methylpiperazin-1-yl)phenylamino)-2-oxoethylthio)-1H-benzo[d]imidazol-6-yl)benzamide (1f). Faint yellow solid. Yield: 90 mg, 81.08%. m.p. 132.3–132.6 °C. 1H-NMR (DMSO-d6) δ: 2.21 (s, 3H), 2.43 (t, 4H, J = 4 Hz), 3.06 (t, 4H, J = 4 Hz), 3.90 (s, 3H), 4.21 (s, 2H), 6.88 (d, 2H, J = 8.8 Hz), 7.07 (t, 1H, J = 7.2 Hz), 7.18 (d, 1H, J = 8 Hz), 7.30 (q, 1H, J = 8.4 Hz), 7.43 (t, 3H, J = 8.8 Hz), 7.50 (t, 1H, J = 7.6 Hz), 7.64 (d, 1H, J = 7.2 Hz), 8.07 (d, 1H, J = 33.2 Hz), 10.08 (d, 1H, J = 32.8 Hz), 10.27 (s, 1H), 12.60 (s, 1H); ESI-MS (m/z, %): 531.3 (M+H)+.
2-Methoxy-N-(2-(2-(4-(4-methylpiperazin-1-yl)phenylamino)-2-oxoethylthio)benzo[d]oxazol-6-yl)-benzamide (1g). Yellow solid. Yield: 100 mg, 75.75%. m.p. 210.4–210.8 °C. 1H-NMR (DMSO-d6) δ: 2.24 (s, 3H), 2.40 (t, 4H, J = 4.4 Hz), 3.14 (t, 4H, J = 4.8 Hz), 3.89 (s, 3H), 4.23 (s, 2H), 6.91 (d, 2H, J = 8.8 Hz), 7.09 (t, 1H, J = 7.2 Hz), 7.20 (d, 1H, J = 8.4 Hz), 7.48 (d, 2H, J = 8.4 Hz), 7.53 (t, 1H, J = 8 Hz), 7.67 (q, 2H, J = 8.8 Hz), 7.80 (d, 1H, J = 8.4 Hz), 8.26 (s, 1H), 9.98 (s,1H), 10.29 (s,1H); ESI-MS (m/z, %): 532.2 (M+H)+.
3.8. Cell Culture
Cell lines including HepG2, Hela, Skov-3, A375 and A431 were maintained in Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS), penicillin (100 U/mL) and streptomycin (10 mg/L). Cell lines including HCT116, PC-9, A549 and H460 were maintained in RPMI 1640 containing 10% fetal bovine serum (FBS), penicillin (100 U/mL) and streptomycin (10 mg/L). Cells were grown in a 5% CO2 incubator at 37 °C.
3.9. Cell Proliferation Assay (MTT Assay)
Cells (2 × 103/well) were seeded in 96-well plates and cultured for 24 h, followed by compounds which dissolved in dimethylsulfoxide (DMSO) treatment for 48 h. Ten microliters of 10 mg/mL MTT was added and cultured for 4 h, the medium was removed and 150 μL of DMSO was added to dissolve formazan crystals. Absorbance was measured at 570 nm using an SpectraMAX M5 microplate spectrophotometer (Molecular Devices). The proliferation inhibitory effects of the compounds on cancer cells were expressed as IC50.
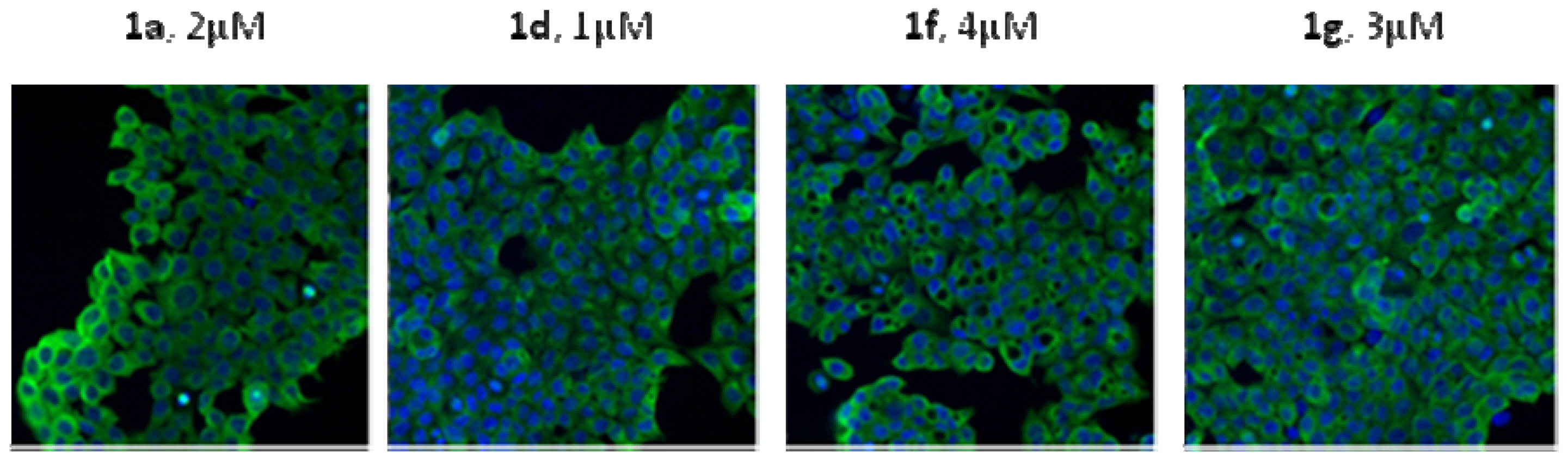
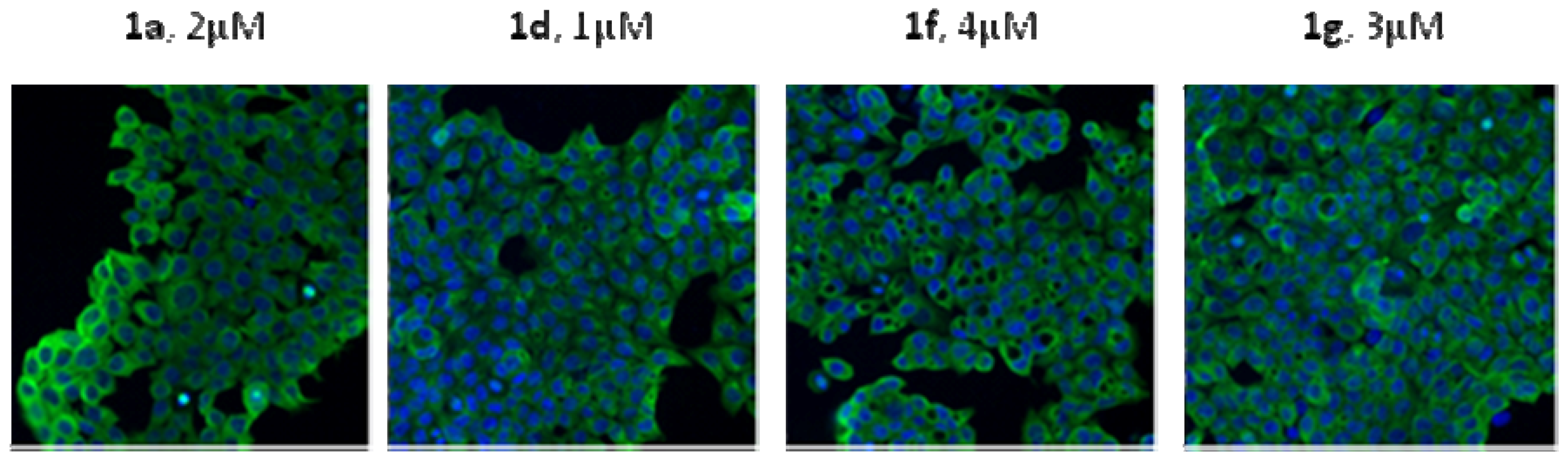
3.10. Immunofluorescence
Cells (1 ×10
5) were plated in 6-well plates. Following drug treatment for 24 h, cells were washed with PBS, punched by Triton X-100. Subsequently cells were probed by the primary antibodies. Then cells were incubated with the secondary antibody for 30 min at room temperature and examined under a fluorescence microscope [
8].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
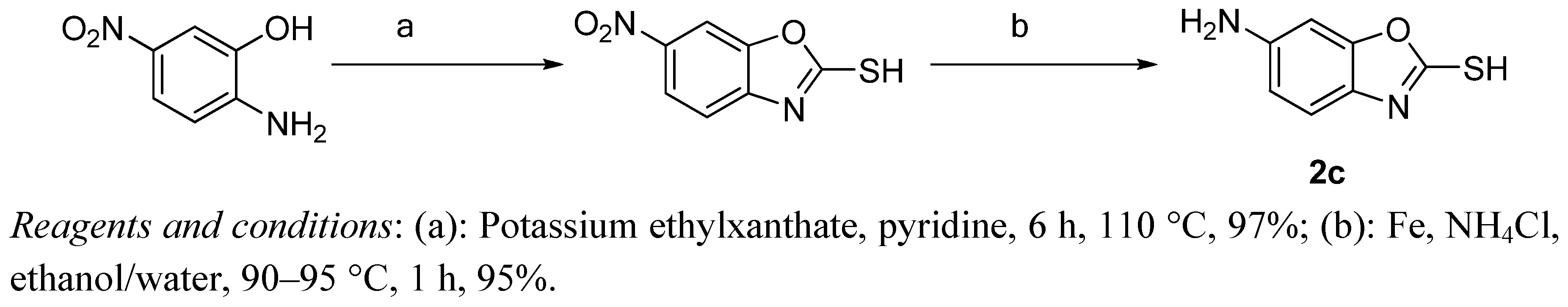{kind=link}
{kind=link}