- Article

Investigation of the Sintering Behavior of Nanoparticulate UN via Molecular Dynamics Simulation
- Wentao Liu,
- Hui Feng and
- Qihong Fang
- + 4 authors
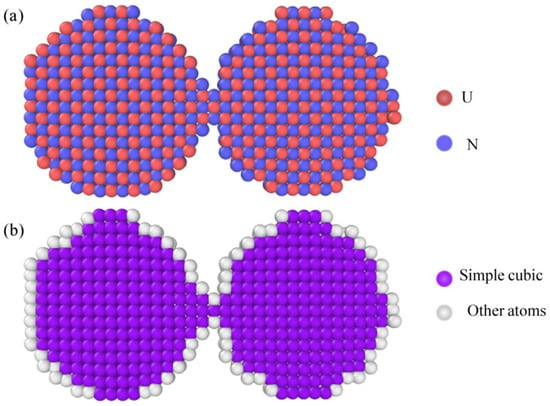
Sintering is a key processing route to consolidate nuclear fuel powders into dense compacts, yet the atomic-level mechanisms governing the sintering of actinide compounds remain poorly understood. Herein, the sintering kinetics and structural evolution of uranium mononitride (UN) nanoparticles are investigated using molecular dynamics (MD) simulations. A three-stage sintering mechanism is revealed based on the symmetrical dual nanoparticle models: initial surface diffusion and neck formation, followed by interface amorphization driven by shear stress, and finally, lattice reconstruction and recrystallization, which peak during the cooling process. By studying the effect of sintering temperature, we find that near-complete densification with good structural integrity is achieved at 1900 K, whereas further increasing the temperature (to 2000 K) led to microstructural instability and near-overburning. In addition, holding time exhibits a clear saturation effect, with variations in holding time showing no significant impact on sintering morphology or density. Therefore, sintering temperature is the dominant factor determining sintering quality. The atomic level insights provided by this work reveal the nonlinear temperature dependence and time saturation effect of UN nanoparticle sintering, and provide a theoretical basis for the prediction, design, and optimization of nuclear fuel sintering process.
20 January 2026