Computational Antibody and Antigen Design
A topical collection in Antibodies (ISSN 2073-4468).
Viewed by 126126
Share This Topical Collection
Editor
 Prof. Dr. Buyong Ma
Prof. Dr. Buyong Ma
Prof. Dr. Buyong Ma
E-Mail
Website
Collection Editor
School of Pharmacy, Shanghai Jiaotong University, Shanghai, China
Interests: computational chemistry and biology; computational immunology; protein-protein interaction; protein aggregation diseases; neurodegenerative disease; Alzheimer’s disease; cancer and inflammation; antigen
Special Issues, Collections and Topics in MDPI journals
Topical Collection Information
Dear Colleagues,
Using computational modeling to help antibody engineering is complementary to traditional animal models and phage display methodologies, hopefully speeding up and providing additional variations into antibody design. Starting from antibody/antigen sequences, one can model the structures of antibody and antigen, predict antibody–antigen interactions through docking and/or molecular dynamics simulations, and provide computationally designed/optimized systems for experimental verification. Computational antibody design can lead to high affinity, high stability, and high specificity. In vaccine design, computational modeling can generate small and stable protein scaffolds, mimicking antigen epitopes to induce potent neutralizing antibodies. Even with this promising power, widely-integrated application of computational modeling in antibody engineering is still yet to arrive. This Special Issue will showcase various aspects of computational antibody and antigen design.
This Special Issue of Antibodies focuses on: (1) computational/structural characterization of antibody structures; (2) bioinformatics study of antibody and antigens; (3) computational algorithms in antibody and antigen design; and (4) applications of computational methods in antibody/antigen development.
Dr. Buyong Ma
Guest Editor
Manuscript Submission Information
Manuscripts should be submitted online at www.mdpi.com by registering and logging in to this website. Once you are registered, click here to go to the submission form. All submissions that pass pre-check are peer-reviewed. Accepted papers will be published continuously in the journal (as soon as accepted) and will be listed together on the collection website. Research articles, review articles as well as short communications are invited. For planned papers, a title and short abstract (about 250 words) can be sent to the Editorial Office for assessment.
Submitted manuscripts should not have been published previously, nor be under consideration for publication elsewhere (except conference proceedings papers). All manuscripts are thoroughly refereed through a single-blind peer-review process. A guide for authors and other relevant information for submission of manuscripts is available on the Instructions for Authors page. Antibodies is an international peer-reviewed open access semimonthly journal published by MDPI.
Please visit the Instructions for Authors page before submitting a manuscript.
The Article Processing Charge (APC) for publication in this open access journal is 1800 CHF (Swiss Francs).
Submitted papers should be well formatted and use good English. Authors may use MDPI's
English editing service prior to publication or during author revisions.
Keywords
- antibody design
- antibody engineering
- vaccine design
- protein-protein interaction
- computational biology
Published Papers (11 papers)
Open AccessEditor’s ChoiceArticle
Computational Prediction of Single-Domain Immunoglobulin Aggregation Propensities Facilitates Discovery and Humanization of Recombinant Nanobodies
by
Felix Klaus Geyer, Julian Borbeck, Wiktoria Palka, Xueyuan Zhou, Jeffrey Takimoto, Brian Rabinovich, Bernd Reifenhäuser, Karlheinz Friedrich and Harald Kolmar
Cited by 2 | Viewed by 4612
Abstract
Background/Objectives: Single-domain immunoglobulins are small protein modules with specific affinities. Among them, the variable domains of heavy chains of heavy-chain-only antibodies (VHH) as the antigen-binding fragment of heavy-chain-only antibodies (also termed nanobodies) have been widely investigated for their applicability, e.g., therapeutics and immunodiagnostics.
[...] Read more.
Background/Objectives: Single-domain immunoglobulins are small protein modules with specific affinities. Among them, the variable domains of heavy chains of heavy-chain-only antibodies (VHH) as the antigen-binding fragment of heavy-chain-only antibodies (also termed nanobodies) have been widely investigated for their applicability, e.g., therapeutics and immunodiagnostics. However, despite their advantageous biochemical and biophysical characteristics, protein aggregation throughout recombinant synthesis is a serious drawback in the development of nanobodies with application perspectives. Therefore, we aimed to develop a computational method to predict the aggregation propensity of VHH antibodies for the selection of promising candidates in early discovery. Methods: We employed a deep learning-based structure prediction for VHHs and derived from it likely biophysical and biochemical properties of the framework region 2 with relevance for aggregation. A total of 106 nanobody variants were produced by recombinant expression and characterized for their aggregation behavior using size exclusion chromatography (SEC). Results: Quantitative characteristics of framework region 2 patches were combined into a function that defines an aggregation score (AS) predicting the aggregation propensities of VHH variants. AS was evaluated for its capability to forecast recombinant VHH aggregation by experimentally studying VHH Fc-fusion proteins for their aggregation. We observed a clear correlation between the calculated aggregation score and the actual aggregation propensities of biochemically characterized VHHs Fc-fusion proteins. Moreover, we implemented an easily accessible pipeline of software modules to design nanobodies with desired solubility properties. Conclusions: AI-based prediction of VHH structures, followed by analysis of framework region 2 properties, can be used to predict the aggregation propensities of VHHs, providing a convenient and efficient tool for selecting stable recombinant nanobodies.
Full article
►▼
Show Figures
Open AccessEditor’s ChoiceArticle
The Accurate Prediction of Antibody Deamidations by Combining High-Throughput Automated Peptide Mapping and Protein Language Model-Based Deep Learning
by
Ben Niu, Benjamin Lee, Lili Wang, Wen Chen and Jeffrey Johnson
Cited by 1 | Viewed by 5585
Abstract
Therapeutic antibodies such as monoclonal antibodies (mAbs), bispecific and multispecific antibodies are pivotal in therapeutic protein development and have transformed disease treatments across various therapeutic areas. The integrity of therapeutic antibodies, however, is compromised by sequence liabilities, notably deamidation, where asparagine (N) and
[...] Read more.
Therapeutic antibodies such as monoclonal antibodies (mAbs), bispecific and multispecific antibodies are pivotal in therapeutic protein development and have transformed disease treatments across various therapeutic areas. The integrity of therapeutic antibodies, however, is compromised by sequence liabilities, notably deamidation, where asparagine (N) and glutamine (Q) residues undergo chemical degradations. Deamidation negatively impacts the efficacy, stability, and safety of diverse classes of antibodies, thus necessitating the critical need for the early and accurate identification of vulnerable sites. In this article, a comprehensive antibody deamidation-specific dataset (n = 2285) of varied modalities was created by using high-throughput automated peptide mapping followed by supervised machine learning to predict the deamidation propensities, as well as the extents, throughout the entire antibody sequences. We propose a novel chimeric deep learning model, integrating protein language model (pLM)-derived embeddings with local sequence information for enhanced deamidation predictions. Remarkably, this model requires only sequence inputs, eliminating the need for laborious feature engineering. Our approach demonstrates state-of-the-art performance, offering a streamlined workflow for high-throughput automated peptide mapping and deamidation prediction, with the potential of broader applicability to other antibody sequence liabilities.
Full article
►▼
Show Figures
Open AccessFeature PaperArticle
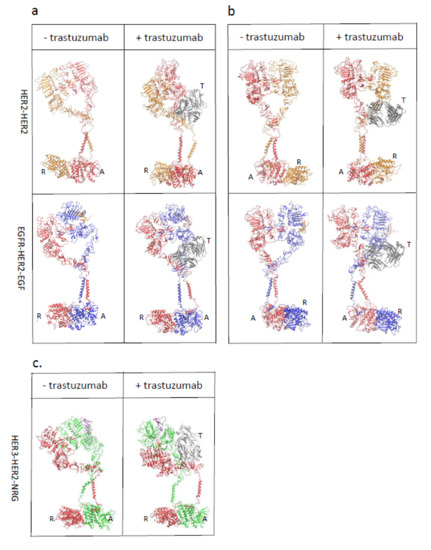
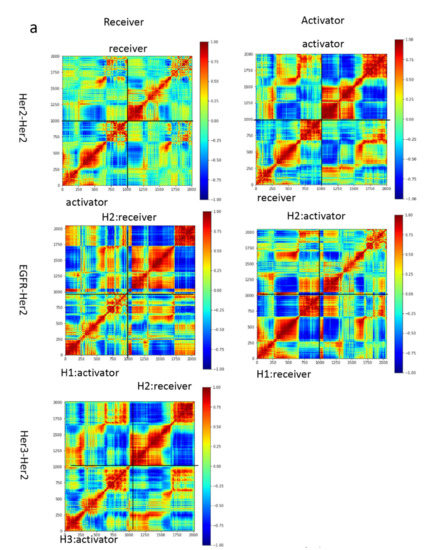
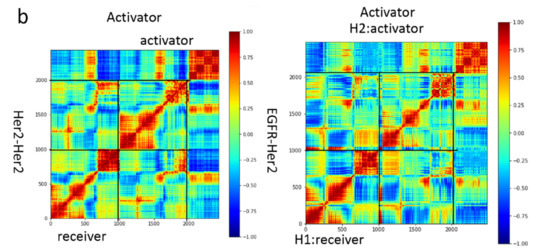
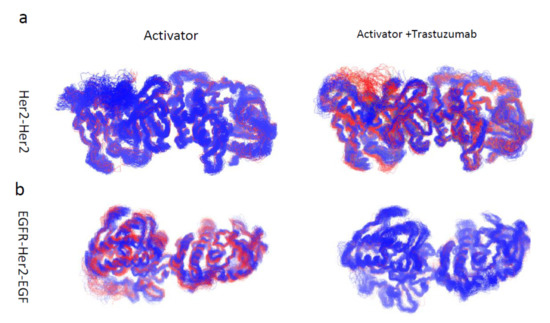
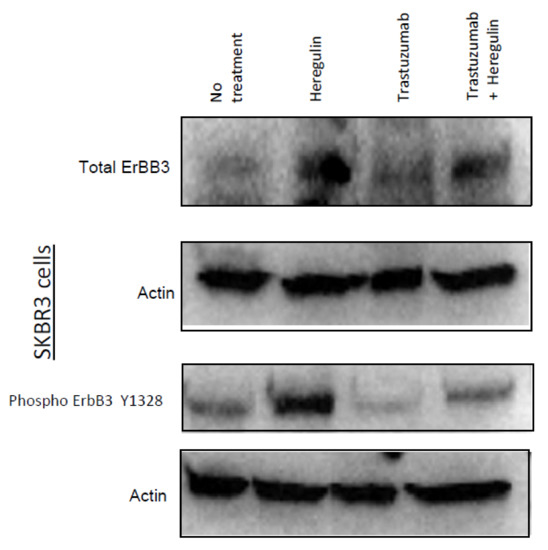
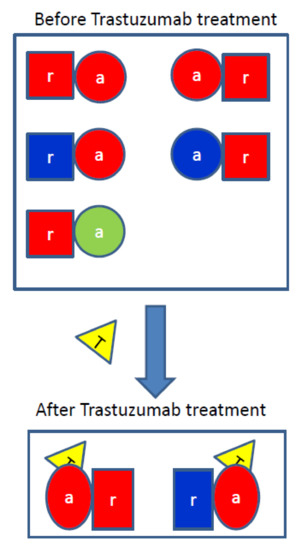
Trastuzumab Blocks the Receiver Function of HER2 Leading to the Population Shifts of HER2-Containing Homodimers and Heterodimers
by
Jun Zhao, Nishant Mohan, Ruth Nussinov, Buyong Ma and Wen Jin Wu
Cited by 23 | Viewed by 8172
Abstract
HER2, a member of the Erythroblastosis Protein B/Human Epidermal Growth Factor Receptor (ErbB/HER) family of receptor tyrosine kinase, is overexpressed in 20~30% of human breast cancers. Trastuzumab, a HER2-targeted therapeutic monoclonal antibody, was developed to interfere with the homodimerization of HER2 in HER2-overexpressing
[...] Read more.
HER2, a member of the Erythroblastosis Protein B/Human Epidermal Growth Factor Receptor (ErbB/HER) family of receptor tyrosine kinase, is overexpressed in 20~30% of human breast cancers. Trastuzumab, a HER2-targeted therapeutic monoclonal antibody, was developed to interfere with the homodimerization of HER2 in HER2-overexpressing breast cancer cells, which attenuates HER2-mediated signaling. Trastuzumab binds to the domain IV of the HER2 extracellular domain and does not directly block the dimerization interface of HER2-HER2 molecules. The three-dimensional structures of the tyrosine kinase domains of ErbB/HER family receptors show asymmetrical packing of the two monomers with distinct conformations. One monomer functions as an activator, whereas the other acts as a receiver. Once activated, the receiver monomer phosphorylates the activator or other proteins. Interestingly, in our previous work, we found that the binding of trastuzumab induced phosphorylation of HER2 with the phosphorylation pattern of HER2 that is different from that mediated by epidermal growth factor (EGF) in human epidermal growth factor receptor 2 (HER2)-positive breast cancer. Binding of trastuzumab to HER2 promoted an allosteric effect of HER2, in both tyrosine kinase domain and ectodomain of HER2 although details of allosteric regulation were missing. In this study, we utilized molecular dynamics (MD) simulations to model the allosteric consequences of trastuzumab binding to HER2 homodimers and heterodimers, along with the apo forms as controls. We focused on the conformational changes of HER2 in its monomeric and dimeric forms. The data indicated the apparent dual role of trastuzumab as an antagonist and an agonist. The molecular details of the simulation provide an atomic level description and molecular insight into the action of HER2-targeted antibody therapeutics.
Full article
►▼
Show Figures
Open AccessReview
A Review of Deep Learning Methods for Antibodies
by
Jordan Graves, Jacob Byerly, Eduardo Priego, Naren Makkapati, S. Vince Parish, Brenda Medellin and Monica Berrondo
Cited by 65 | Viewed by 19656
Abstract
Driven by its successes across domains such as computer vision and natural language processing, deep learning has recently entered the field of biology by aiding in cellular image classification, finding genomic connections, and advancing drug discovery. In drug discovery and protein engineering, a
[...] Read more.
Driven by its successes across domains such as computer vision and natural language processing, deep learning has recently entered the field of biology by aiding in cellular image classification, finding genomic connections, and advancing drug discovery. In drug discovery and protein engineering, a major goal is to design a molecule that will perform a useful function as a therapeutic drug. Typically, the focus has been on small molecules, but new approaches have been developed to apply these same principles of deep learning to biologics, such as antibodies. Here we give a brief background of deep learning as it applies to antibody drug development, and an in-depth explanation of several deep learning algorithms that have been proposed to solve aspects of both protein design in general, and antibody design in particular.
Full article
►▼
Show Figures
Open AccessArticle
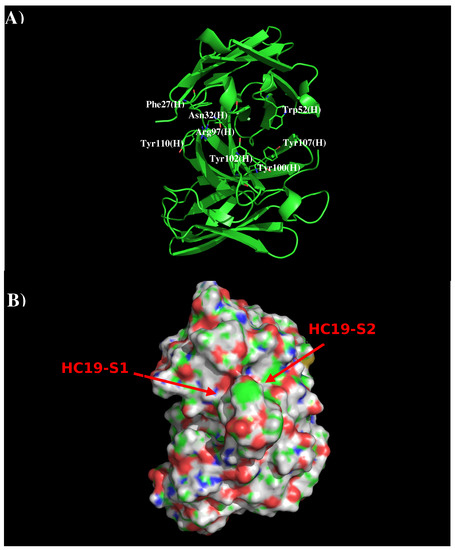
Computational Prediction of the Epitopes of HA1 Protein of Influenza Viruses to its Neutralizing Antibodies
by
Xiaoyan Zeng, Fiona S. Legge, Chao Huang, Xiao Zhang, Yongjun Jiao, Herbert R. Treutlein and Jun Zeng
Cited by 4 | Viewed by 7743
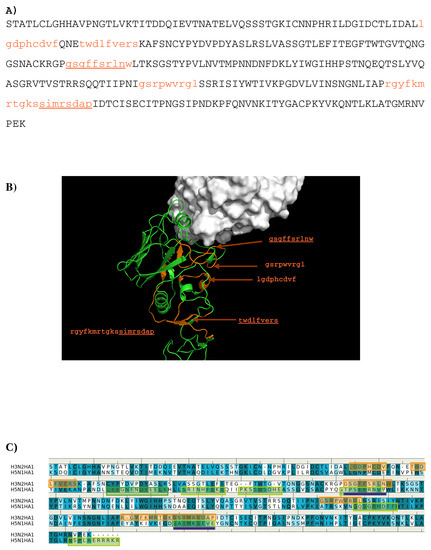
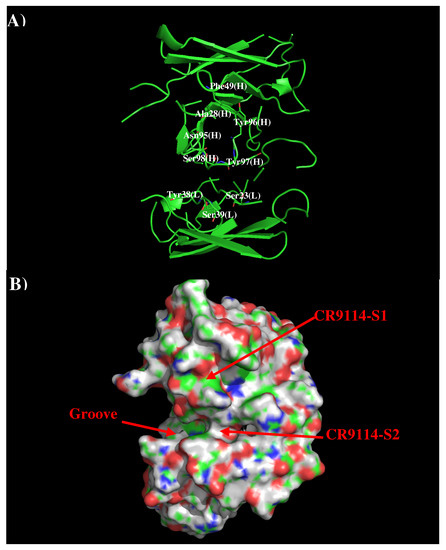
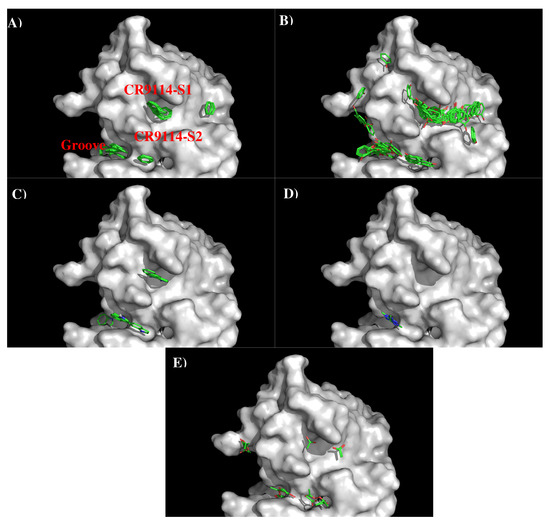
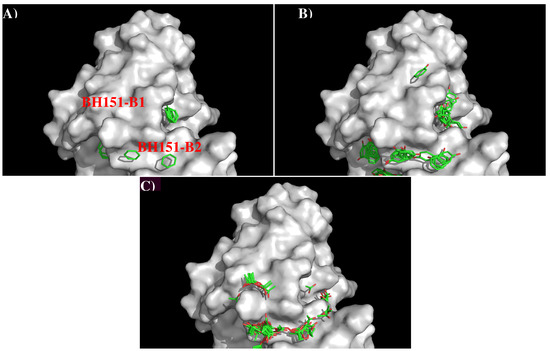
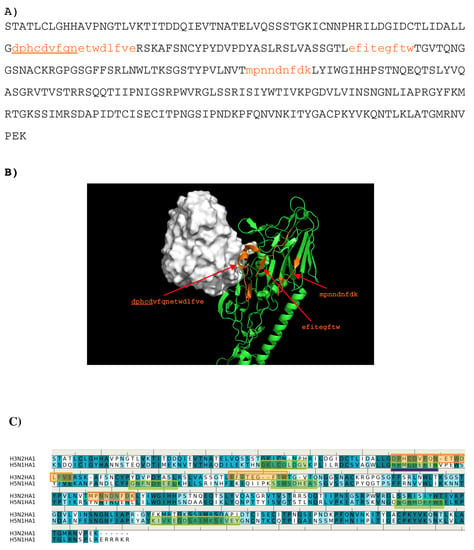
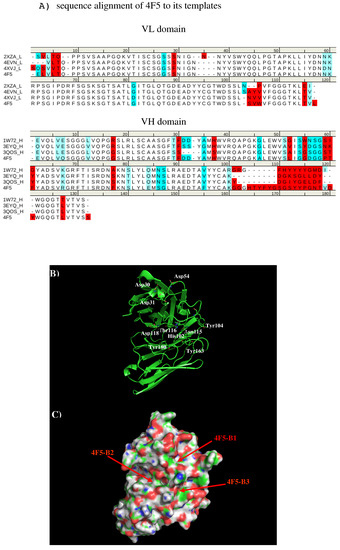
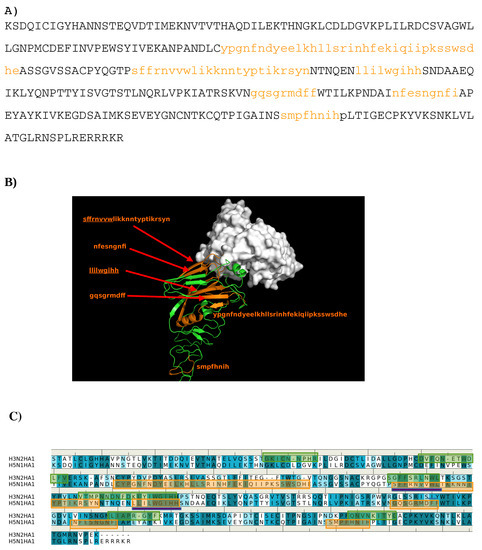
Abstract
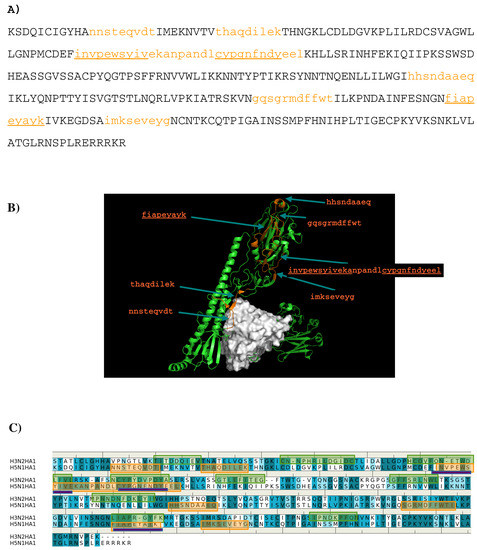
In this work, we have used a new method to predict the epitopes of HA1 protein of influenza virus to several antibodies HC19, CR9114, BH151 and 4F5. While our results reproduced the binding epitopes of H3N2 or H5N1 for the neutralizing antibodies HC19,
[...] Read more.
In this work, we have used a new method to predict the epitopes of HA1 protein of influenza virus to several antibodies HC19, CR9114, BH151 and 4F5. While our results reproduced the binding epitopes of H3N2 or H5N1 for the neutralizing antibodies HC19, CR9114, and BH151 as revealed from the available crystal structures, additional epitopes for these antibodies were also suggested. Moreover, the predicted epitopes of H5N1 HA1 for the newly developed antibody 4F5 are located at the receptor binding domain, while previous study identified a region 76-WLLGNP-81 as the epitope. The possibility of antibody recognition of influenza virus via different mechanism by binding to different epitopes of an antigen is also discussed.
Full article
►▼
Show Figures
Open AccessFeature PaperArticle
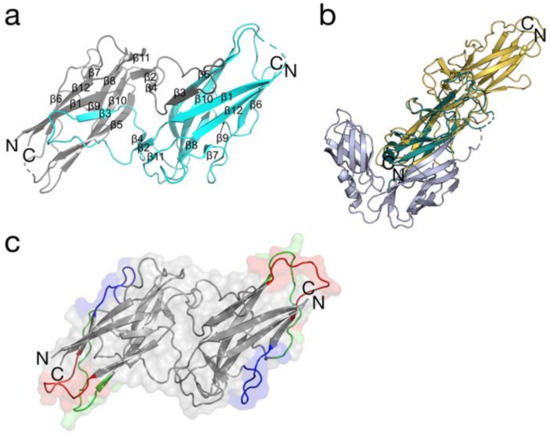
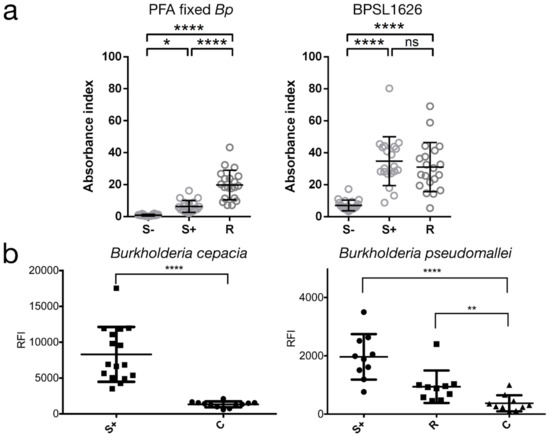
BPSL1626: Reverse and Structural Vaccinology Reveal a Novel Candidate for Vaccine Design against Burkholderia pseudomallei
by
Riccardo Capelli, Claudio Peri, Riccardo Villa, Arnone Nithichanon, Oscar Conchillo-Solé, Daniel Yero, Paola Gagni, Marcella Chiari, Ganjana Lertmemongkolchai, Marina Cretich, Xavier Daura, Martino Bolognesi, Giorgio Colombo and Louise J. Gourlay
Cited by 17 | Viewed by 8941
Abstract
Due to significant advances in computational biology, protein prediction, together with antigen and epitope design, have rapidly moved from conventional methods, based on experimental approaches, to in silico-based bioinformatics methods. In this context, we report a reverse vaccinology study that identified a panel
[...] Read more.
Due to significant advances in computational biology, protein prediction, together with antigen and epitope design, have rapidly moved from conventional methods, based on experimental approaches, to in silico-based bioinformatics methods. In this context, we report a reverse vaccinology study that identified a panel of 104 candidate antigens from the Gram-negative bacterial pathogen
Burkholderia pseudomallei, which is responsible for the disease melioidosis.
B. pseudomallei can cause fatal sepsis in endemic populations in the tropical regions of the world and treatment with antibiotics is mostly ineffective. With the aim of identifying potential vaccine candidates, we report the experimental validation of predicted antigen and type I fimbrial subunit, BPSL1626, which we show is able to recognize and bind human antibodies from the sera of
Burkholderia infected patients and to stimulate T-lymphocytes in vitro. The prerequisite for a melioidosis vaccine, in fact, is that both antibody- and cell-mediated immune responses must be triggered. In order to reveal potential antigenic regions of the protein that may aid immunogen re-design, we also report the crystal structure of BPSL1626 at 1.9 Å resolution on which structure-based epitope predictions were based. Overall, our data suggest that BPSL1626 and three epitope regions here-identified can represent viable candidates as potential antigenic molecules.
Full article
►▼
Show Figures
Open AccessArticle
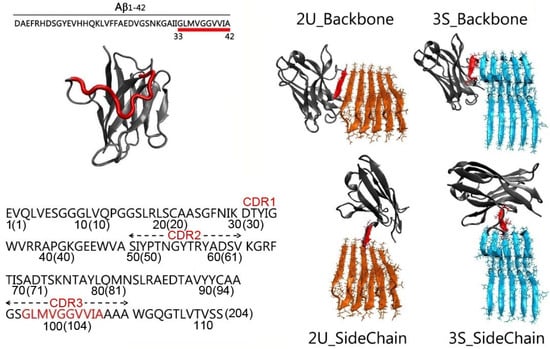
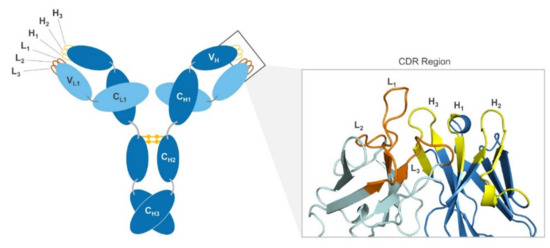
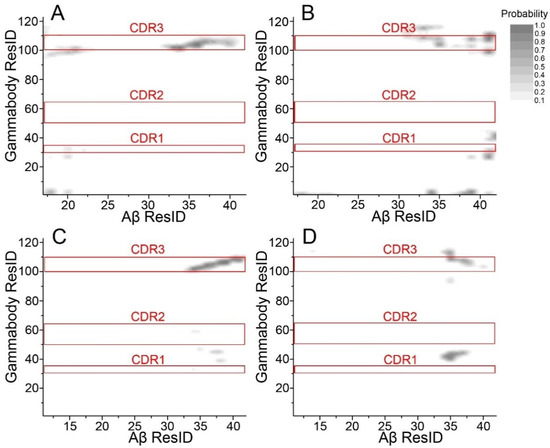
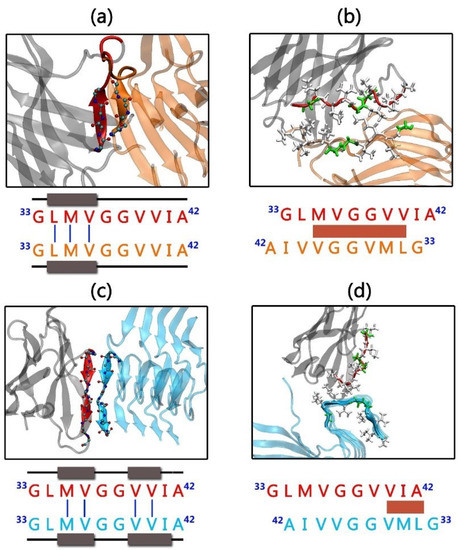
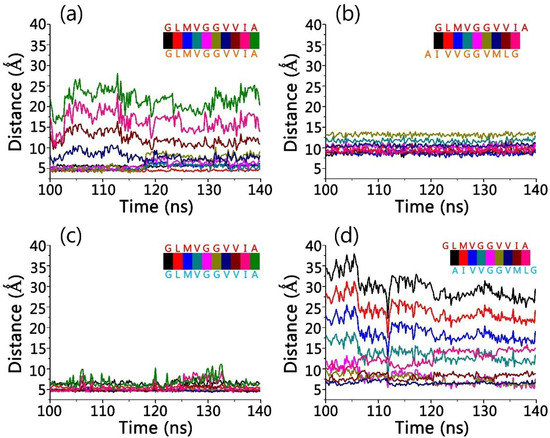
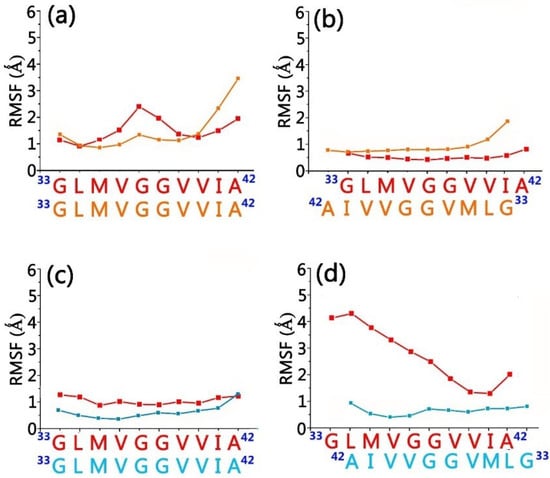
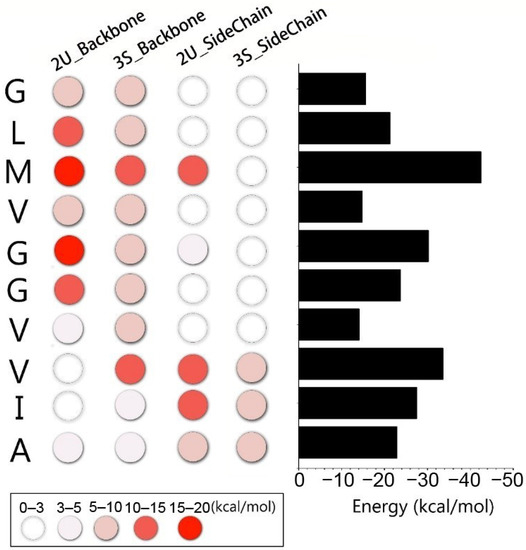
Molecular Recognition between Aβ-Specific Single-Domain Antibody and Aβ Misfolded Aggregates
by
Mingzhen Zhang, Jie Zheng, Ruth Nussinov and Buyong Ma
Cited by 49 | Viewed by 8552
Abstract
Aβ is the toxic amyloid polypeptide responsible for Alzheimer’s disease (AD). Prevention and elimination of the Aβ misfolded aggregates are the promising therapeutic strategies for the AD treatments. Gammabody, the Aβ-Specific Single-domain (VH) antibody, recognizes Aβ aggregates with high affinity and specificity and
[...] Read more.
Aβ is the toxic amyloid polypeptide responsible for Alzheimer’s disease (AD). Prevention and elimination of the Aβ misfolded aggregates are the promising therapeutic strategies for the AD treatments. Gammabody, the Aβ-Specific Single-domain (VH) antibody, recognizes Aβ aggregates with high affinity and specificity and reduces their toxicities. Employing the molecular dynamics simulations, we studied diverse gammabody-Aβ recognition complexes to get insights into their structural and dynamic properties and gammabody-Aβ recognitions. Among many heterogeneous binding modes, we focused on two gammabody-Aβ recognition scenarios: recognition through Aβ β-sheet backbone and on sidechain surface. We found that the gammabody primarily uses the complementarity-determining region 3 (CDR3) loop with the grafted Aβ sequence to interact with the Aβ fibril, while CDR1/CDR2 loops have very little contact. The gammabody-Aβ complexes with backbone binding mode are more stable, explaining the gammabody’s specificity towards the C-terminal Aβ sequence.
Full article
►▼
Show Figures
Open AccessFeature PaperArticle
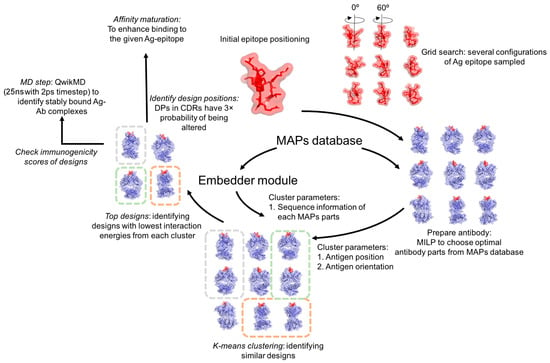
OptMAVEn-2.0: De novo Design of Variable Antibody Regions against Targeted Antigen Epitopes
by
Ratul Chowdhury, Matthew F. Allan and Costas D. Maranas
Cited by 45 | Viewed by 15671
Abstract
Monoclonal antibodies are becoming increasingly important therapeutic agents for the treatment of cancers, infectious diseases, and autoimmune disorders. However, laboratory-based methods of developing therapeutic monoclonal antibodies (e.g., immunized mice, hybridomas, and phage display) are time-consuming and are often unable to target a specific
[...] Read more.
Monoclonal antibodies are becoming increasingly important therapeutic agents for the treatment of cancers, infectious diseases, and autoimmune disorders. However, laboratory-based methods of developing therapeutic monoclonal antibodies (e.g., immunized mice, hybridomas, and phage display) are time-consuming and are often unable to target a specific antigen epitope or reach (sub)nanomolar levels of affinity. To this end, we developed Optimal Method for Antibody Variable region Engineering (OptMAVEn) for de novo design of humanized monoclonal antibody variable regions targeting a specific antigen epitope. In this work, we introduce OptMAVEn-2.0, which improves upon OptMAVEn by (1) reducing computational resource requirements without compromising design quality; (2) clustering the designs to better identify high-affinity antibodies; and (3) eliminating intra-antibody steric clashes using an updated set of clashing parts from the Modular Antibody Parts (MAPs) database. Benchmarking on a set of 10 antigens revealed that OptMAVEn-2.0 uses an average of 74% less CPU time and 84% less disk storage relative to OptMAVEn. Testing on 54 additional antigens revealed that computational resource requirements of OptMAVEn-2.0 scale only sub-linearly with respect to antigen size. OptMAVEn-2.0 was used to design and rank variable antibody fragments targeting five epitopes of Zika envelope protein and three of hen egg white lysozyme. Among the top five ranked designs for each epitope, recovery of native residue identities is typically 45–65%. MD simulations of two designs targeting Zika suggest that at least one would bind with high affinity. OptMAVEn-2.0 can be downloaded from our GitHub repository and webpage as (links in Summary and Discussion section).
Full article
►▼
Show Figures
Open AccessReview
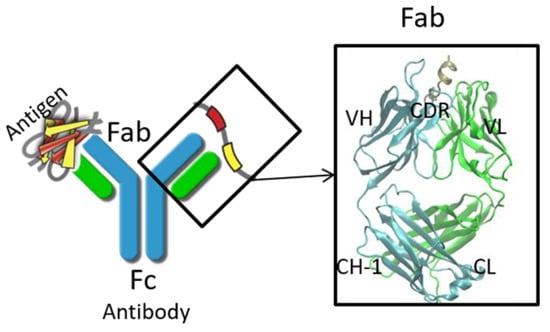
In Silico Methods in Antibody Design
by
Jun Zhao, Ruth Nussinov, Wen-Jin Wu and Buyong Ma
Cited by 34 | Viewed by 19266
Abstract
Antibody therapies with high efficiency and low toxicity are becoming one of the major approaches in antibody therapeutics. Based on high-throughput sequencing and increasing experimental structures of antibodies/antibody-antigen complexes, computational approaches can predict antibody/antigen structures, engineering the function of antibodies and design antibody-antigen
[...] Read more.
Antibody therapies with high efficiency and low toxicity are becoming one of the major approaches in antibody therapeutics. Based on high-throughput sequencing and increasing experimental structures of antibodies/antibody-antigen complexes, computational approaches can predict antibody/antigen structures, engineering the function of antibodies and design antibody-antigen complexes with improved properties. This review summarizes recent progress in the field of in silico design of antibodies, including antibody structure modeling, antibody-antigen complex prediction, antibody stability evaluation, and allosteric effects in antibodies and functions. We listed the cases in which these methods have helped experimental studies to improve the affinities and physicochemical properties of antibodies. We emphasized how the molecular dynamics unveiled the allosteric effects during antibody-antigen recognition and antibody-effector recognition.
Full article
►▼
Show Figures
Open AccessArticle
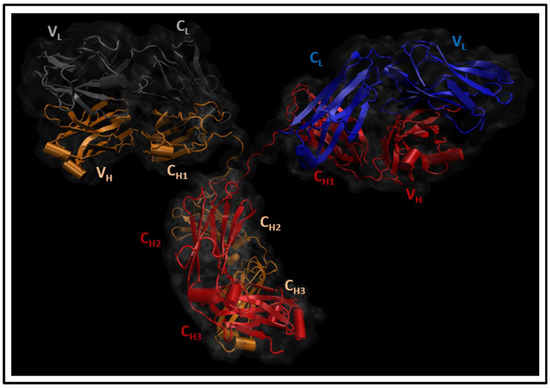
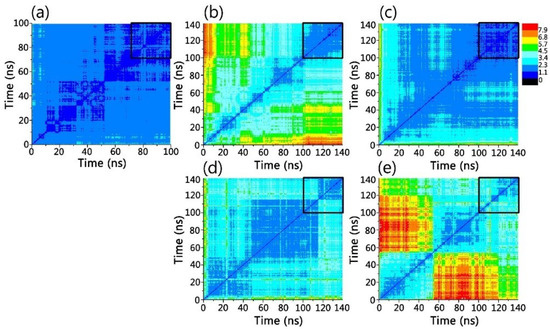
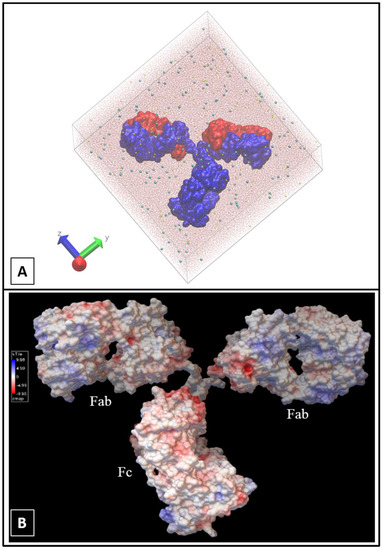
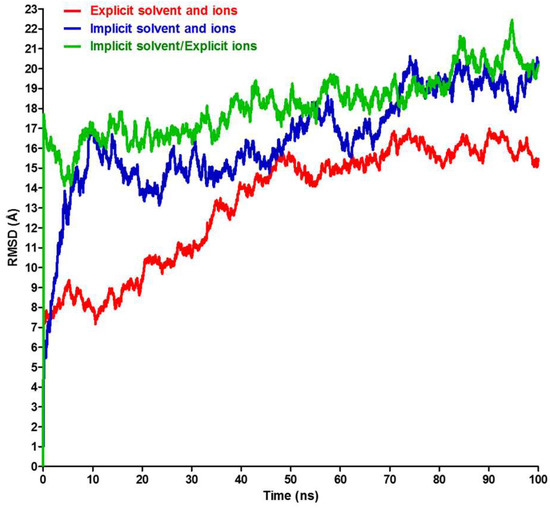
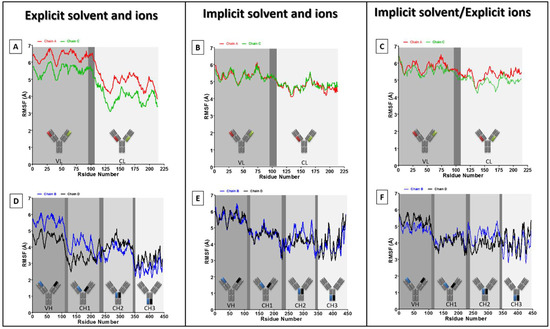
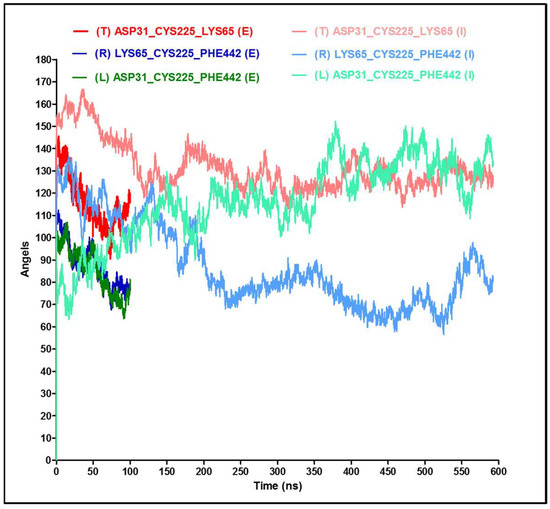
Thinking outside the Laboratory: Analyses of Antibody Structure and Dynamics within Different Solvent Environments in Molecular Dynamics (MD) Simulations
by
Mohammed M. Al Qaraghuli, Karina Kubiak-Ossowska and Paul A. Mulheran
Cited by 31 | Viewed by 10616
Abstract
Monoclonal antibodies (mAbs) have revolutionized the biomedical field, directly influencing therapeutics and diagnostics in the biopharmaceutical industry, while continuing advances in computational efficiency have enabled molecular dynamics (MD) simulations to provide atomistic insight into the structure and function of mAbs. Despite the success
[...] Read more.
Monoclonal antibodies (mAbs) have revolutionized the biomedical field, directly influencing therapeutics and diagnostics in the biopharmaceutical industry, while continuing advances in computational efficiency have enabled molecular dynamics (MD) simulations to provide atomistic insight into the structure and function of mAbs. Despite the success of MD tools, further optimizations are still required to enhance the computational efficiency of complex mAb simulations. This issue can be tackled by changing the way the solvent system is modelled to reduce the number of atoms to be tracked but must be done without compromising the accuracy of the simulations. In this work, the structure of the IgG
2a antibody was analyzed in three solvent systems: explicit water and ions, implicit water and ions, and implicit water and explicit ions. Root-mean-square distance (RMSD), root-mean-square fluctuations (RMSF), and interchain angles were used to quantify structural changes. The explicit system provides the most atomistic detail but is ~6 times slower in its exploration of configurational space and required ~4 times more computational time on our supercomputer than the implicit simulations. Overall, the behavior of the implicit and explicit simulations is quantifiably similar, with the inclusion of explicit ions in the implicit simulation stabilizing the antibody to reproduce well the statistical fluctuations of the fully explicit system. Therefore, this approach holds promise to maximize the use of computational resources to explore antibody behavior.
Full article
►▼
Show Figures
Open AccessArticle
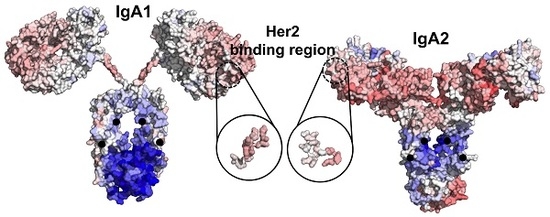
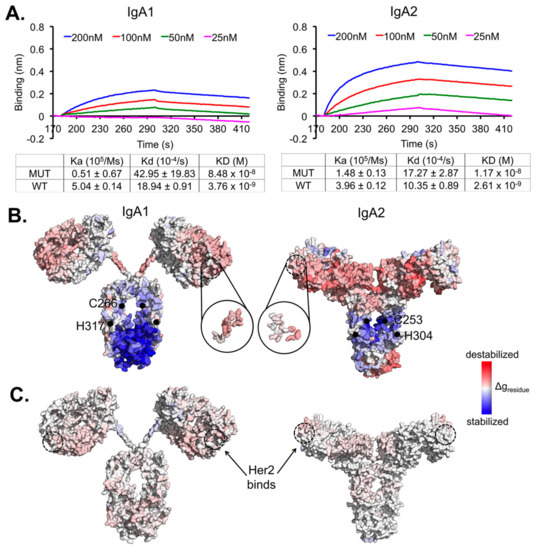
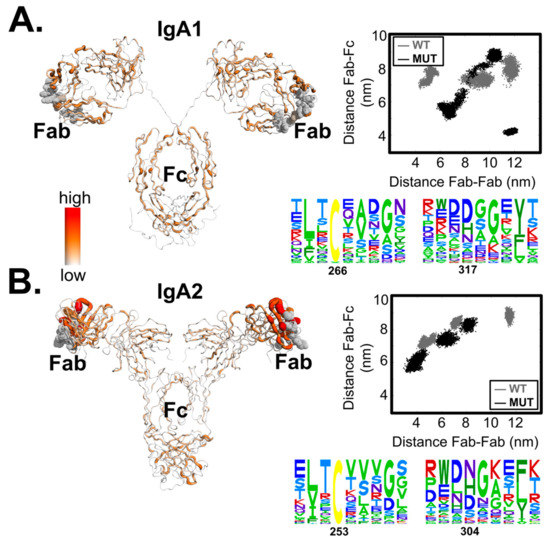
Allosteric Effects between the Antibody Constant and Variable Regions: A Study of IgA Fc Mutations on Antigen Binding
by
Chinh Tran-To Su, Wai-Heng Lua, Wei-Li Ling and Samuel Ken-En Gan
Cited by 40 | Viewed by 11962
Abstract
Therapeutic antibodies have shifted the paradigm of disease treatments from small molecules to biologics, especially in cancer therapy. Despite the increasing number of antibody candidates, much remains unknown about the antibody and how its various regions interact. Recent findings showed that the antibody
[...] Read more.
Therapeutic antibodies have shifted the paradigm of disease treatments from small molecules to biologics, especially in cancer therapy. Despite the increasing number of antibody candidates, much remains unknown about the antibody and how its various regions interact. Recent findings showed that the antibody constant region can govern localization effects that are useful in reducing side effects due to systemic circulation by the commonly used IgG isotypes. Given their localized mucosal effects, IgA antibodies are increasingly promising therapeutic biologics. While the antibody Fc effector cell activity has been a focus point, recent research showed that the Fc could also influence antigen binding, challenging the conventional idea of region-specific antibody functions. To investigate this, we analysed the IgA antibody constant region and its distal effects on the antigen binding regions using recombinant Pertuzumab IgA1 and IgA2 variants. We found that mutations in the C-region reduced Her2 binding experimentally, and computational structural analysis showed that allosteric communications were highly dependent on the antibody hinge, providing strong evidence that we should consider antibodies as whole proteins rather than a sum of functional regions.
Full article
►▼
Show Figures





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}