Abstract
Considering the potential bioactivities of natural product and natural product-like compounds with highly complex and diverse structures, the screening of collections and small-molecule libraries for high-throughput screening (HTS) and high-content screening (HCS) has emerged as a powerful tool in the development of novel therapeutic agents. Herein, we review the recent advances in divergent synthetic approaches such as complexity-to-diversity (Ctd) and biomimetic strategies for the generation of structurally complex and diverse indole-based natural product and natural product-like small-molecule libraries.
1. Introduction
The inherent biological activities of natural products are induced by specific and selective interactions with macromolecules in living organisms, which makes them potential bioactive compounds and drug candidates [1]. To allow facile access to natural product-based small-molecule modulators and potential therapeutic agents, natural product screening collections are essential [2]. Extensive investigations have been conducted for the isolation and synthesis of such screening collections [3,4,5,6]. However, the available collections of natural products are insufficient for processes such as high-throughput screening (HTS) and high-content screening (HCS), as they require hundreds of thousands of compounds [7].
To address the unmet needs associated with the construction of libraries which have a large number of natural products and related compounds, two different approaches, namely, combinatorial chemistry and diversity-oriented synthesis (DOS), have been broadly applied [8]. Natural product-based combinatorial chemical libraries are usually constructed by adding various building blocks to natural products with bioactivity and high synthetic accessibility in order to reduce the time and cost of drug candidate development [9]. Combinatorial chemistry has significantly contributed to the discovery and optimization of therapeutic compounds. However, the deficiency of skeletal diversity in natural product-based combinatorial chemical libraries could limit the application of undruggable targets and unbiased phenotypic screening [10,11]. Consequently, substantial synthetic efforts have been devoted to the construction of natural product-based libraries with a high degree of molecular skeletal and stereochemical diversity and complexity through the DOS strategy.
A synthetic approach based on the DOS strategy was introduced by Schreiber et al., allowing the construction of small-molecule libraries containing natural product-like scaffolds from simple starting materials through rapid synthetic transformations in a highly divergent and efficient fashion [12,13,14,15,16,17,18,19,20,21,22]. Various bioactive compounds were identified for undruggable targets and from unbiased phenotypic screening by screening DOS libraries of natural products and related structurally complex and diverse synthetic compounds, hence proving the robustness of this strategy.
Despite considerable success in the generation of natural product-based small-molecule libraries via the DOS strategy, there remains a demand for complementary synthetic approaches to various natural products and related compounds that provides greater structural diversity and complexity in a highly efficient manner. Thus, significant efforts towards the development of innovative synthetic approaches such as complexity-to-diversity (Ctd) and biomimetic synthetic strategies are underway. The purpose of this review is to briefly overview these two novel divergent synthetic strategies based on studies reported between 2014 and 2021. The discussion of DOS approaches for the generation of diverse natural-like compounds and discovery of bioactive compounds, found in the outstanding reviews of [14,18,20], are outside the scope of this review. In particular, indole is a biologically relevant structural heteroaromatic scaffold which is found in complex natural products and synthetic drugs [23,24,25,26,27,28]. The indole scaffold is a major target for the construction of small-molecule libraries for the discovery of therapeutic leads [29]. Several innovative synthetic methods and strategies have been developed to construct indole-based compound libraries to date. Thus, in this review we focus mainly on the divergent generation of indole-based natural products or related compounds via the Ctd and biomimetic synthetic strategies.
2. Complexity-to-Diversity Strategy for Rapid Generation of Indole-Based Natural Products and Related Compounds
In the typical total synthesis of natural products and the DOS approach for the generation of diverse natural products and related compounds, complex and intriguing structures of natural products are the goal and final target, not the starting point. However, the Ctd strategy considers the complexity and diversity in the synthetic scaffolds of the starting points. The Ctd strategy, introduced by Hergenrother et al. in 2013, employs systematic ring-distortion reactions for the construction of natural-product-like small-molecule collections using easily accessible complex natural products as starting points [30,31,32,33,34]. The Ctd approach achieves skeletal and stereochemical diversity via various chemoselective synthetic operations on the core ring region of the starting natural products while retaining their inherent stereochemistry and complexity. Biosynthetic pathways, with their delicate and efficient enzymatic routes which create complex and diverse natural products from common intermediates, inspired the development of the Ctd strategy. Moreover, owing to the structural complexity and uniqueness of the naturally-occurring skeletons in Ctd, diverse and discrete core structures can be easily generated through relatively simple chemical reactions such as ring expansion, contraction, breaking, and aromatization, as compared to the total synthesis of each natural product (Figure 1). Therefore, this strategy allows for the exploration of new biologically relevant chemical spaces. Initial reports have demonstrated structurally complex and commercially available natural products from diverse structural classes, such as abietic acid, adrenosterone, gibberellic acid, and quinine, as starting points for the Ctd strategy. The diversified core skeletons obtained via chemo-, regio-, and stereoselective chemical transformations have distinct 3D structural features which could have potential selective interactions with biopolymers. Indole alkaloids have been considered as a target and starting point for the Ctd strategy as well.
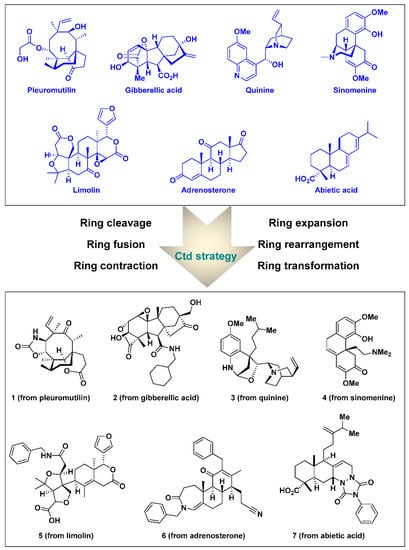
Figure 1.
Natural products as starting points in the complexity-to-diversity (Ctd) strategy along with representative compounds generated via ring-distortion reactions.
2.1. Ctd Strategy Based on Yohimbine (8)
Yohimbine (8) is a complex pentacyclic indole alkaloid extracted from the bark of the Pausinystalia tree [35]. It can act as an α2-adrenoceptor antagonist and is used to treat erectile dysfunction in men [36]. In 2017, Huigens et al. selected yohimbine (8) as the starting point for Ctd after considering its high complexity and easy availability [37].
In order to systematically and efficiently alter the core ring of yohimbine (8), they devised a ring-distortion strategy based on two known synthetic transformations, namely, C-N bond cleavage [38] and oxidative rearrangement of tryptoline, a substructure of yohimbine [39] (Scheme 1). Chemoselective ring cleavage reactions are powerful tools for ring distortion. To this end, cyanogen bromide (CNBr) was used to activate the tertiary amine of yohimbine (8) to ammonium ion 9, which could undergo orthogonal ring cleavage by different nucleophile attacks. Selective ring cleavage (pathway A, Scheme 1) occurred only in N,N-dimethylformamide (DMF) solvent, followed by bromide attack on the less hindered carbon via the SN2 pathway, resulting in the major product 10. In contrast, the ammonium ion intermediary 9 formed the diastereomeric mixture of 11 and 12 by simultaneous SN1 and SN2 processes (pathway B, Scheme 1) in a chloroform/alcohol solvent mixture. Interestingly, after the introduction of 2-iodobenzyl alcohol, the diastereomers (11 and 12) underwent a Cu(I)-catalyzed C–N coupling reaction to afford the ring fusion products 13 and 14, respectively. The ring rearrangement scaffold 15 was obtained via another tryptoline ring-distortion reaction involving oxidation and subsequent alkyl migration. Upon treatment with CNBr, spirooxindole 15 underwent selective C–N cleavage to afford compound 17.
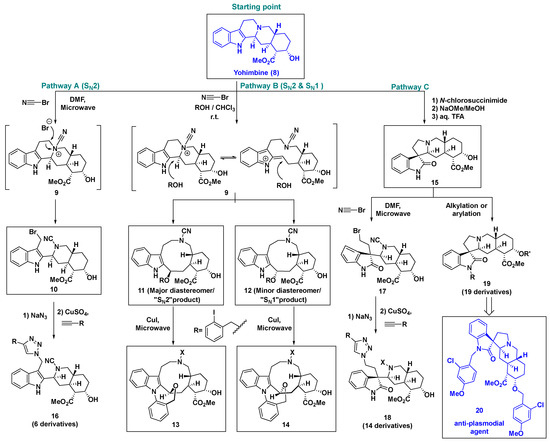
Scheme 1.
Ring distortion of yohimbine (8) for rapid access to complex and diverse indole-based natural product-like small molecules and identification of new anti-plasmodial agent compound 20.
This Ctd strategy based on ring-distortion transformations of yohimbine (8) is effective for the rapid generation of new natural product-like scaffolds with high complexity. The subsequent typical diversification pathways, such as functional group introductions or click chemistry (often used in combinatorial chemistry), afforded as many as seventy products. The phenotype-based screening of this yohimbine-based library has successfully led to the identification of a new bioactive compound demonstrating antiproliferative activity against hypoxia-inducible factor (HIF)-dependent cancer cells [37]. In addition, compound 20 (Scheme 1) was recently found to be potent against chloroquine-resistant Plasmodium falciparum Dd2 cells (EC50 = 0.33 μM) [40].
2.2. Ctd Strategy Based on Vincamine (21)
Vincamine (21) is a commercially available monoterpenoid indole alkaloid administered to patients to enhance global or regional blood flow [41,42,43]. In 2020, Huigens et al. developed a remarkable Ctd strategy for vincamine (21), synthesizing eight stereochemically and structurally complex natural product-like scaffolds via the systematic application of a ring-distortion pathway including ring cleavage, ring rearrangement, and ring fusion reactions (Scheme 2) [44]. First, two reported ring cleavage reactions were employed to afford two new natural product-like compounds, 22 and 23. Methyl propiolate was used to activate the tertiary amine of vincamine (21) to the ammonium ion, which was subsequently subjected to selective ring cleavage by methanol attack to form 23. Notably, 22 became another useful starting point for the Ctd strategy. The carboxylic acid of 22 could be converted to either the ester, 24, or the amide, 25. Stereo- and regioselective oxidation of C3 in the indole core ring of 24 by meta-chloroperbenzoic acid (m-CPBA) afforded the hydroxylated compound 26, which underwent base-promoted semipinacol rearrangement to yield the spirooxindole 27. In contrast, the amide 25 underwent unexpected ring fusion upon treatment with m-CPBA to afford the novel compound 29. It has been proposed that the selective oxidation of compound 25 to a 3-hydroxyindolenine intermediate followed by dehydration yields intermediate 28, which undergoes 5-exo-trig cyclization to furnish 29. In addition, CNBr-induced ring cleavage reactions yielded two new scaffolds, 30 and 31, from compound 24 and vincamine (21), respectively. Subsequently, a typical diversified synthetic method was applied to the newly synthesized eight natural product-like compounds, which produced an eighty-member small-molecule library with distinct natural product-like complex scaffolds. The robustness of this Ctd strategy for vincamine (21) has been successfully validated by the identification of a new bioactive compound, 33, which has antagonistic activity against hypocretin type 2 receptor (HCRTR2) and hence possesses therapeutic potential against opioid abuse [44].
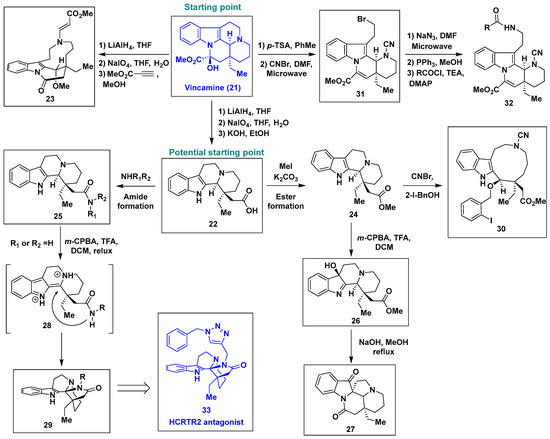
Scheme 2.
Ring distortion of vincamine (21) for rapid access to complex and diverse indole-based natural product-like small molecules and identification of a new HCRTR2 antagonist compound, 33. m-CPBA, meta-chloroperbenzoic acid; 2-I-BnOH, 2-iodobenzyl alcohol.
2.3. Ctd Strategy Based on Nature-Inspired Indole Monoterpenoids
More recently, the Al-Tel group reported a new Ctd strategy for the nature-inspired indole monoterpenoid compounds 36 and 37 [45], prepared from compounds 34 and 35 [46] (Scheme 3). The compounds 36 and 37 have inherent complexity and transformable moieties such as cyclohexanone, piperidine, and indole rings, which makes them efficient starting points for the Ctd strategy. Using 36 and 37, novel compounds such as indole [2,3-a]quinolizines (42), canthine-type alkaloids (39), and arborescidine-type alkaloids (44) were generated. Access to canthine-type scaffolds (39) was efficiently achieved by an unexpected reaction pathway upon treatment with electron-withdrawing-group-bearing hydrazines (38) in acetic acid. The proposed reaction mechanism involved the condensation reaction of compound 36 with phenylhydrazine (38) to yield the hydrazone intermediate 40. Subsequent ring cleavage via the cascade reactions of tautomerization, ring-opening, and elimination of the methoxy group afforded the intermediate 41. The diastereoselective intramolecular aza-Michael addition produced the canthine-type scaffold 39. In contrast, the reaction of 36 with electron-donating-group-bearing hydrazines (38) proceeded via the regioselective [3,3] sigmatropic rearrangement of the hydrazone intermediates, thus generating octahydroindole[2,3-a]quinolizine scaffolds (42) via the typical Fischer–Indole synthetic pathway. When compound 37 was exposed to oxidation conditions, unexpected oxidative rearrangement occurred followed by the 7-exo-trig aza-Michael addition reaction, which afforded the new arborescidine-like indole alkaloid 44.
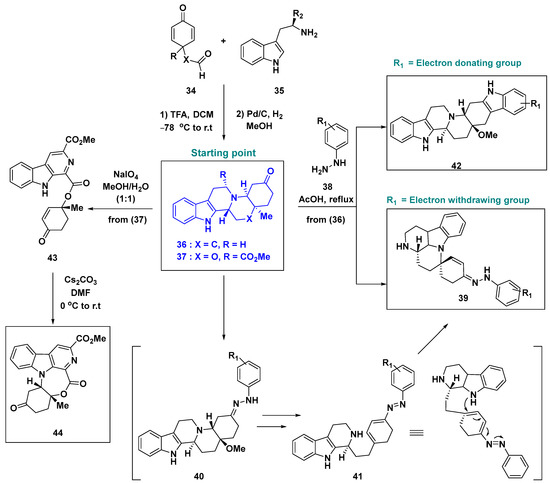
Scheme 3.
Ctd strategy based on nature-inspired indole monoterpenoids (36 and 37). Pd/C, palladium on carbon.
3. Biomimetic Synthetic Divergent Approaches to Indole-Based Natural Product and Natural Product-Like Small Molecules
Biologically active natural products are among the best targets for the introduction of biological assay systems to explore unknown biological pathways, which may lead to potential therapeutic candidates [3,4,5,6]. However, synthetic accessibility is an obstacle owing to the stereo- and skeletal-complexity of natural products, despite notable advances in total synthesis and methodology. In contrast, natural product biosynthesis in living organisms allows facile access to structurally complex and distinct skeletons from relatively simple common intermediates through enzyme-catalyzed processes. This tremendous efficiency and diversity-generating power of the biosynthetic pathways allows for the rapid generation of diverse natural products and related compounds, including indole-based compounds [45,46,47,48,49,50,51,52].
A biosynthetic reaction of tryptamine (45) and secologanin (46) yields the key intermediate and a biosynthetic branching point strictosidine (47), a precursor to other structurally diverse and biologically significant monoterpene indole alkaloids formed via the biosynthetic pathways of structural rearrangement, oxidation, and intramolecular cyclization [53]. Thus, strictosidine (47) makes an attractive biomimetic branching point to furnish structurally distinct and complex skeletons. Consequently, substantial research efforts were aimed at developing new biomimetic branching points. The Kozmin group developed a new pluripotent synthetic branching point, indoloquinolizidine (55), which was readily assembled from tryptamine derivatives (52), 1,3-dicarbonyl compounds (53), and itaconic anhydride (54) (Scheme 4) [54]. Key intermediates (55) were designed based on strictosidine (47), which could undergo a highly controlled diversification process to afford complex skeletal natural products including hydroxy indolenine (48), spirocyclic geissoschizine oxindole (49), ketoamide (50), and pumiloside (51) (Scheme 4a). The biosynthetic pathways of strictosidine (47) were mimicked by transforming the indoloquinolizidines (55) to skeletally diverse scaffolds such as hydroxyindoline (56), dihydroindolone (57), tricyclic ketoamide (58), and pyrroloquinolone (59) (Scheme 4b). This bioinspired synthetic strategy successfully generated an 847-member small-molecule library. More such biomimetic pathways have been reported in the literature [29].
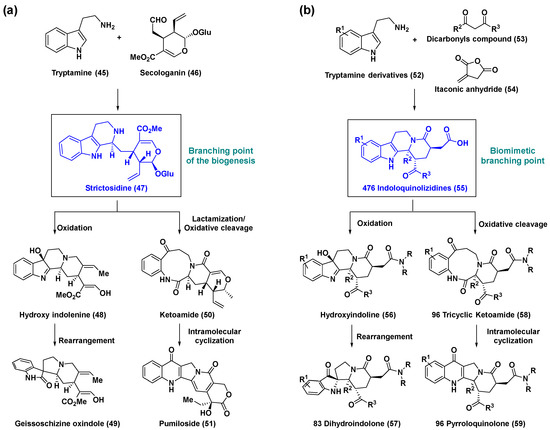
Scheme 4.
(a) Biosynthetic transformation of strictosidine (47), branching point of the biogenesis, and subsequent formation of monoterpenoid indole alkaloids. (b) Divergent synthetic pathways for the construction of an 847-member skeletally complex natural product-like compound library using a new biomimetic branching point, indoloquinolizidines (55).
3.1. Development of a New Biomimetic Divergent Branching Point from Dehydrosecodine (61)
Preakuammicine (60), synthesized from strictosidine (47), may be rearranged by two C–C bond cleavages to provide one of the most important intermediates in biosynthesis, dehydrosecodine (61), which comprises a dihydropyridine and a vinylindole. This intermediate, 61, allows easy access to generate the aspidosperma-type alkaloid tabersonine (62), iboga-type catharanthine (63), and andranginine (64), respectively, through distinct modes of [4 + 2] cycloaddition reactions (Scheme 5a) [55,56]. Despite its utility, dehydrosecodine (61) has not been isolated or synthesized because of its high reactivity and instability under oxidation conditions, which restrict its practical applications. Therefore, dehydrosecodine derivatives are an attractive option to design desired biomimetic pathways.
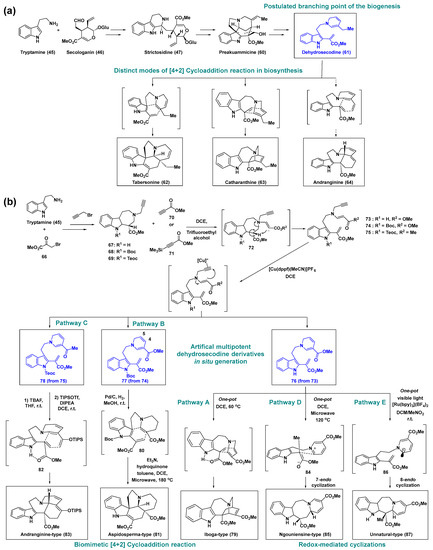
Scheme 5.
(a) Biosynthetic transformation of preakuammicine (60) via the postulated branching point, i.e., intermediate 61 to monoterpenoid indole alkaloids. (b) Biomimetic route for the preparation of monoterpenoid indole-type alkaloids via the transformation of dehydrosecodine derivatives (76–78), which act as biomimetic branching points. dppf, 1,1′-bis(diphenylphosphino)ferrocene; bpy, 2,2′-bipyridine; Teoc, 2-(trimethylsilyl)-ethoxycarbonyl.
Oguri et al. developed new artificial pluripotent dehydrosecodine derivatives (73–75), which were subjected to biomimetic [4 + 2] cycloadditions and redox-mediated cyclizations to generate five architecturally complex scaffolds (Scheme 5b) [57]. The introduction of additional electron-withdrawing substituents such as carbonyl groups allows feasible access to dehydrosecodine derivatives (73–75) and controls their regio- and stereoselectivity in the systematic intramolecular synthetic progress. First, the reactions of tryptamine (45) with other building blocks yielded tricyclic compounds (67–69), which were then converted to en-yne compounds (73–75) upon treatment with methyl propiolate (70) or 4-(trimethylsilyl)-3-butyn-2-one (71), respectively, via the formation of zwitterionic intermediate (72) and subsequent regioselective Hofmann elimination. The en-yne compounds (73–75) underwent 6-endo cyclization to generate dihydropyridine-vinylindole intermediates (76–78). This reaction pathway mimicked that of dehydrosecodine (61), without further intra- and intermolecular reactions of the resultant π-conjugated system in the presence of a Cu(I) catalyst. Interestingly, the en-yne compound 73 was transformed to the iboga-type compound 79 upon treatment with a Cu(I) catalyst in 1,2-Dichloroethene (DCE) at 60 °C via a one-pot cascade dihydropyridine formation and subsequent [4 + 2] cycloaddition reaction (Pathway A).
In contrast, compound 74, possessing a Boc group at the indole N1 position, was transformed to the dehydrosecodine derivative 77 by Cu(I)-catalyzed dihydropyridine formation without a subsequent cycloaddition. It was suggested that the hydrogen at the indole N1 position forms a hydrogen bond with the carbonyl oxygen of the α,β-unsaturated methylester, resulting in the increased electrophilicity of the dienophile and fixed orientation, thus rendering the subsequent [4 + 2] cycloaddition reaction more pronounced. Based on this observation, compound 74 was subjected to Cu(I)-catalyzed dihydropyridine formation; the subsequent site-selective hydrogenation of the C4-C5 double bond furnished an isolable tetrahydropyridine intermediate, 80, which did not lead to in situ iboga-skeleton formation or dihydropyridine oxidation (Pathway B). Finally, in the presence of hydroquinone and trimethylamine, the tetrahydropyridine intermediate 80 underwent a distinct mode of [4 + 2] cycloaddition followed by thermal decomposition of the Boc group (180 °C in a microwave) to furnish the aspidosperma-type skeleton compound 81.
Furthermore, the tricyclic compound 69, with a 2-(trimethylsilyl)-ethoxycarbonyl (Teoc) group on the indole nitrogen, was converted upon treatment with 4-(trimethylsilyl)-3-butyn-2-one (71) into en-yne compound 75, possessing a methyl ketone group. The resultant en-yne compound, 75, underwent Cu(I)-catalyzed dihydropyridine formation to afford the dehydrosecodine derivative intermediate 78, which was then transformed to silyl enol ether intermediate, 82, when the Teoc group was removed. The distinct mode of [4 + 2] cycloaddition spontaneously produced the andranginine-type scaffold 83 (Pathway C).
Notably, the dihydropyridine-vinylindole intermediate 76 was found to be capable of undergoing redox-mediated cyclization reactions as well as [4 + 2] cycloaddition. The ngouniensine-type alkaloid 85 was produced by microwave-assisted heating of the dihydropyridine-vinylindole intermediate 76 at 120 °C. Based on the experimental results of deuterium-labeled dihydropyridine-vinylindole intermediate 76 and subsequent structural confirmation, a mechanism was proposed to the effect that the hydride of the dihydropyridine-vinylindole intermediate 76 shifts to form the zwitterionic intermediate 84, which possesses a pyridinium cation and an enolate anion, and subsequently undergoes 7-endo cyclization to furnish the ngouniensine-type alkaloid 85. Furthermore, upon treatment with the photo-redox catalyst [Ru(bpy)3](BF4)2 under visible-light irradiation at 0 °C, the dihydropyridine ring of 76 underwent single-electron oxidation and was converted to the radical species 86, which generated an unnatural tetracyclic scaffold, 87, via redox-mediated 8-endo cyclization (Pathway E). This study successfully validated the utility of dehydrosecodine derivatives acting as biomimetic branching points to generate natural product-like small-molecule libraries without structural simplification.
3.2. Development of New Biomimetic Divergent Branching Points to Form Monoterpene Indole Alkaloids
Dai et al. developed a new pluripotent intermediate, 90, for a divergent synthetic approach to natural or unnatural monoterpene indole alkaloids [58] (Scheme 6). Compound 89 engaged in a cascade involving Witkop–Winterfeldt oxidative indole cleavage and transannular cyclization upon treatment with m-CPBA, leading to the key intermediate 90, which served as a synthetic branching point. Intermediate 90 was unstable in nature and was immediately subjected to the divergent synthetic pathway without purification or with rapid purification. For example, in situ-generated intermediate 90 could be subjected to the Staudinger-aza-Wittig process in the presence of triphenylphosphane (PPh3), yielding mersicarpine (91). Furthermore, following the Bestmann ketene lactonization of in situ-generated intermediate 90, the resultant lactam 92 was, upon treatment with PPh3, transformed into the unprecedented indole-based skeleton compound 93.
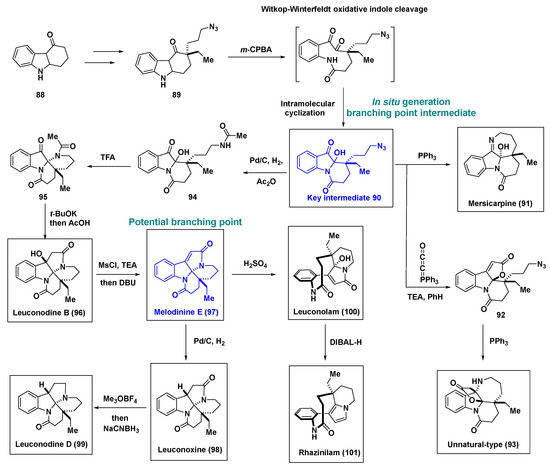
Scheme 6.
Divergent synthetic pathways to generate seven natural product and natural product-like monoterpene indole alkaloids using a new biomimetic branching point, intermediate 90. DBU, 1,8-diazabicyclo 5.4.0 undec-7-ene; DIBAL-H, diisobutylaluminium hydride; MsCl, methanesulfonyl chloride.
To generate different monoterpene indole alkaloids, melodinine E (97) was used as another pluripotent branching point. To synthesize melodinine E (97), the azide of intermediate 90 was first converted to the acetamide of compound 94. Upon treatment with TFA, cyclization occurred to afford compound 95, which was then converted into leuconodine B (96) via an intramolecular aldol reaction. Leuconodine B (96) underwent dehydration to afford melodinine E (97), which yielded leuconoxine (98) upon catalytic hydrogenation. Chemoselective reduction of the five-membered lactam upon treatment with Meerwein’s salt (Me3OBF4) and sodium cyanoborohydride furnished leuconodine D (99). In contrast, the addition of H2SO4 induced the rearrangement of melodinine E (97) to leuconolam (100), which on chemoselective DIBAL-H reduction of the 5-hydroxy-pyrrolone yielded rhazinilam (101). Notably, this study provided new insights into the potential biosynthetic pathways for terpene indole alkaloids and the feasibility of constructing indole-based natural products and natural product-like small-molecule libraries via the development of artificial pluripotent intermediates.
3.3. Biomimetic Divergent Synthesis to Generate Iboga and “Post-Iboga” Alkaloids
Iboga alkaloids are a large family of monoterpenoid indole alkaloids that contain indole, tetrahydroazepine, and isoquinuclidine scaffolds. Following the 1901 isolation of ibogaine, an iboga alkaloid possessing anti-addictive activity, considerable progress has been made in the identification of new ibogaine-type natural products over the past 100 years, leading to the identification of hundreds of iboga alkaloids and the development of remarkable synthetic pathways for them [59,60,61,62]. In addition, secondary metabolites produced from iboga alkaloids via biosynthetic pathways have sparked great interest. In 2019, inspired by the efficient biosynthesis of secondary metabolites with iboga alkaloids as the starting point, Han et al. developed a biomimetic method for the transformation of (+)-catharanthine (63) to various iboga alkaloids and secondary metabolites coined “post-iboga” alkaloids (Scheme 7) [63]. In that study, (+)-catharanthine (63), a starting material for the synthesis of the anticancer drug navelbine, was used as a biomimetic branching point with high accessibility.
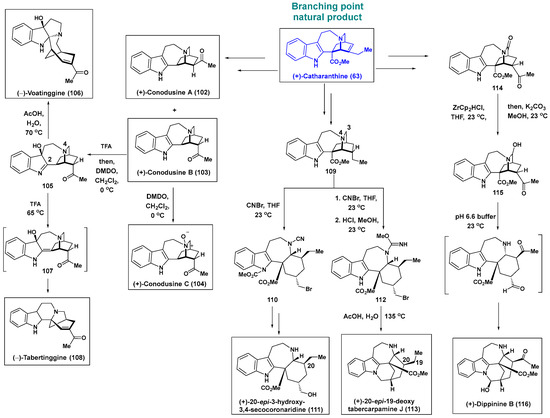
Scheme 7.
Biomimetic routes for the preparation of various iboga and “post-iboga” alkaloids from (+)-catharanthine (63) as the biomimetic branching point. ZrCp2HCl, bis(cyclopentadienyl)zirconium chloride hydride; DMDO, dimethyldioxirane.
First, (+)-catharanthine (63) was converted to (−)-conodusine A (102) and (+)-conodusine B (103). Dimethyldioxirane (DMDO) conveniently and selectively oxidized the tertiary amine group of (+)-conodusine B (103) to produce (+)-conodusine C (104). In contrast, when the tertiary amine group of 103 was protected by TFA, as well as upon subsequent addition of DMDO, hydroxyindolenine 105 was obtained. Interestingly, it was demonstrated that hydroxyindolenine, 105, is converted into “post-iboga” alkaloids such as (−)-voatinggine (106) and (−)-tabertinggine (108) through distinct structural reorganizations under acidic conditions. Hydroxyindolenine 105 was transformed into (−)-voatinggine (106) upon treatment with acetic acid. The proposed reaction mechanism entails acid-mediated β-elimination of N4 in hydroxyindolenine 105, followed by nucleophilic attack on C2. In contrast, when hydroxyindolenine 105 was subjected to TFA at 65 °C, (−)-tabertinggine was generated. The proposed mechanism involves the tautomerization of hydroxyindolenine 105 to the enamine derivative 107. The subsequent protonation of the tertiary alcohol group and azafulvenium ion formation and reorganization yielded(−)-tabertinggine (108).
To advance different “post-iboga” alkaloids, CNBr was introduced to cleave the C3–N4 bond. Compound 109, synthesized from (+)-catharanthine (63), underwent selective C3–N4 bond cleavage upon treatment with CNBr, producing the ring cleavage product 110. Compound 110 was transformed into the C20-epimer (+)-3-hydroxy-3,4-secocoronaridine (111), one of the “post-iboga” alkaloids. CNBr-induced ring cleavage of 109 yielded the carbamimidate compound 112, which underwent hydrolysis intramolecular alkylation to furnish pentacyclic chippiine- and dippinine-type “post-iboga” alkaloids such as 20-epi-19-deoxytabercarpamine J (113). Finally, selective reduction of the lactam moiety in 114 (generated from (+)-catharanthine (63) through a synthetic sequence) using a Schwartz reagent led to the formation of the unstable hemiaminal compound 115, which underwent rearrangement to form (+)-dippinine B (116) in a pH 6.6 buffer solution. The skeletally diverse and complex secondary metabolites of iboga alkaloids and “post-iboga” alkaloids obtained from the starting point of (+)-catharanthine (63) confirms the robustness of this strategy. In addition, the study provided new insights into the potential biosynthetic pathways for “post-iboga” alkaloids.
4. Conclusions
Natural products with highly complex and diverse structures are desirable candidates for screening bioactivity owing to their remarkable specificity toward biomolecular targets. However, the current natural product screening libraries are insufficient for processes such as HTS and HCS. To address this issue, a DOS strategy has been used to generate natural products and drug-like compounds with high skeletal and stereochemical diversity. Despite significant advances in DOS strategies, efficient synthetic methods for the rapid and easy preparation of small-molecule libraries comprising complex and diverse natural products remain challenging. Herein, we briefly reviewed Ctd and biomimetic synthetic strategies for the efficient generation of indole-based natural product and related small-molecule libraries. These two strategies enable high molecular diversity and structural complexity in the small-molecule libraries, thus distinguishing themselves as powerful synthetic tools. However, high demand for more starting points and branching points will always remain. The synthesis and identification of new pluripotent synthetic starting points could be a promising way to advance new complex natural products and related small-molecule libraries.
Author Contributions
T.K., M.W.H. and J.K. selected publications and analyzed published data; T.K., M.W.H. and J.K. prepared the manuscript draft, M.W.H. and J.K. reviewed and edited the text. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Korean government (MSIT) (2021R1A6A1A10044154 and 2020R1G1A1011397).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.A.P.; Thornburg, C.C.; Henrich, C.J.; Grkovic, T.; O’Keefe, B.R. Creating and screening natural product libraries. Nat. Prod. Rep. 2020, 37, 893–918. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Orhan, I.E. Natural Products as Enzyme Inhibitors in Drug Design and Discovery. Curr. Top Med. Chem. 2022, 22, 188. [Google Scholar] [CrossRef]
- Lachance, H.; Wetzel, S.; Kumar, K.; Waldmann, H. Charting, Navigating, and Populating Natural Product Chemical Space for Drug Discovery. J. Med. Chem. 2012, 55, 5989–6001. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T.; Taskforce, I.N.P.S. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Janzen, W.P. Screening Technologies for Small Molecule Discovery: The State of the Art. Chem. Biol. 2014, 21, 1162–1170. [Google Scholar] [CrossRef]
- O’Connor, C.J.; Laraia, L.; Spring, D.R. Chemical genetics. Chem. Soc. Rev. 2011, 40, 4332–4345. [Google Scholar] [CrossRef]
- Geysen, H.M.; Schoenen, F.; Wagner, D.; Wagner, R. Combinatorial compound libraries for drug discovery: An ongoing challenge. Nat. Rev. Drug Discov. 2003, 2, 222–230. [Google Scholar] [CrossRef]
- Schreiber, S.L. Organic chemistry: Molecular diversity by design. Nature 2009, 457, 153–154. [Google Scholar] [CrossRef]
- Swinney, D.C.; Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011, 10, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 2000, 287, 1964–1969. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.D.; Schreiber, S.L. A planning strategy for diversity-oriented synthesis. Angew. Chem. Int. Ed. 2004, 43, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Galloway, W.R.J.D.; Isidro-Llobet, A.; Spring, D.R. Diversity-oriented synthesis as a tool for the discovery of novel biologically active small molecules. Nat. Commun. 2010, 1, 80–92. [Google Scholar] [CrossRef]
- Ibbeson, B.M.; Laraia, L.; Alza, E.; O’Connor, C.J.; Tan, Y.S.; Davies, H.M.L.; McKenzie, G.; Venkitaraman, A.R.; Spring, D.R. Diversity-oriented synthesis as a tool for identifying new modulators of mitosis. Nat. Commun. 2014, 5, 3155–3163. [Google Scholar] [CrossRef]
- Beckmann, H.S.G.; Nie, F.L.; Hagerman, C.E.; Johansson, H.; Tan, Y.S.; Wilcke, D.; Spring, D.R. A strategy for the diversity-oriented synthesis of macrocyclic scaffolds using multidimensional coupling. Nat. Chem. 2013, 5, 861–867. [Google Scholar] [CrossRef]
- Garcia-Castro, M.; Zimmermann, S.; Sankar, M.G.; Kumar, K. Scaffold Diversity Synthesis and Its Application in Probe and Drug Discovery. Angew. Chem. Int. Ed. 2016, 55, 7586–7605. [Google Scholar] [CrossRef]
- Gerry, C.J.; Schreiber, S.L. Chemical probes and drug leads from advances in synthetic planning and methodology. Nat. Rev. Drug Discov. 2018, 17, 333–352. [Google Scholar] [CrossRef]
- Kato, N.; Comer, E.; Sakata-Kato, T.; Sharma, A.; Sharma, M.; Maetani, M.; Bastien, J.; Brancucci, N.M.; Bittker, J.A.; Corey, V.; et al. Diversity-oriented synthesis yields novel multistage antimalarial inhibitors. Nature 2016, 538, 344–349. [Google Scholar] [CrossRef]
- Kim, J.; Kim, H.; Park, S.B. Privileged Structures: Efficient Chemical "Navigators" toward Unexplored Biologically Relevant Chemical Spaces. J. Am. Chem. Soc. 2014, 136, 14629–14638. [Google Scholar] [CrossRef]
- Kim, J.; Jung, J.; Koo, J.; Cho, W.; Lee, W.S.; Kim, C.; Park, W.; Park, S.B. Diversity-oriented synthetic strategy for developing a chemical modulator of protein-protein interaction. Nat. Commun. 2016, 7, 13196–13205. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.H.; Cho, H.N.; Choi, B.Y.; Kim, J.; Ha, J.; Suh, S.W.; Park, S.B. Phenotypic Discovery of Neuroprotective Agents by Regulation of Tau Proteostasis via Stress-Responsive Activation of PERK Signaling. Angew. Chem. Int. Ed. 2021, 60, 1831–1838. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, S.L.; Yang, F.; Dong, S. Marine Indole Alkaloids-Isolation, Structure and Bioactivities. Mar. Drugs 2021, 19, 658. [Google Scholar] [CrossRef] [PubMed]
- Wibowo, J.T.; Ahmadi, P.; Rahmawati, S.I.; Bayu, A.; Putra, M.Y.; Kijjoa, A. Marine-Derived Indole Alkaloids and Their Biological and Pharmacological Activities. Mar. Drugs 2022, 20, 3. [Google Scholar] [CrossRef] [PubMed]
- Taliani, S.; Da Settimo, F.; Martini, C.; Laneri, S.; Novellino, E.; Greco, G. Exploiting the Indole Scaffold to Design Compounds Binding to Different Pharmacological Targets. Molecules 2020, 25, 2331. [Google Scholar] [CrossRef]
- Dhuguru, J.; Skouta, R. Role of Indole Scaffolds as Pharmacophores in the Development of Anti-Lung Cancer Agents. Molecules 2020, 25, 1615. [Google Scholar] [CrossRef]
- Pacheco, P.A.F.; Santos, M.M.M. Recent Progress in the Development of Indole-Based Compounds Active against Malaria, Trypanosomiasis and Leishmaniasis. Molecules 2022, 27, 319. [Google Scholar] [CrossRef]
- Zhang, M.Z.; Chen, Q.; Yang, G.F. A review on recent developments of indole-containing antiviral agents. Eur. J. Med. Chem. 2015, 89, 421–441. [Google Scholar] [CrossRef]
- Norwood, V.M.; Huigens, R.W. Harnessing the Chemistry of the Indole Heterocycle to Drive Discoveries in Biology and Medicine. Chembiochem 2019, 20, 2273–2297. [Google Scholar] [CrossRef]
- Huigens, R.W.; Morrison, K.C.; Hicklin, R.W.; Flood, T.A.; Richter, M.F.; Hergenrother, P.J. A ring-distortion strategy to construct stereochemically complex and structurally diverse compounds from natural products. Nat. Chem. 2013, 5, 195–202. [Google Scholar] [CrossRef]
- Rafferty, R.J.; Hicklin, R.W.; Maloof, K.A.; Hergenrother, P.J. Synthesis of Complex and Diverse Compounds through Ring Distortion of Abietic Acid. Angew. Chem. Int. Ed. 2014, 53, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.; Drown, B.S.; Hergenrother, P.J. Access to a Structurally Complex Compound Collection via Ring Distortion of the Alkaloid Sinomenine. Org. Lett. 2016, 18, 4852–4855. [Google Scholar] [CrossRef] [PubMed]
- Llabani, E.; Hicklin, R.W.; Lee, H.Y.; Motika, S.E.; Crawford, L.A.; Weerapana, E.; Hergenrother, P.J. Diverse compounds from pleuromutilin lead to a thioredoxin inhibitor and inducer of ferroptosis. Nat. Chem. 2019, 11, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Furiassi, L.; Tonogai, E.J.; Hergenrother, P.J. Limonin as a Starting Point for the Construction of Compounds with High Scaffold Diversity. Angew. Chem. Int. Ed. 2021, 60, 16119–16128. [Google Scholar] [CrossRef]
- Morales, A. Yohimbine in erectile dysfunction: The facts. Int. J. Impot. Res. 2000, 12, S70–S74. [Google Scholar] [CrossRef]
- Abdel-Zaher, A.O.; Ahmed, I.T.; El-Koussi, A.D. The potential antidiabetic activity of some alpha-2 adrenoceptor antagonists. Pharmacol. Res. 2001, 44, 397–409. [Google Scholar] [CrossRef]
- Paciaroni, N.G.; Ratnayake, R.; Matthews, J.H.; Norwood, V.M.; Arnold, A.C.; Dang, L.H.; Luesch, H.; Huigens, R.W. A Tryptoline Ring-Distortion Strategy Leads to Complex and Diverse Biologically Active Molecules from the Indole Alkaloid Yohimbine. Chem. Eur. J. 2017, 23, 4327–4335. [Google Scholar] [CrossRef]
- Albright, J.D.; Goldman, L. Alkaloid studies. V. Reaction of tertiary amines with cyanogen bromide under solvolytic conditions. J. Am. Chem. Soc. 1969, 91, 4317–4318. [Google Scholar] [CrossRef]
- Stahl, R.; Borschberg, H. A reinvestigation of the oxidative rearrangement of yohimbane-type alkaloids. part B. formation of oxindol (=1,3-dihydro-2H-indol-2-one) derivatives. Helv. Chim. Acta 1996, 79, 1361–1378. [Google Scholar] [CrossRef]
- Paciaroni, N.G.; Perry, D.L.; Norwood, V.M.; Murillo-Solano, C.; Collins, J.; Tenneti, S.; Chakrabarti, D.; Huigens, R.W. Re-Engineering of Yohimbine’s Biological Activity through Ring Distortion: Identification and Structure-Activity Relationships of a New Class of Antiplasmodial Agents. ACS Infect. Dis. 2020, 6, 159–167. [Google Scholar] [CrossRef]
- Dany, F.; Goudoud, J.C. Torsades de pointes et arythmies ventriculaires graves par administration parenterale de vincamine. Arch. Mal. Coeur. 1980, 73, 298–306. [Google Scholar] [PubMed]
- Rassat, J.; Robenek, H.; Themann, H. Changes in mouse hepatocytes caused by vincamin. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1982, 318, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Fawzy, M.A.; Maher, S.A.; El-Rehany, M.A.; Welson, N.N.; Albezrah, N.K.A.; Batiha, G.E.; Fathy, M. Vincamine Modulates the Effect of Pantoprazole in Renal Ischemia/Reperfusion Injury by Attenuating MAPK and Apoptosis Signaling Pathways. Molecules 2022, 27, 1383. [Google Scholar] [CrossRef]
- Norwood, V.M.T.; Brice-Tutt, A.C.; Eans, S.O.; Stacy, H.M.; Shi, G.; Ratnayake, R.; Rocca, J.R.; Abboud, K.A.; Li, C.; Luesch, H.; et al. Preventing Morphine-Seeking Behavior through the Re-Engineering of Vincamine’s Biological Activity. J. Med. Chem. 2020, 63, 5119–5138. [Google Scholar] [CrossRef] [PubMed]
- Srinivasulu, V.; Sieburth, S.M.; Khanfar, M.A.; Abu-Yousef, I.A.; Majdalawieh, A.; Ramanathan, M.; Sebastian, A.; Al-Tel, T.H. Stereoselective Late-Stage Transformations of Indolo [2,3-a]quinolizines Skeleta to Nature-Inspired Scaffolds. J. Org. Chem. 2021, 86, 12872–12885. [Google Scholar] [CrossRef] [PubMed]
- Srinivasulu, V.; Schilf, P.; Ibrahim, S.; Khanfar, M.A.; Sieburth, S.M.; Omar, H.; Sebastian, A.; AlQawasmeh, R.A.; O’Connor, M.J.; Al-Tel, T.H. Multidirectional desymmetrization of pluripotent building block en route to diastereoselective synthesis of complex nature-inspired scaffolds. Nat. Commun. 2018, 9, 4989–5002. [Google Scholar] [CrossRef]
- Bauer, R.A.; Wenderski, T.A.; Tan, D.S. Biomimetic diversity-oriented synthesis of benzannulated medium rings via ring expansion. Nat. Chem. Biol. 2013, 9, 21–29. [Google Scholar] [CrossRef]
- Yasir, A.; Ishtiaq, S.; Jahangir, M.; Ajaib, M.; Salar, U.; Khan, K.M. Biology-Oriented Synthesis (BIOS) of Piperine Derivatives and their Comparative Analgesic and Antiinflammatory Activities. Med. Chem. 2018, 14, 269–280. [Google Scholar] [CrossRef]
- Svenda, J.; Sheremet, M.; Kremer, L.; Maier, L.; Bauer, J.O.; Strohmann, C.; Ziegler, S.; Kumar, K.; Waldmann, H. Biology-Oriented Synthesis of a Withanolide-Inspired Compound Collection Reveals Novel Modulators of Hedgehog Signaling. Angew. Chem. Int. Ed. 2015, 54, 5596–5602. [Google Scholar] [CrossRef]
- Day, A.J.; Sumby, C.J.; George, J.H. Biomimetic Synthetic Studies on the Bruceol Family of Meroterpenoid Natural Products. J. Org. Chem. 2020, 85, 2103–2117. [Google Scholar] [CrossRef]
- Day, A.J.; Lee, J.H.Z.; Phan, Q.D.; Lam, H.C.; Ametovski, A.; Sumby, C.J.; Bell, S.G.; George, J.H. Biomimetic and Biocatalytic Synthesis of Bruceol. Angew. Chem. Int. Ed. 2019, 58, 1427–1431. [Google Scholar] [CrossRef] [PubMed]
- Stocking, E.M.; Williams, R.M. Chemistry and biology of biosynthetic Diels-Alder reactions. Angew. Chem. Int. Ed. 2003, 42, 3078–3115. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, S.E.; Maresh, J. Chemistry and biology of monoterpeneindole alkaloid biosynthesis. J. Nat. Prod. Rep. 2006, 23, 532–547. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Scotti, J.S.; Kozmin, S.A. Emulating the Logic of Monoterpenoid Alkaloid Biogenesis to Access a Skeletally Diverse Chemical Library. J. Org. Chem. 2013, 78, 8645–8654. [Google Scholar] [CrossRef][Green Version]
- Finefield, J.M.; Sherman, D.H.; Kreitman, M.; Williams, R.M. Enantiomeric natural products: Occurrence and biogenesis. Angew. Chem. Int. Ed. 2012, 51, 4802–4836. [Google Scholar] [CrossRef]
- Dong, C.; Mo, F.; Wang, J. Highly diastereoselective addition of the lithium enolate of alpha-diazoacetoacetate to N-sulfinyl imines: Enantioselective synthesis of 2-Oxo and 3-Oxo pyrrolidines. J. Org. Chem. 2008, 73, 1971–1974. [Google Scholar] [CrossRef]
- Mizoguchi, H.; Oikawa, H.; Oguri, H. Biogenetically inspired synthesis and skeletal diversification of indole alkaloids. Nat. Chem. 2014, 6, 57–64. [Google Scholar] [CrossRef]
- Yang, Y.; Bai, Y.; Sun, S.; Dai, M. Biosynthetically inspired divergent approach to monoterpene indole alkaloids: Total synthesis of mersicarpine, leuconodines B and D, leuconoxine, melodinine E, leuconolam, and rhazinilam. Org. Lett. 2014, 16, 6216–6219. [Google Scholar] [CrossRef]
- Iyer, R.N.; Favela, D.; Zhang, G.; Olson, D.E. The iboga enigma: The chemistry and neuropharmacology of iboga alkaloids and related analogs. Nat. Prod. Rep. 2021, 38, 307–329. [Google Scholar] [CrossRef]
- Lavaud, C.; Massiot, G. The Iboga Alkaloids. Prog. Chem. Org. Nat. Prod. 2017, 105, 89–136. [Google Scholar]
- Tang, B.Q.; Wang, W.J.; Huang, X.J.; Li, G.Q.; Wang, L.; Jiang, R.W.; Yang, T.T.; Shi, L.; Zhang, X.Q.; Ye, W.C. Iboga-Type Alkaloids from Ervatamia officinalis. J. Nat. Prod. 2014, 77, 1839–1846. [Google Scholar] [CrossRef] [PubMed]
- Nge, C.E.; Gan, C.Y.; Low, Y.Y.; Thomas, N.F.; Kam, T.S. Voatinggine and tabertinggine, pentacyclic indole alkaloids derived from an iboga precursor via a common cleavamine-type intermediate. Org. Lett. 2013, 15, 4774–4777. [Google Scholar] [CrossRef] [PubMed]
- Seong, S.; Lim, H.; Han, S. Biosynthetically Inspired Transformation of Iboga to Monomeric Post-iboga Alkaloids. Chem 2019, 5, 353–363. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).