Acylated Flavone Glycosides from the Roots of Saussurea lappa and Their Antifungal Activity

Abstract
:Introduction
Results and Discussion
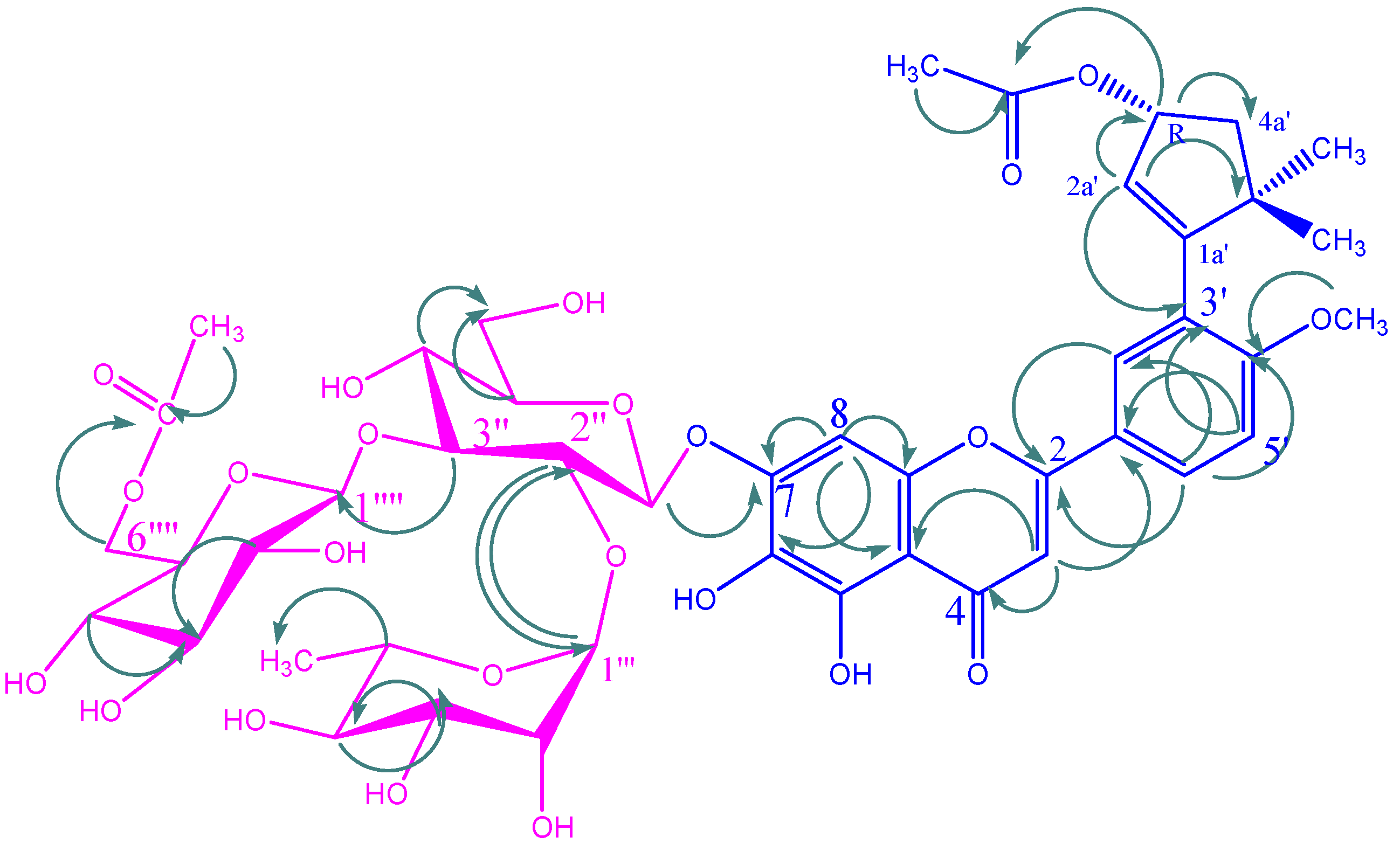
KSR1
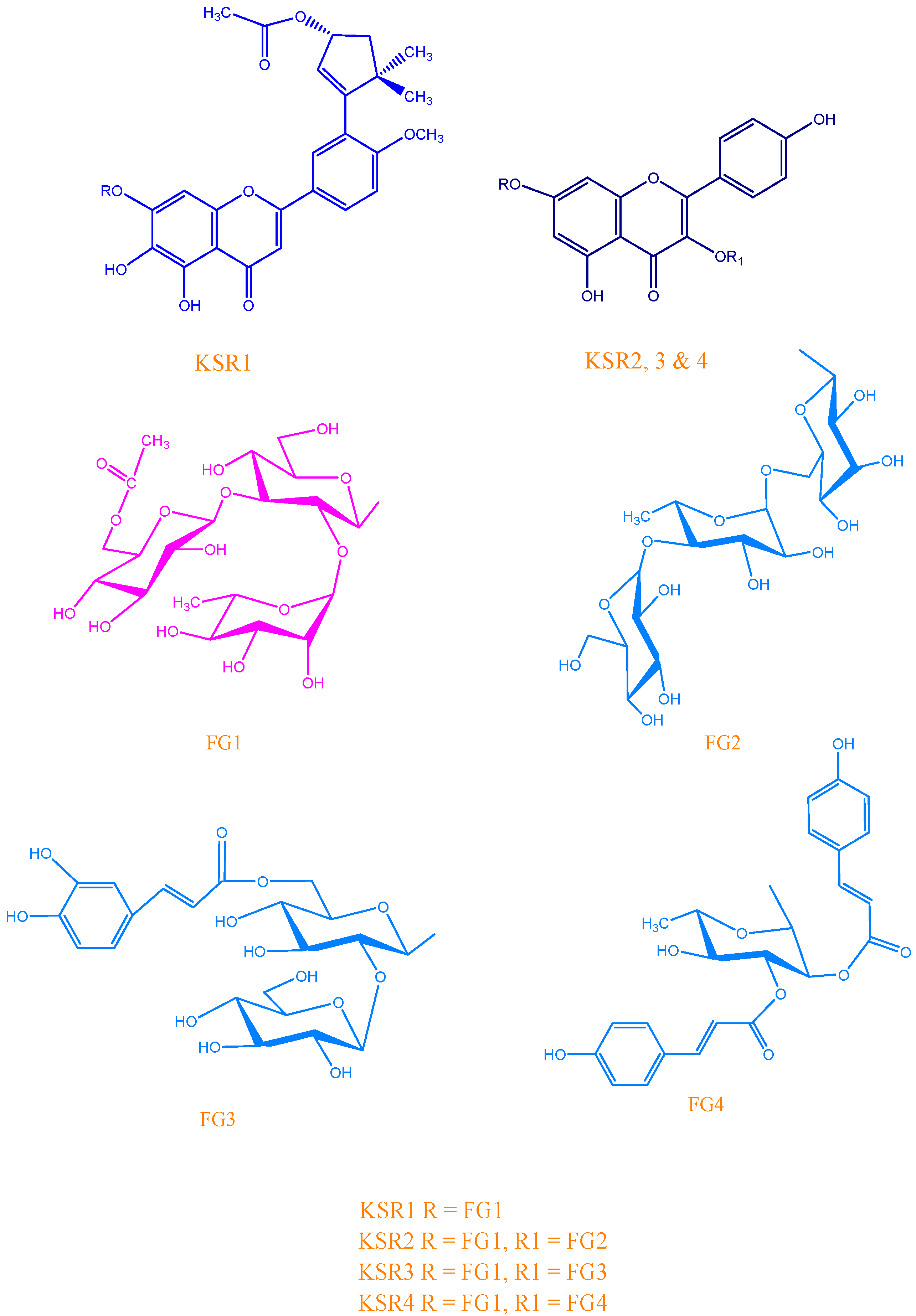

{kind=link}
{kind=link}
{kind=link}
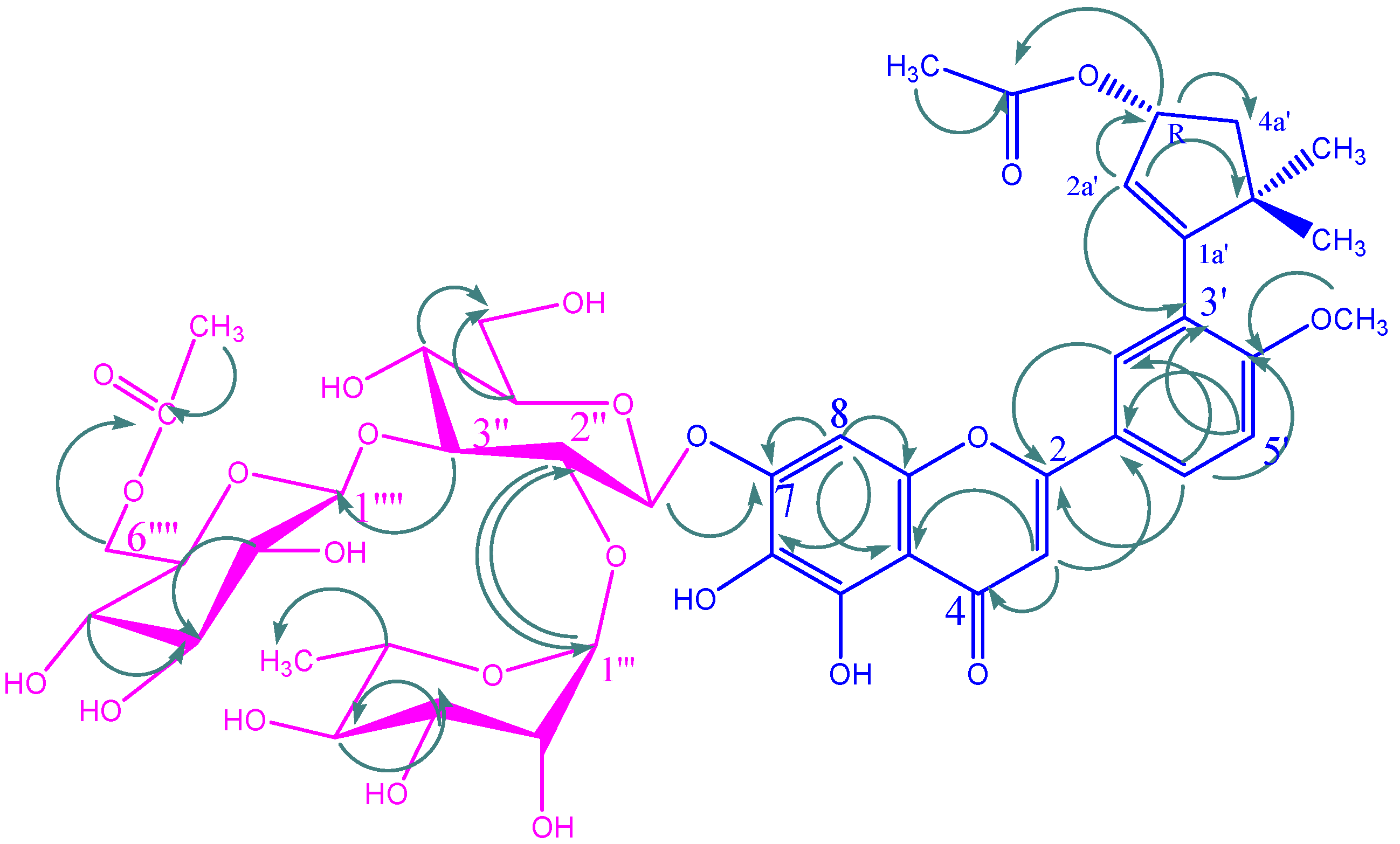
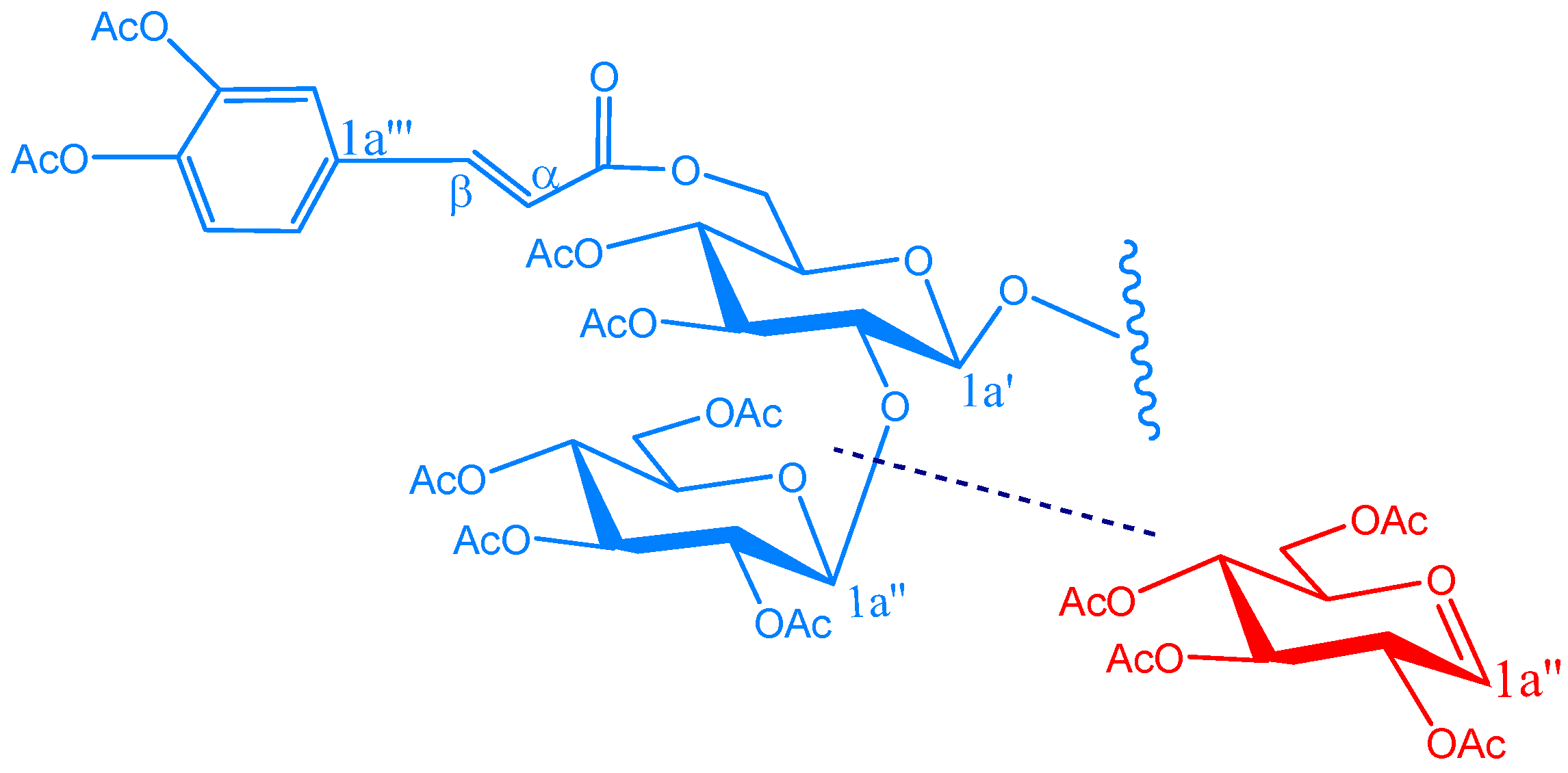{kind=link}
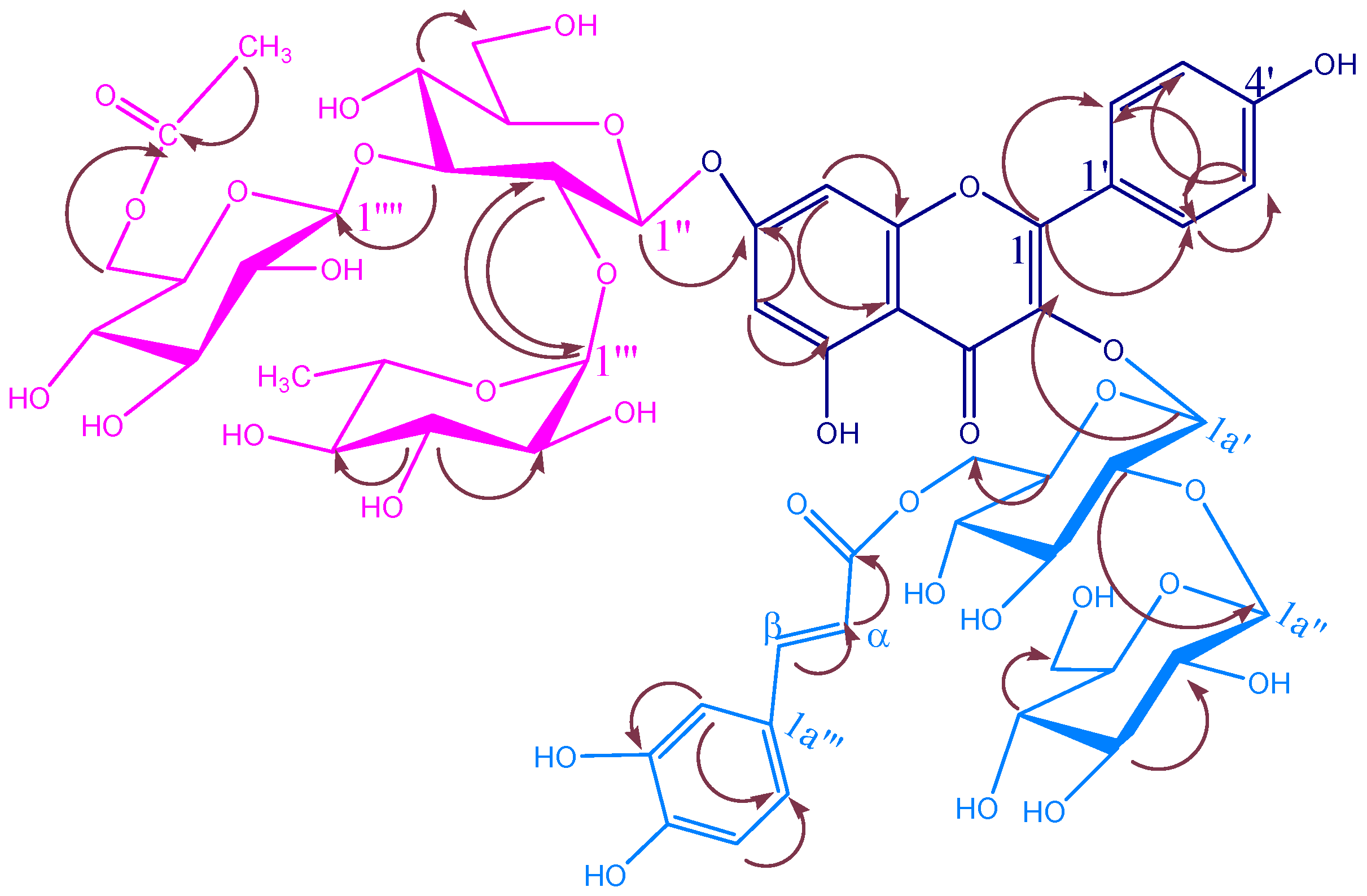{kind=link}
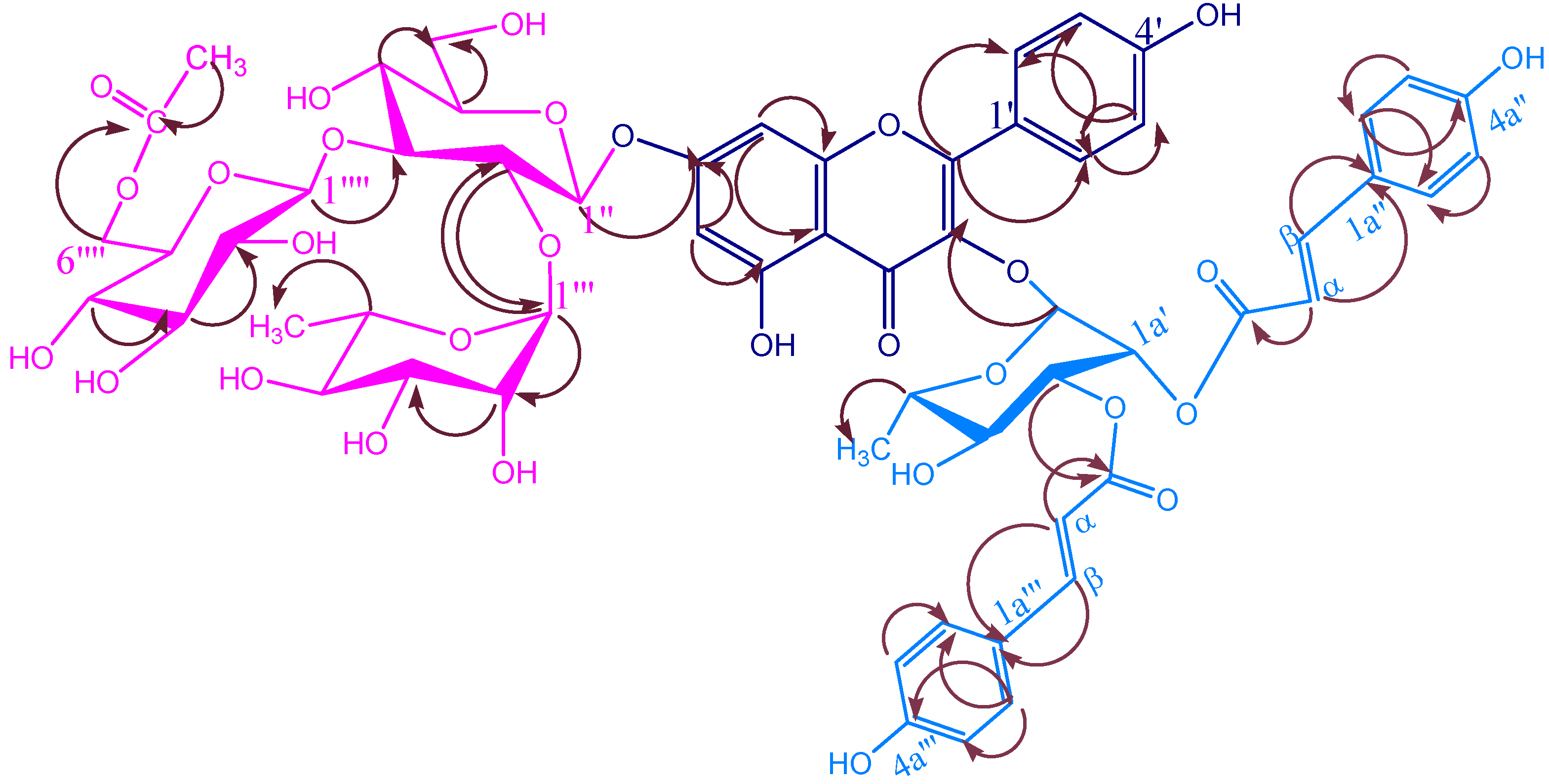{kind=link}
| C/H | δC | δH (m, J Hz) | C/H | δC | δH (m, J Hz) |
|---|---|---|---|---|---|
| Aglycone | |||||
| 2 | 163.84 | 8a | 105.58 | ||
| 3 | 102.89 | 6.79 (s) | 1′ | 123.01 | |
| 4 | 182.12 | 2′ | 112.86 | 7.44 (d, 2.3) | |
| 4a | 148.78 | 3′ | 146.78 | ||
| 5 | 146.78 | 4′ | 151.10 | ||
| 6 | 130.75 | 5′ | 112.01 | 7.12 (d, 8.7) | |
| 7 | 150.87 | 6′ | 118.60 | 7.55 (dd, 8.7, 2.3) | |
| 8 | 93.70 | 6.98 (s) | OCH3 | 55.71 | 3.88 (s) |
| 3′-cyclopent-1-ene | |||||
| 1a′ | 158.20 | 5a′ | 26.30 | ||
| 2a′ | 132.70 | 5.02 (d, 7.2 ) | 3a′-CO | 168.3 | |
| 3a′ | 74.20 | 5.12 (ddd,12.8, 4.9, 5) | CO-CH3 | 18.7 | 2.06 (s) |
| 4aα′ | 29.60 | 1.94 (m, dd, 12.8) | 5a′-αCH3 | 16.5 | 1.12 (s) |
| 4aβ′ | 29.60 | 1.66 (m, dd) | 5a′-βCH3 | 16.5 | 1.12 (s) |
| 7-O-glucopyranosyl | |||||
| 1′′ | 97.27 | 5.49 (d, 7.3) | 5′′ | 76.85 | 3.25 (t, 9.8) |
| 2′′ | 76.53 | 3.81 (d, 9.4) | 6′′ | 60.89 | 3.44 (dd, 12.0, 6.0) |
| 3′′ | 86.63 | 3.73 (t, 9.4) | OCOCH3 | 20.38 | 1.94 (s) |
| 4′′ | 68.14 | 3.44† | OCOCH3 | 170.04 | |
| 2′′-O-rhamnopyranosyl | |||||
| 1′′′ | 101.03 | 5.21 (d, 1.5) | 4′′′ | 71.97 | 3.18 (t, 9.8) |
| 2′′′ | 70.20 | 3.85† | 5′′′ | 68.89 | 3.64 (dd, 9.5, 6.1) |
| 3′′′ | 70.20 | 3.37 (m) | 6′′′ | 17.78 | 1.04 (d, 6.1) |
| 3′′-O-glucopyranosyl | |||||
| 1′′′′ | 103.01 | 4.42 (d, 7.6) | 4′′′′ | 69.93 | 3.13 (t, 9.8) |
| 2′′′′ | 73.33 | 3.13 (t, 9.8) | 5′′′′ | 72.91 | 3.85† |
| 3′′′′ | 76.53 | 3.22 (t, 9.2) | 6′′′′ | 62.89 | 4.10 (dd, 11.9, 6.7), 4.32 (brd, 9.8) |
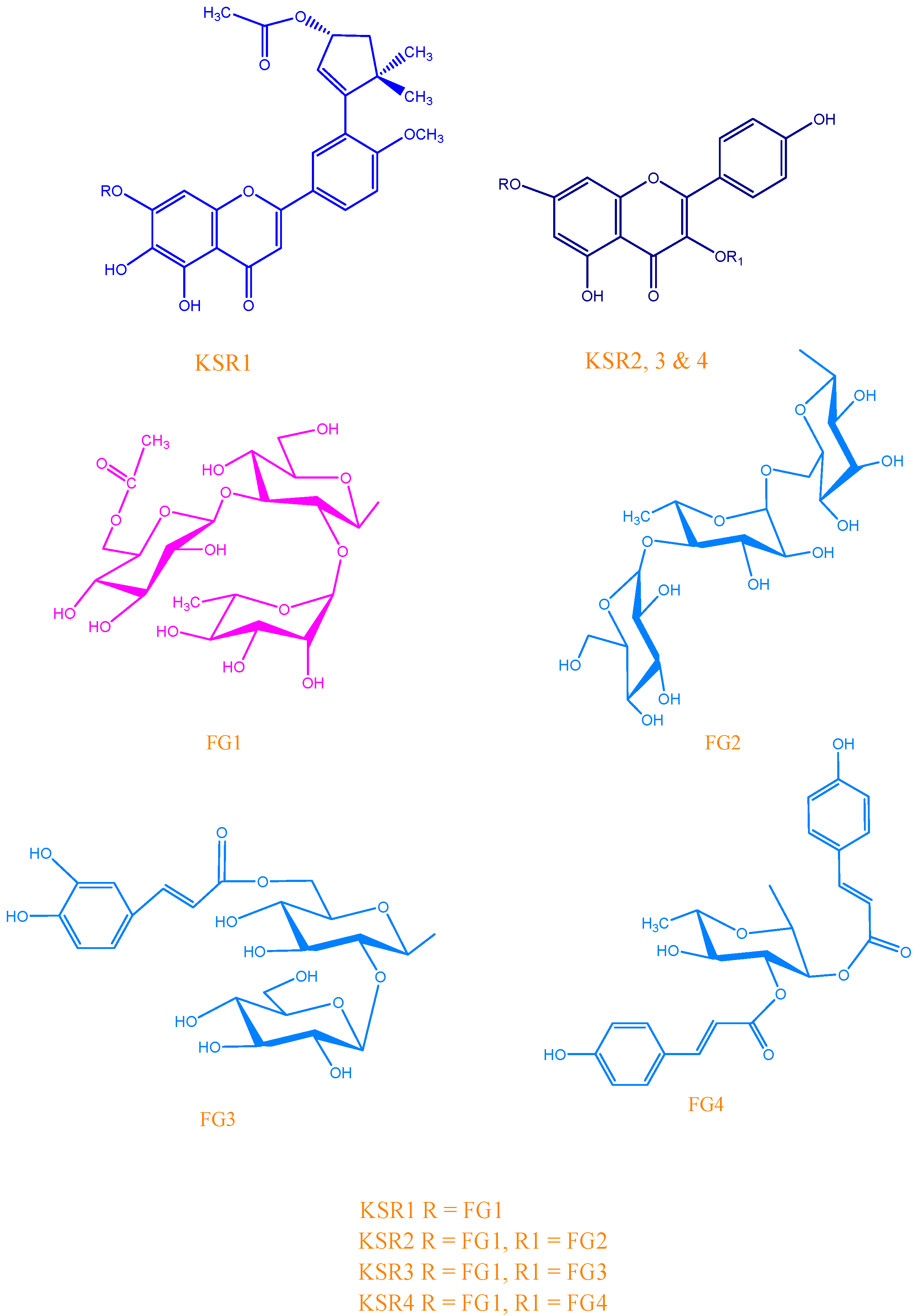
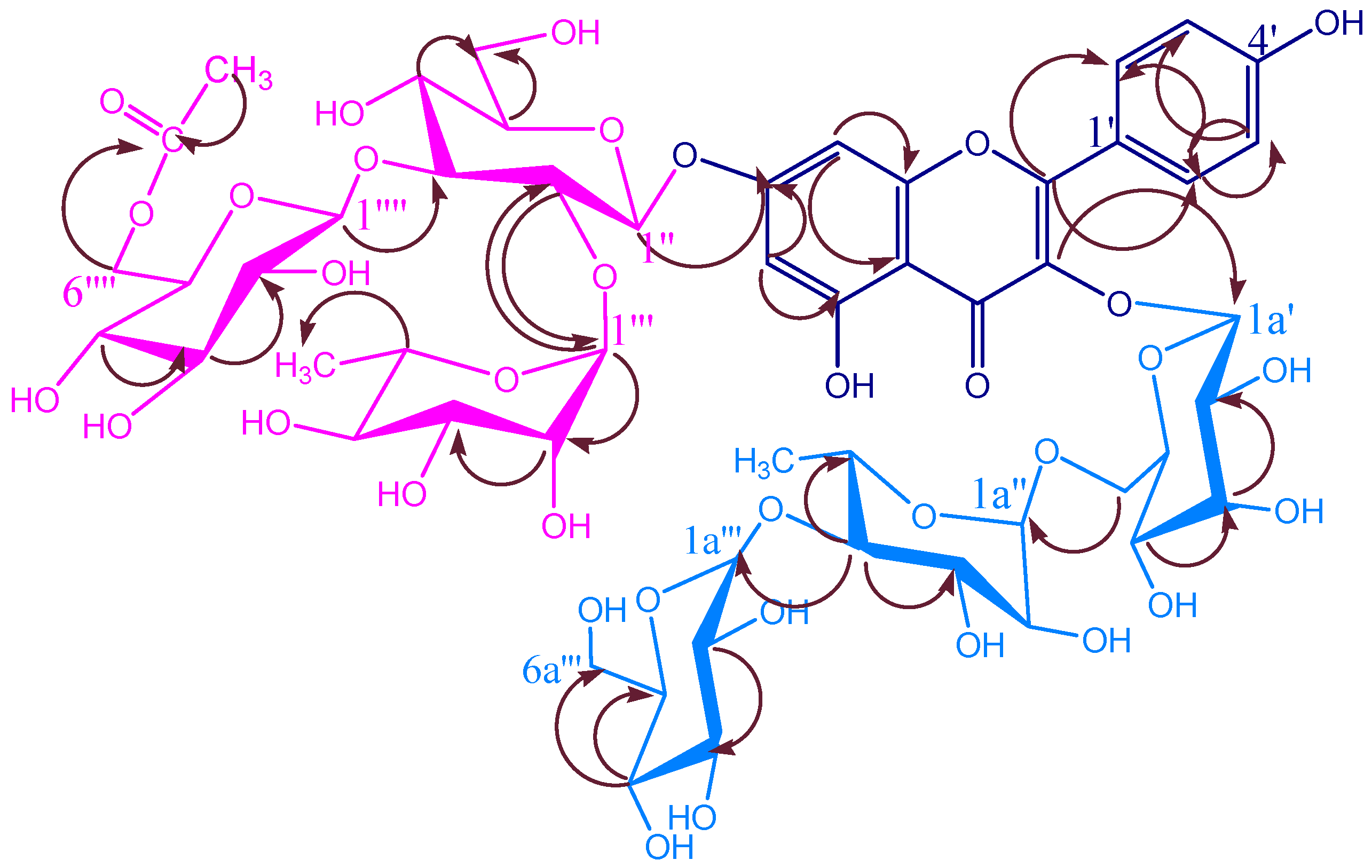
KSR2
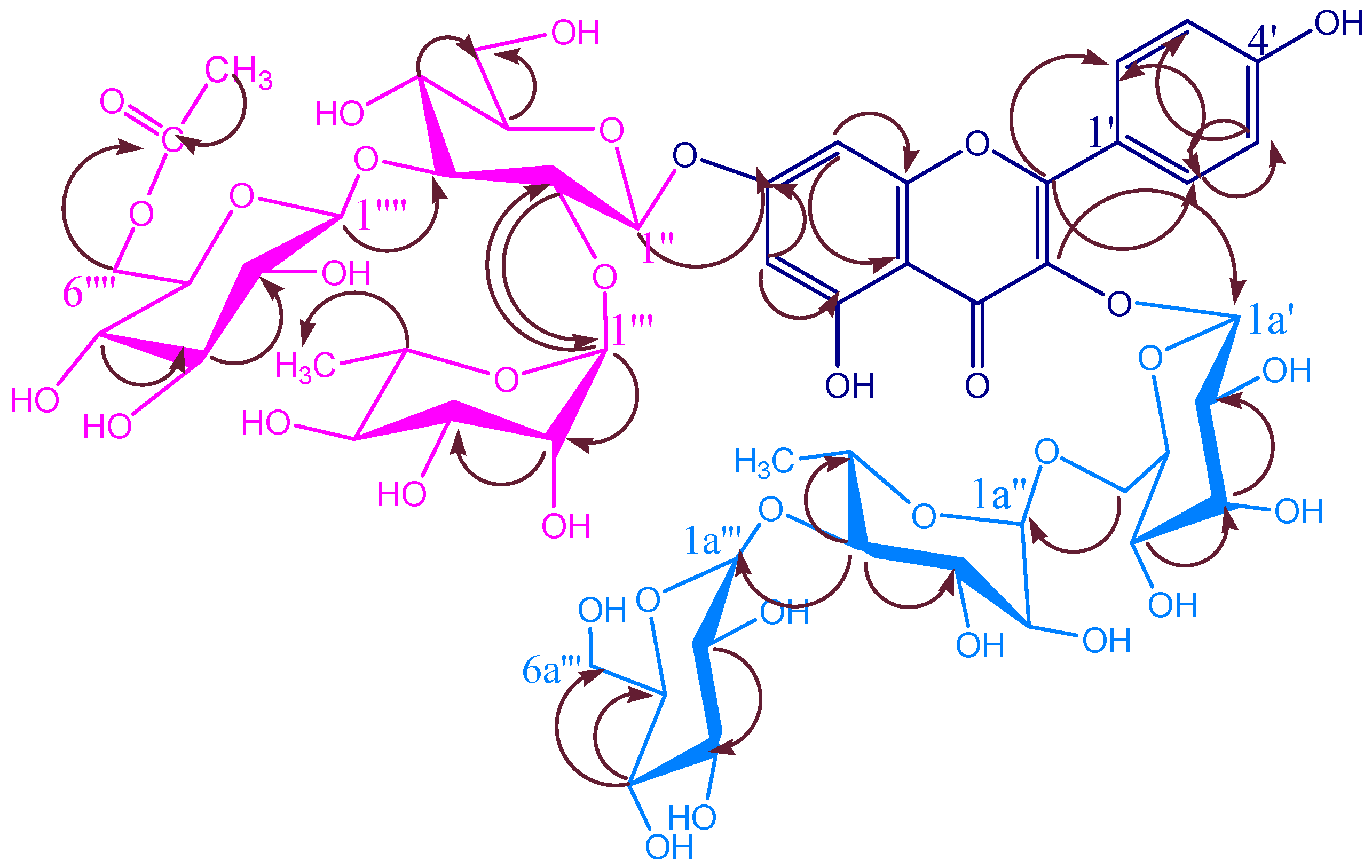
| C/H | KSR2 | KSR3 | KSR4 | |||
|---|---|---|---|---|---|---|
| δC | δH (m, J) | δC | δH (m, J) | δC | δH (m, J) | |
| Aglycone | ||||||
| 2 | 156.7 | 156.5 | 156.7 | |||
| 3 | 133.5 | 133.1 | 133.5 | |||
| 4 | 177.5 | 177.3 | 177.5 | |||
| 4a | 104.0 | 104.1 | 104.0 | |||
| 5 | 161.3 | 161.2 | 161.3 | |||
| 6 | 99.0 | 6.20 (s) | 98.8. | 6.22 (s) | 99.0 | 6.20 (s) |
| 7 | 164.6 | 164.1 | 164.6 | |||
| 8 | 93.9 | 6.42 (s) | 93.7 | 6.41 (s) | 93.9 | 6.42 (s) |
| 8a | 156.6 | 156.3 | 156.6 | |||
| 1′ | 121.0 | 121.6 | 121.0 | |||
| 2′ | 131.1 | 8.05 (d, 8.6) | 131.1 | 8.04 (d, 8.6) | 131.1 | 8.05 (d, 8.6) |
| 3′ | 115.2 | 6.87 (d, 8.6) | 115.2 | 6.9 (d, 8.6) | 115.2 | 6.87 (d, 8.6) |
| 4′ | 160.1 | 160.3 | 160.1 | |||
| 5′ | 115.2 | 6.87 (d, 8.6) | 115.3 | 6.9 (d, 8.6) | 115.2 | 6.87 (d, 8.6) |
| 6′ | 131.1 | 8.05 (d, 8.6) | 131.2 | 8.04 (d, 8.6) | 131.1 | 8.05 (d, 8.6) |
| 7-O-glucopyranosyl | ||||||
| 1′′ | 97.27 | 5.49 (d, 7.3) | 98.20 | 5.46 (d, 7.3) | 97.30 | 5.49 (d, 7.3) |
| 2′′ | 76.53 | 3.81 (d, 9.4) | 76.55 | 3.84 (d, 9.4) | 76.50 | 3.87 (d, 9.4) |
| 3′′ | 86.63 | 3.73 (t, 9.4) | 86.62 | 3.72 (t, 9.4) | 86.77 | 3.76 (t, 9.4) |
| 4′′ | 68.14 | 4.08 (dd, 3.3, 1.8) | 68.15 | 4.68 (dd, 3.3, 1.8) | 68.14 | 4.07 (dd, 3.3, 1.8) |
| 5′′ | 76.85 | 3.25 (t, 9.8) | 76.85 | 3.52 (t, 9.8) | 76.88 | 3.30 (t, 9.8) |
| 6′′ | 60.89 | 3.44 (dd, 12.0, 6.0) | 60.89 | 3.46 (dd, 12.0, 6.0) | 60.86 | 3.43 (dd, 12.0, 6.0) |
| OCOCH3 | 20.38 | 1.94 (s) | 20.38 | 1.95 (s) | 20.50 | 2.00 (s) |
| OCOCH3 | 170.04 | 170.50 | 170.60 | |||
| 2′′-O-rhamnopyranosyl | ||||||
| 1′′′ | 101.03 | 5.21 (d, 1.5) | 101.01 | 5.20 (d, 1.5) | 101.00 | 5.25 (d, 1.5) |
| 2′′′ | 70.20 | 3.85† | 70.24 | 3.85† | 70.20 | 3.85† |
| 3′′′ | 70.20 | 3.37 (m) | 70.20 | 3.37 (m) | 70.20 | 3.38 (m) |
| 4′′′ | 71.97 | 3.18 (t, 9.8) | 71.99 | 3.20 (t, 9.8) | 72.03 | 3.19 (t, 9.8) |
| 5′′′ | 68.89 | 3.64 (dd, 9.5, 6.1) | 68.88 | 3.60 (dd, 9.5, 6.1) | 68.89 | 3.64 (dd, 9.5, 6.1) |
| 6′′′ | 17.78 | 1.04 (d, 6.1) | 17.80 | 1.06 (d, 6.1) | 17.77 | 1.05 (d, 6.1) |
| 3′′-O-glucopyranosyl | ||||||
| 1′′′′ | 103.01 | 4.42 (d, 7.6) | 103.05 | 4.46 (d, 7.6) | 103.04 | 4.45 (d, 7.6) |
| 2′′′′ | 73.33 | 3.13 (t, 9.8) | 73.33 | 3.15 (t, 9.8) | 73.35 | 3.17 (t, 9.8) |
| 3′′′′ | 76.53 | 3.24 (t, 9.2) | 76.53 | 3.22 (t, 9.2) | 76.57 | 3.26 (t, 9.2) |
| 4′′′′ | 69.93 | 3.16 (t, 9.8) | 69.93 | 3.14 (t, 9.8) | 69.90 | 3.13 (t, 9.8) |
| 5′′′′ | 72.91 | 3.85(m) | 72.96 | 3.89 (m) | 72.98 | 3.88(m) |
| 6′′′′ | 62.89 | 4.15 (dd, 11.9, 6.7) 4.35 (brd, 9.8) | 62.89 | 4.10 (dd, 11.9, 6.7) 4.36 (brd, 9.8) | 62.86 | 4.10 (dd, 11.9, 6.7) 4.38 (brd, 9.8) |
KSR3
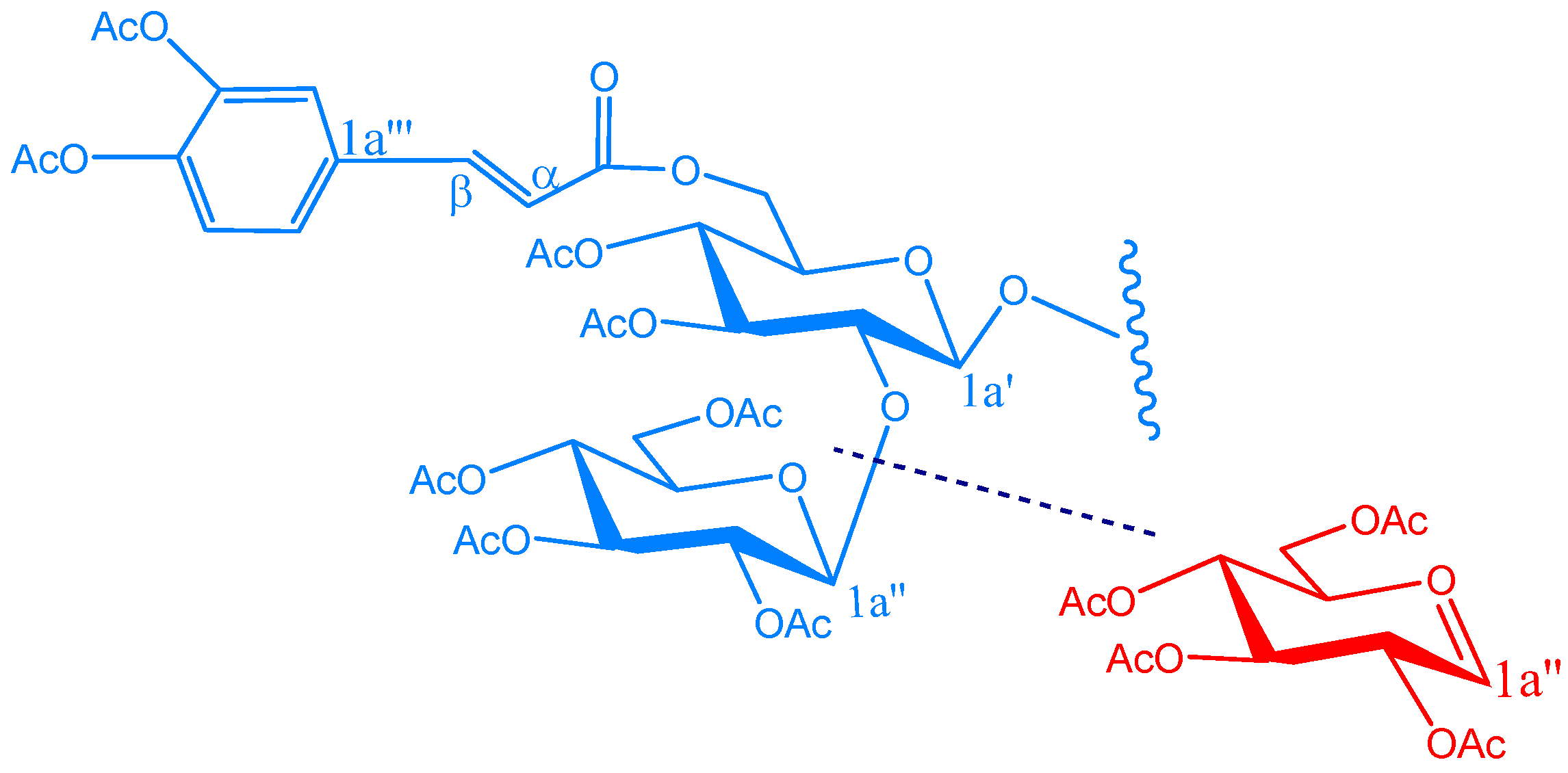
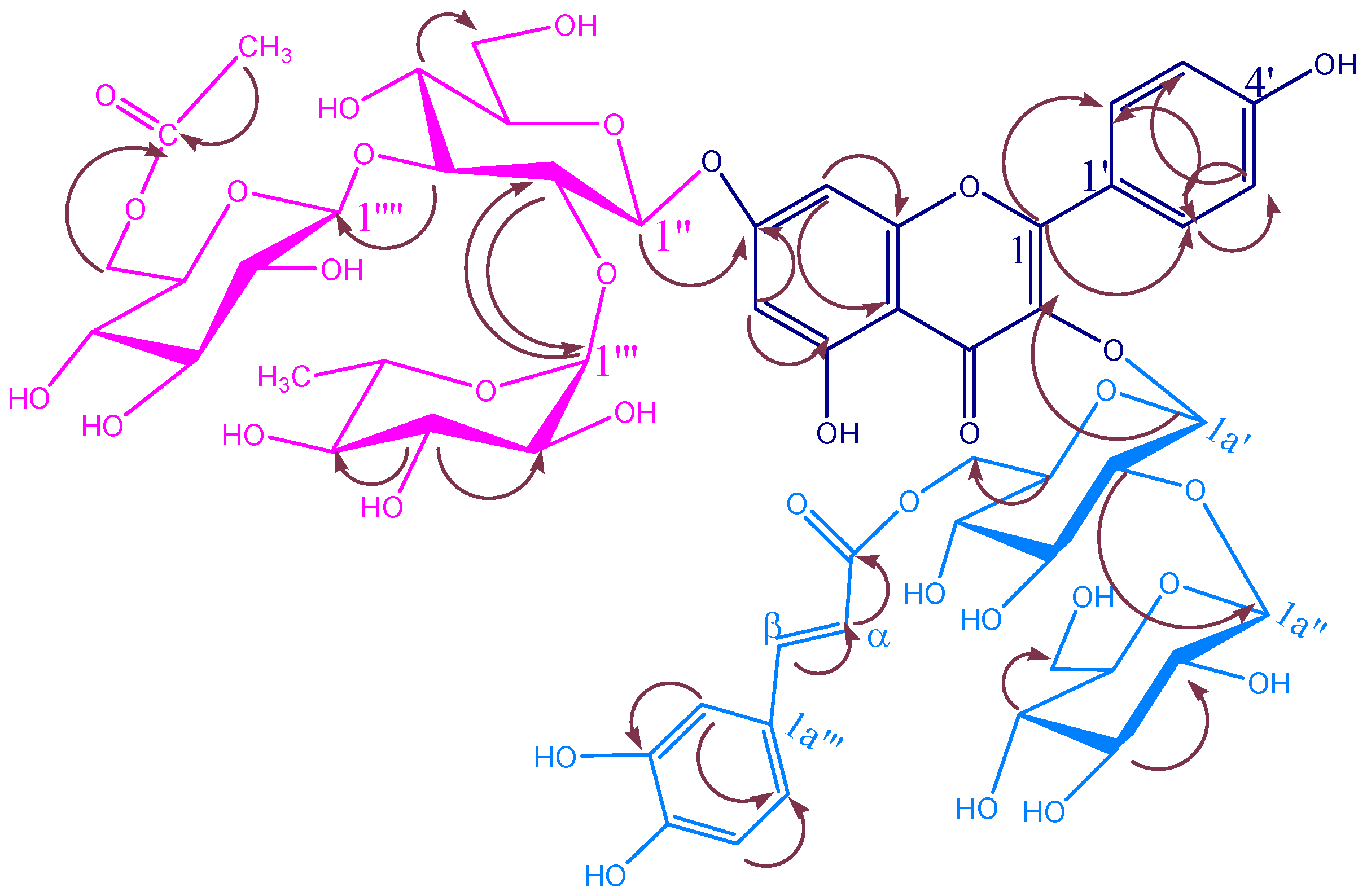
| KSR2 | |||||
|---|---|---|---|---|---|
| C/H | δC | δH (m, J Hz) | C/H | δC | δH (m, J Hz) |
| 3-O-galactopyranosyl | |||||
| 1a′ | 102.3 | 5.30 (d, 7.7 ) | 4a′ | 68.1 | 3.83 (dm, 3.3) |
| 2a′ | 71.2 | 3.57 (dd, 9.8,7.4) | 5a′ | 73.6 | 3.47 (ddm, 7.5, 5) |
| 3a′ | 73.1 | 3.51 (dd, 9.8, 3.3) | 6a′ | 65.4 | 3.23 (dd, 11.7, 7.5) 3.62 (dd, 11.7, 5) |
| 4a′′-O-glucopyranosyl | |||||
| 1a′′′ | 104.6 | 4.34 (d, 7.8) | 4a′′′ | 70.2 | 3.02 (m) |
| 2a′′′ | 74.6 | 2.99 (dd, 9.8, 7.7) | 5a′′′ | 77.0 | 3.55 (m) |
| 3a′′′ | 76.7 | 3.15 (dd, 8.7, 7.4) | 6a′′′ | 61.3 | 3.44 (dd, 11.5, 5.2) 3.67 (d, 10.9) |
| 6a′-O-rhamnopyranosyl | |||||
| 1a′′ | 100.0 | 4.42 (brd, 1.8) | 4a′′ | 82.3 | 3.37 (d, 9.5) |
| 2a′′ | 70.2 | 3.48 (dd, 3.3, 1.8) | 5a′′ | 66.8 | 3.47 (dq, 9.5,6.2) |
| 3a′′ | 70.7 | 3.56 (dd, 9.5, 3.3) | 6a′′ | 17.9 | 1.16 (s) |
| KSR3 | |||||
| C/H | δC | δH (m, J Hz) | C/H | δC | δH (m, J Hz) |
| 3-O-glucopyranosyl | |||||
| 1a′ | 101.5 | 4.30 (d, 7.5) | 4a′ | 70.0 | 3.03 (dm, 3.9) |
| 2a′ | 82.2 | 3.61 (dd, 8.9, 7.5) | 5a′ | 75.8 | 3.55 (ddm, 8.7, 5.8) |
| 3a′ | 75.7 | 3.15 (8.6, 3.5) | 6a′ | 64.6 | 3.44 (dd, 11.6, 7.5) 3.67 (dd, 11.7, 5) |
| 6a′-O-caffeoyl | |||||
| αa′′ | 114.5 | 5.59 (d, 15.6) | 4a′′ | 146.9 | |
| βa′′ | 146.4 | 7.30 (d, 15.6) | 5a′′ | 116.3 | 6.62 (d, 8.1) |
| 1a′′ | 127.4 | 6a′′ | 122.8 | 6.69 (d, 8.1) | |
| 2a′′ | 115.0 | 6.79 (bs) | COO | 169.1 | |
| 3a′′ | 149.3 | 6.75 (d, 8.7) | |||
| 2a′-O-glucopyranosyl | |||||
| 1a′′ | 105.6 | 4.81 (d, 7.8) | 4a′′ | 71.7 | 3.53 (d, 8.9) |
| 2a′′ | 74.7 | 3.19 (dd, 8.1,3.3) | 5a′′ | 77.7 | 3.23 (m) |
| 3a′′ | 76.7 | 3.38 (dd, 8.9,3.4) | 6a′′ | 61.7 | 3.78 (m) 3.80 (m) |
| KSR4 | |||||
| C/H | δC | δH (m, J Hz) | C/H | δC | δH (m, J Hz) |
| 3-O-rhamnopyranosyl | |||||
| 1a′ | 100.1 | 5.60 (d, 1.7) | 4a′ | 70.9 | 3.64 (t, 9.7) |
| 2a′ | 70.8 | 5.83 (d, 1.7) | 5a′ | 72.1 | 3.56 (m ) |
| 3a′ | 72.9 | 5.30 (dd, 9.7, 3.4) | 6a′ | 17.7 | 1.06 (d, 6.3) |
| 2a′-O-E-p-coumaroyl | |||||
| αa′′′ | 114.8 | 6.28 (d, 15.9) | 4a′′′ | 161.5 | |
| βa′′′ | 147.6 | 7.60 (d, 15.9) | 5a′′′ | 116.8 | 6.75 (d, 8.7) |
| 1a′′′ | 127.0 | 6a′′′ | 131.3 | 7.37 (d, 8.4) | |
| 2a′′′ | 131.3 | 7.37 (d, 8.4) | COO | 168.4 | |
| 3a′′′ | 116.8 | ||||
| 3a′-O-E-p-coumaroyl | |||||
| αa′′′ | 114.3 | 6.36 (d, 15.9) | 4a′′′ | 161.3 | |
| βa′′′ | 146.9 | 7.62 (d, 15.9) | 5a′′′ | 116.7 | 6.80 (d, 8.7) |
| 1a′′′ | 126.9 | 6a′′′ | 131.1 | 7.44 (d, 8.4) | |
| 2a′′′ | 131.1 | 7.44 (d, 8.4) | COO | 167.7 | |
| 3a′′′ | 116.7 | 6.80 (d, 8.7) | |||
KSR4
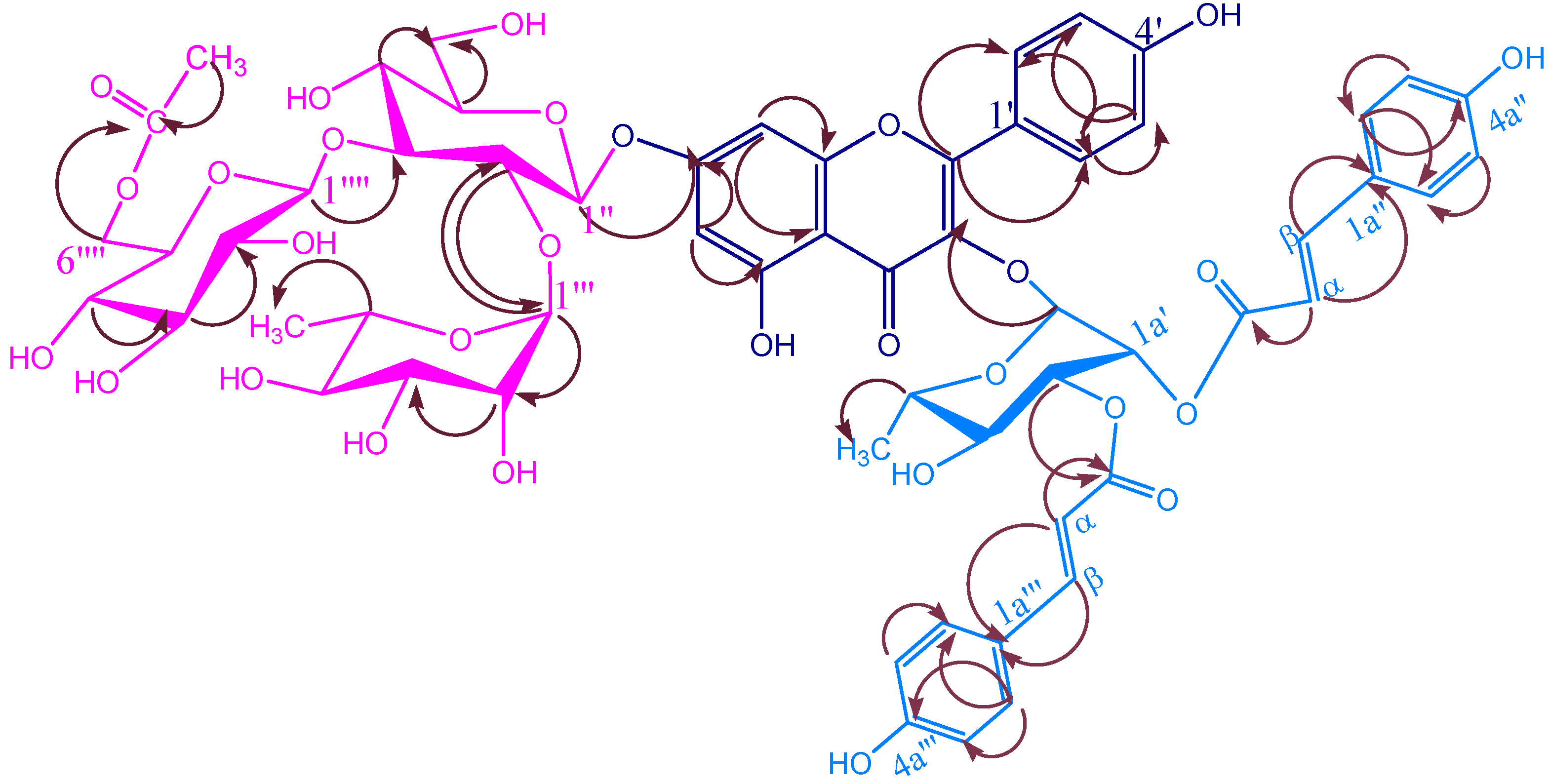
Biological activity
| A. niger | 1.2 (1.2) | 2.5 (2.5) | 0.6 (0.6) | 7.5 (7.5) | 0.6 (0.6) | 3.0 (8.0) | 0.3 (0.3) |
| A. ochraceus | 1.2 (1.2) | 2.5 (2.5) | 0.6 (0.6) | 7.5 (7.5) | 0.6 (0.6) | 3.0 (8.0) | 0.3 (0.3) |
| A. versicolor | 0.6 (1.2) | 2.5 (2.5) | 0.3 (0.6) | 7.5 (7.5) | 0.6 (1.2) | 4.0 (8.0) | 0.3 (0.3) |
| A. flavus | 1.2 (1.2) | 2.5 (2.5) | 0.6 (1.2) | 7.5 (7.5) | 0.3 (0.6) | 1.0 (8.0) | 0.3 (0.3) |
| P. ochrochloron | 1.2 (2.4) | 5.0 (5.0) | 1.2 (2.4) | 10.0 (12.0) | 0.6 (1.2) | 2.0 (10.0) | 0.45 (0.45) |
| P. funiculosum | 1.2 (2.4) | 1.25 (1.25) | 1.2 (2.4) | 10.0 (12.0) | 0.6 (1.2) | 4.0 (10.0) | 0.45 (0.45) |
| T. viride | 1.2 (2.4) | 5.0 (7.5) | 1.2 (1.2) | 10.0 (15.0) | 1.2 (2.4) | 4.0 (4.0) | 0.45 (0.45) |
| C. cladosporioides | 3.45 (3.75) | 2.4 (2.4) | 12.0 (10.0) | 8.0 (4.0) | 5.0 (5.0) | 0.06 (0.06) | 1.25 (1.25) |
| A. alternata | 5.0 (5.0) | 7.5 (7.5) | 6.0 (6.0) | 3.0 (8.0) | 10.0 (12.0) | 1.0 (1.0) | 1.25 (1.25) |
Experimental
General
Plant Material:
Hydrolysis of the isolated compounds
Acetylation of KSR3
Bioassays:
Acknowledgments
References
- Kaul, M.K. Medicinal Plants of Kashmit and Ladakh; Indus Publishing Co.: New Delhi, 1997; p. 144. [Google Scholar]
- Nandkarni, A.K. Indian Materia Medica; Popular Book Depot: Mumbai, 1954; p. 1108. [Google Scholar]
- Chopra, R.N.; Nayar, S.L.; Chopra, I.C. Glossary of Indian Medicinal Plants; Publication and Information Directorate: New Delhi, 1956; p. 222. [Google Scholar]
- Kirtikar, K.; Basu, B.D. Idian Medicinal Plants; Lalit Mohan Basu: Allahabad, 1993; p. 1420. [Google Scholar]
- Sarin, Y.K. Illustrated Manual of Herbal Drugs Used in Ayurveda; CSIR & ICMR: New Delhi, 1996; p. 62. [Google Scholar]
- Chang, M.S; Wan, J.S; Ming, J.D.; Jang, J.L.; Gum, H.L. Cytotoxic Sesquiterpene Lactones from the Root of Saussurea lappa. J. Nat. Prod. 2003, 66, 1175–80. [Google Scholar] [CrossRef]
- Pai, P.P.; Kulkarni, G.H. Isolation of α-amyrin sterate, β-amyrin and lupeol palmitates from the costus leaves. Curr. Sci. 1997, 46, 261–62. [Google Scholar]
- Bruno, M.; Gunther, O. (E)-9-isopropyl-6-methyl-5,9-decadiene-2-one, a terpenoid C18-ketone with a novel skeleton. J. Chem.Soc. 1997, 353–54. [Google Scholar]
- Singh, I.P.; Talwar, K.K.; Arora, J.K.; Chhabra, B.R.; Kalsi, P.S. A biologically active guaianolide from Saussurea lappa. Phytochemistry 1992, 31, 2529–31. [Google Scholar] [CrossRef]
- Talwar, K.; Singh, I.P.; Kalsi, P.S. A sesquiterpenoid with plant growth regulatory activity from Saussurea lappa. Phytochemistry 1992, 31, 336–38. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Hatakeyama, S.; Inoue, Y.; Yamahara. Saussureamines A, B, C, D, and E, new anti-ulcer principles from Chinese Saussureamines Radix. J. Chem. Pharm. Bull. 1993, 41, 214–16. [Google Scholar] [CrossRef]
- Kalsi, P.S; Kumar, S; Jawanda, G.S.; Chhabra, B.R. Guaianolides from Saussurea lappa. Phytochemistry 1995, 40, 1713–15. [Google Scholar] [CrossRef]
- Dirk, C.A; Renee, J.G.; Soren, R.J.; Fevi, O; Nigel, C.V. Acylated flavone glycoside from Veronica. Phytochemistry 2003, 64, 1295–01. [Google Scholar]
- Kemp, W. Organic Spectroscopy; MacMillan: London, New York, 1991; pp. 66, 194. [Google Scholar]
- Mohamd, A. Techniques in Terpenoid identification; Birla Publication: New Delhi, 2001; p. 176. [Google Scholar]
- Tuchinda, P; Udhcacho, J.; Reutrakul, V.; Santisuk, T.; Skeleton, B.W.; White, A.H.; Taylor, W.C. Pimarane Diterpenes from Kaemperia. Phytochemistry 1994, 36, 731–34. [Google Scholar]
- Nasser, A.; Singh, B. Acetylated flavonol triglycosides from Ammi majus L. Phytochemistry 1998, 49, 2177–2180. [Google Scholar] [CrossRef]
- Emam, A.M.; Elias, R; Maussa, A.M.; Faure, R.; Debrauwer, L.; Balansard, G. Two flavonoid triglycosides from buddleja madagascariensis. Phytochemistry 1998, 48, 739–42. [Google Scholar] [CrossRef]
- Joao, F.; Castelao, J.R.; Otto, R.G.; Robexto, A.D.; Antonio, A.L.M. Xanthonolignoids from Kielmeyera and Caraipa Species 13C-NMR spectroscopy for Xanthones. Phytochemistry 1977, 16, 735–40. [Google Scholar] [CrossRef]
- Amaro-Luis, J.M.; Moser, J.; Yuen, D.A.; Larsen, T.B.; Matyska, C.; Li, T.-M.; Li, W.-K.; Yu, J.-G. Flavonoids from Artabotrys Hexapetalus. Phytochemistry 1997, 45, 831–33. [Google Scholar] [CrossRef]
- Nkengfack, A.E.; Fomum, Z.T.; Ubillas, R.; Sanson, D.R.; Tempesta, M.S. Extractives from Erythrina Eriotrocha. J. Nat. Prod. 1990, 53, 509–12. [Google Scholar] [CrossRef]
- Saracoglu, I.; Varel, M.; Calis, I.; Donmez, A.A. Neolignan, Flavonoid, Phenylethanoid and Iridoid Glycosides from Phlomis intergrigolia. Turk. J. Chem. 2003, 27, 739–47. [Google Scholar]
- Agrawal, P.K. NMR Spectroscopy in the structural elucidation of oligosaccharides and glylcosides. Phytochemistry 1992, 31, 3307–30. [Google Scholar] [CrossRef]
- Voutquenene, L.; Kokougan, C.; Lavad, C.; Pouny, I.; Litaudon, M. Triterpenoid Saponins and acylated prosapogenins from Harpullia Ausro Caledonica. Phytochemistry 2002, 59, 825–832. [Google Scholar] [CrossRef]
- Alexander, S.S.; Nikolay, E.N.; Vera, Yu. A.; Nikolay, K.K. 1H and 13C-NMR data for 2-O-, 3-O- and 2,3-Di-O-glycosylated methyl α- and β-D-glucopyranosides and β-D-galactopyranosides. Mag. Res. Chem. 1993, 31, 599–613. [Google Scholar] [CrossRef]
- He, Z.; Qiao, C.; Han, Q.; Wang, Y.; Ye, W.; Xu, H. New Triterpenoid Saponins from the roots of Platycodon Grandiflorum. Tetrahedron 2005, 61, 2211–2215. [Google Scholar] [CrossRef]
- Bradbury, H.; Jenkins, J. Determination of the Structure of Trisaccharides by 13C-NMR Spectroscopy. Carbohydr. Res. 1984, 126–156. [Google Scholar]
- Harborne, J.B.; Mabry, T.J. The Flavonoids: Advances in Research; Chapmann & Hall: London, New York, 1982. [Google Scholar]
- Bloor, S.J.; Bradley, J.M.; Lewis, D.H.; Davies, K.M. Identification of flavonol and anthocyanin metabolites in leaves of Petunia Mitchell and its LC Transgenic. Phytochemistry 1998, 49, 1427–30. [Google Scholar]
- Miyase, T.; Iwata, Y.; Ueno, A.; Tenuifolioses, G-P. Oligosaccharide multi-esters form the roots of poygalatenuifolia willd. Chem. Pharm. Bull. 1992, 40, 2741–2748. [Google Scholar] [CrossRef]
- Komori, T.; Ida, Y.; Mutou, Y.; Miyahara, K.; Nohara, T.; Kawasaki, T. Mass spectra of spirostanol and furostanol glycosides. Biomed. Mass Spectrum. 1975, 2, 65–77. [Google Scholar] [CrossRef]
- Tomas-Barberan, F.A.; Gil, M.I.; Ferrenres, F.; Tomas-Lorente, F. Flavonoid P-Coumaroyl and 8-hydroxyflavone Allosylglucosides in Some Labiatae. Phytochemistry 1992, 31, 3097–3102. [Google Scholar] [CrossRef]
- Fiorini, C.; David, B.; Fouraste, I.; Vercauteren, J. Acylated Kaempferol Glycosides from Laurus nobilis leaves. Phytochemistry 1998, 47, 821–25. [Google Scholar] [CrossRef]
- Aritomim, M.; Kawasaki, T. Three Higly Oxygenated Flavone Glucuronides in Leaves of Spinacia oleracea. Phytochemistry 1984, 23, 2043–47. [Google Scholar] [CrossRef]
- Booth, C. Fungal Culture Media. In Methods in Microbiology; Academic Press: London, New York, 1971; pp. 49–94. [Google Scholar]
- Hanel, H.; Raether, W. A more sophisticated method of determining the fungicidal effect of water insolube preparations with a cell harvester, using miconazole as an example. Mycoses 1988, 31, 148–54. [Google Scholar] [CrossRef]
- Daouk, R.K.; Dagher, S.M.; Sattout, E.J. Antifungal activity of the Essential oil of Origanum syriacum L. J. Food Prot. 1995, 58, 1147–49. [Google Scholar]
- Sample Availability: Very small samples are available from the authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Rao, K.S.; Babu, G.V.; Ramnareddy, Y.V. Acylated Flavone Glycosides from the Roots of Saussurea lappa and Their Antifungal Activity. Molecules 2007, 12, 328-344. https://doi.org/10.3390/12030328
Rao KS, Babu GV, Ramnareddy YV. Acylated Flavone Glycosides from the Roots of Saussurea lappa and Their Antifungal Activity. Molecules. 2007; 12(3):328-344. https://doi.org/10.3390/12030328
Chicago/Turabian StyleRao, Kolisetty Sambasiva, Goriparthi Venu Babu, and Yemireddy Venkata Ramnareddy. 2007. "Acylated Flavone Glycosides from the Roots of Saussurea lappa and Their Antifungal Activity" Molecules 12, no. 3: 328-344. https://doi.org/10.3390/12030328
APA StyleRao, K. S., Babu, G. V., & Ramnareddy, Y. V. (2007). Acylated Flavone Glycosides from the Roots of Saussurea lappa and Their Antifungal Activity. Molecules, 12(3), 328-344. https://doi.org/10.3390/12030328