
The Accurate Prediction of Antibody Deamidations by Combining High-Throughput Automated Peptide Mapping and Protein Language Model-Based Deep Learning


Highlights
- Fully automated peptide mapping is used to generate comprehensive and quality antibody sequence liability (deamidation) dataset, overcoming labor and time bottlenecks.
- A novel chimeric model is developed, combining ESM-2 protein language model (pLM)-derived embeddings and antibody local-sequence information for accurate sequence liability occurrence and trend predictions.
- The workflow, which integrates high-throughput peptide mapping with deep learning, can be extended for broader applications to predict other sequence liabilities like oxidation and isomerization, enabling effective and accelerated drug-candidate selections.
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
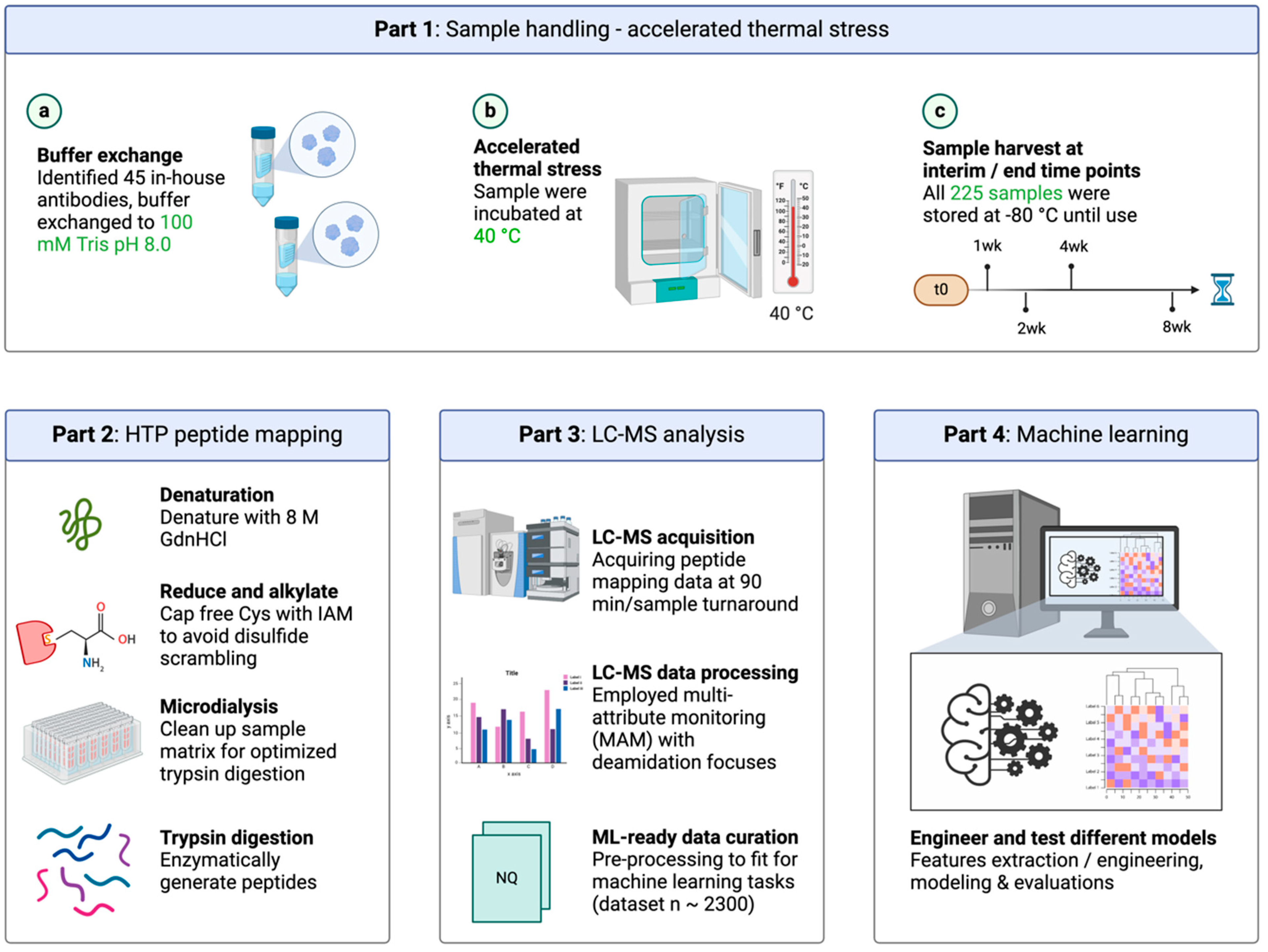
2.2. Accelerated Thermal Stress
2.3. Automated Peptide Mapping
2.4. LC-MS/MS Analysis
3. Results
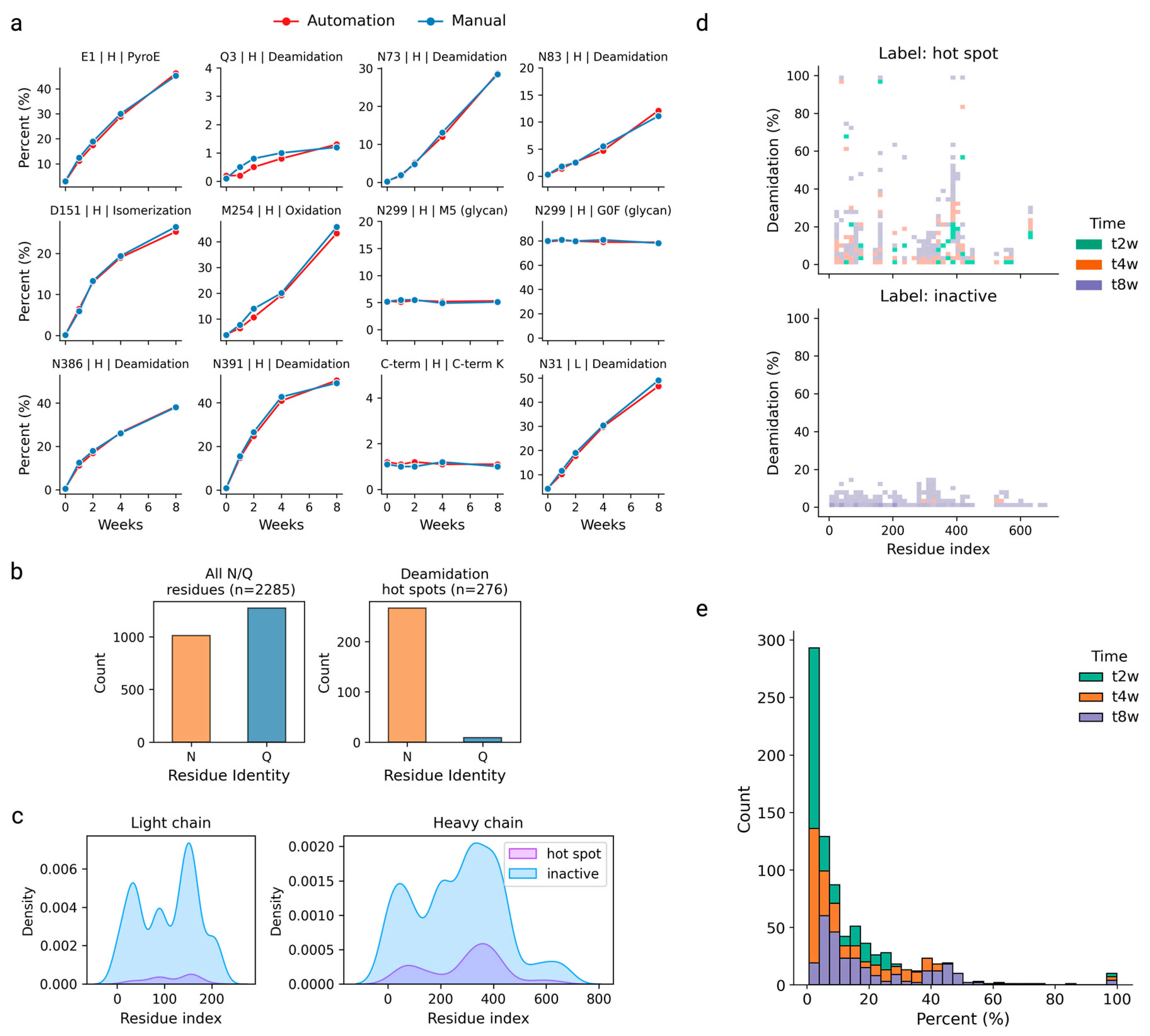
3.1. High-Throughput (HTP) Automated Peptide Mapping
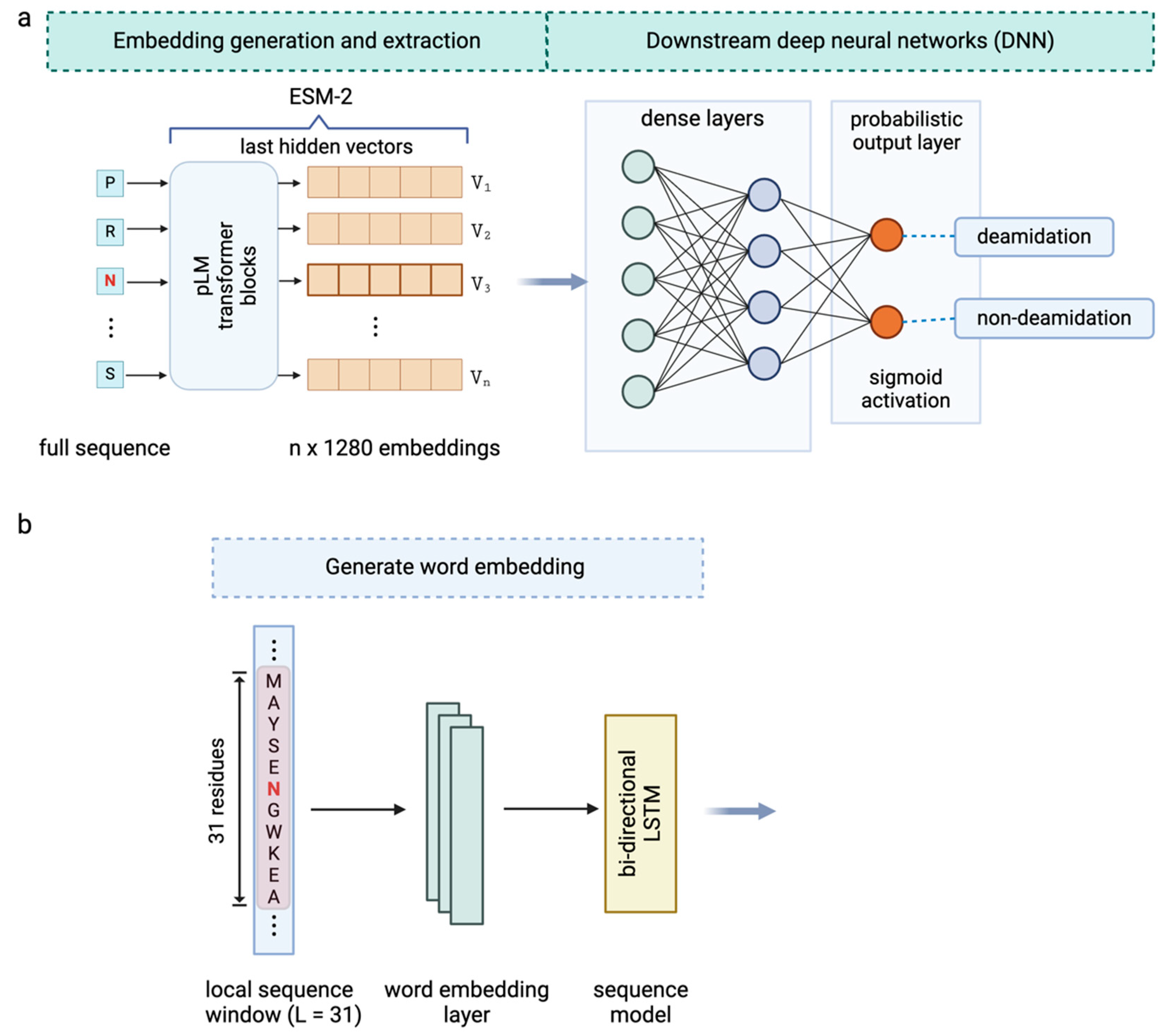
3.2. The Use of ESM-2 Embedding for Deamidation Site Prediction
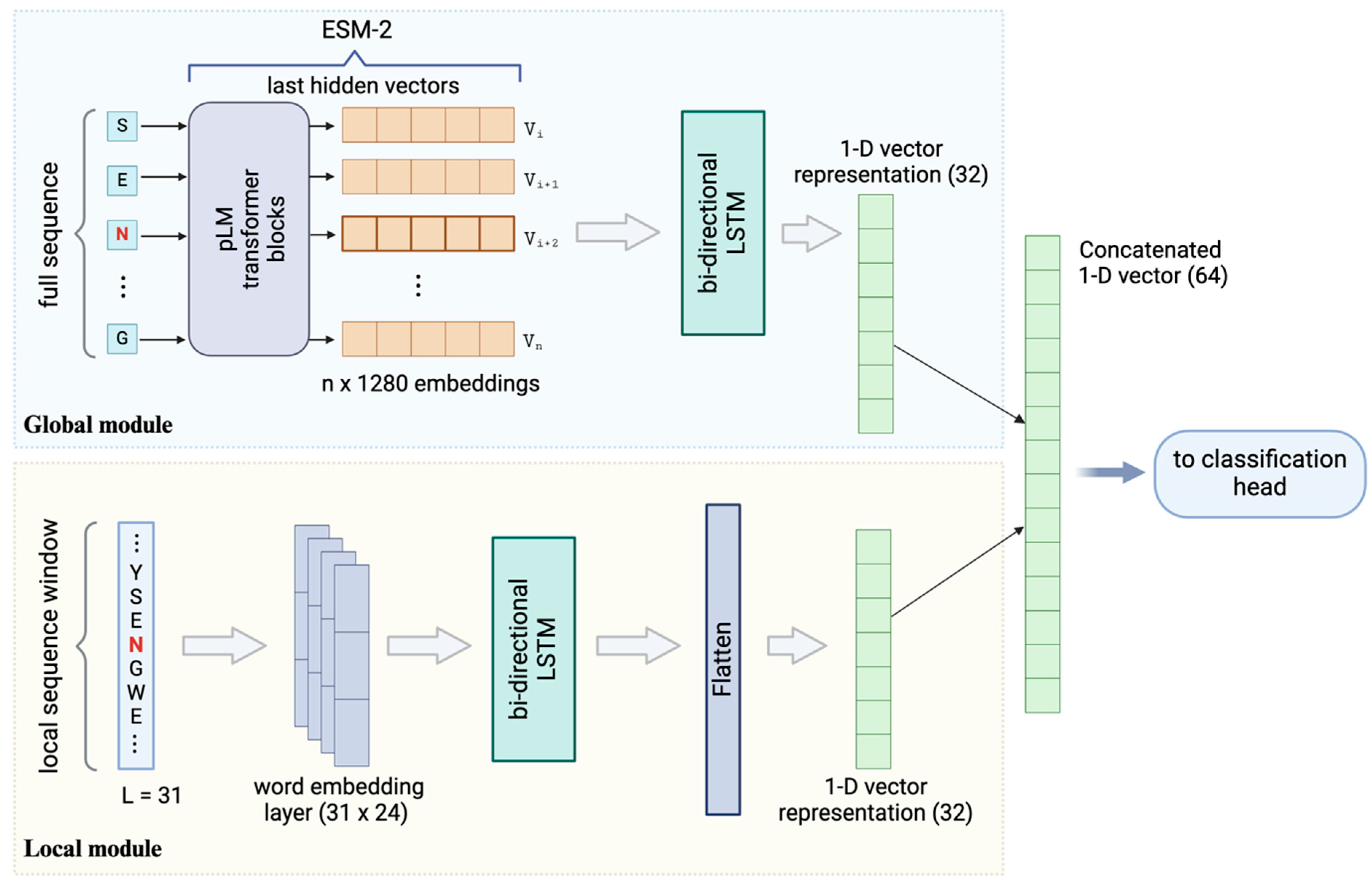
3.3. Enhanced Prediction by Combining ESM-2 Embedding and Local Sequence Information
3.4. Performance Evaluation of Models
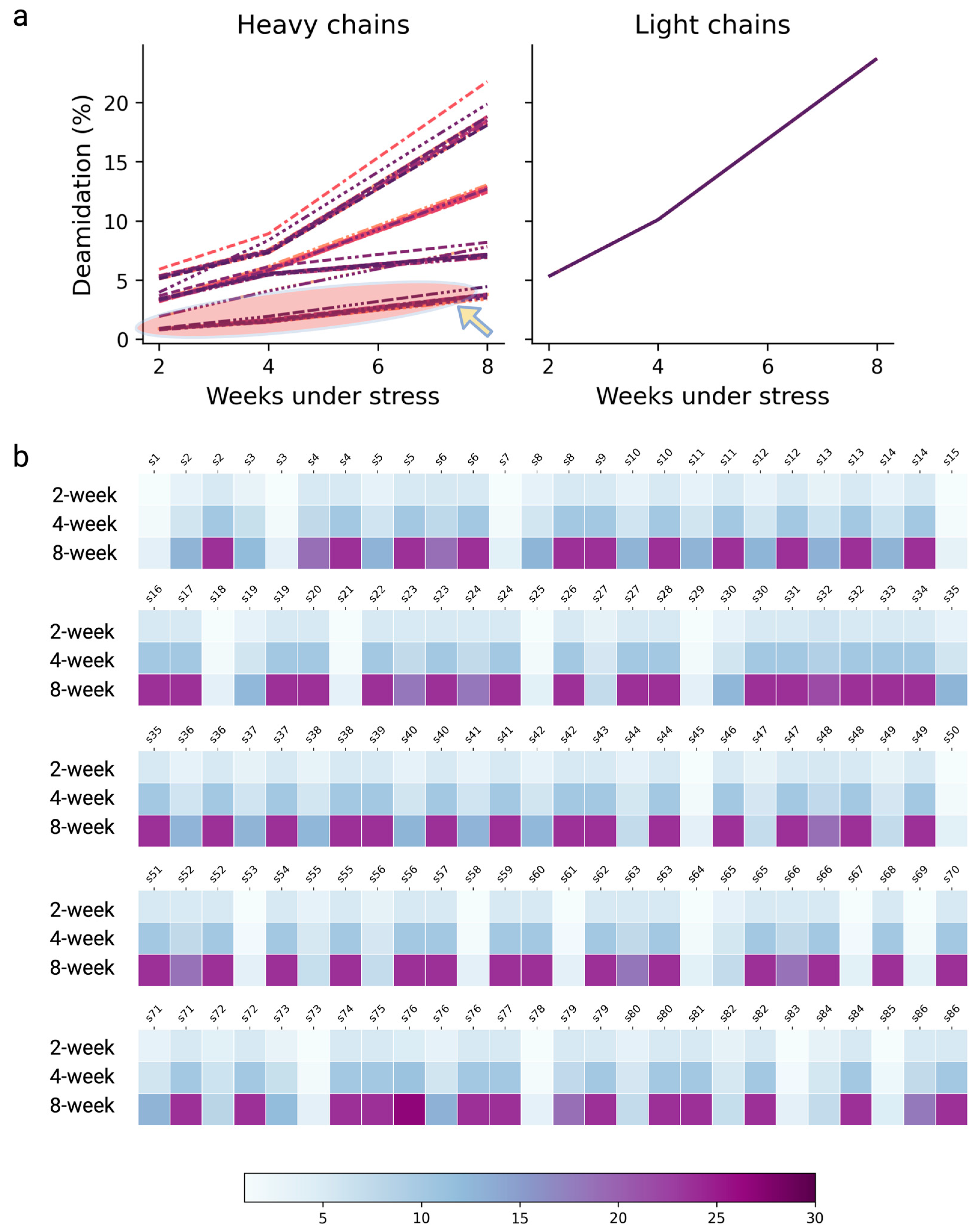
3.5. Independent Dataset Predicting Deamidation Hot Spots
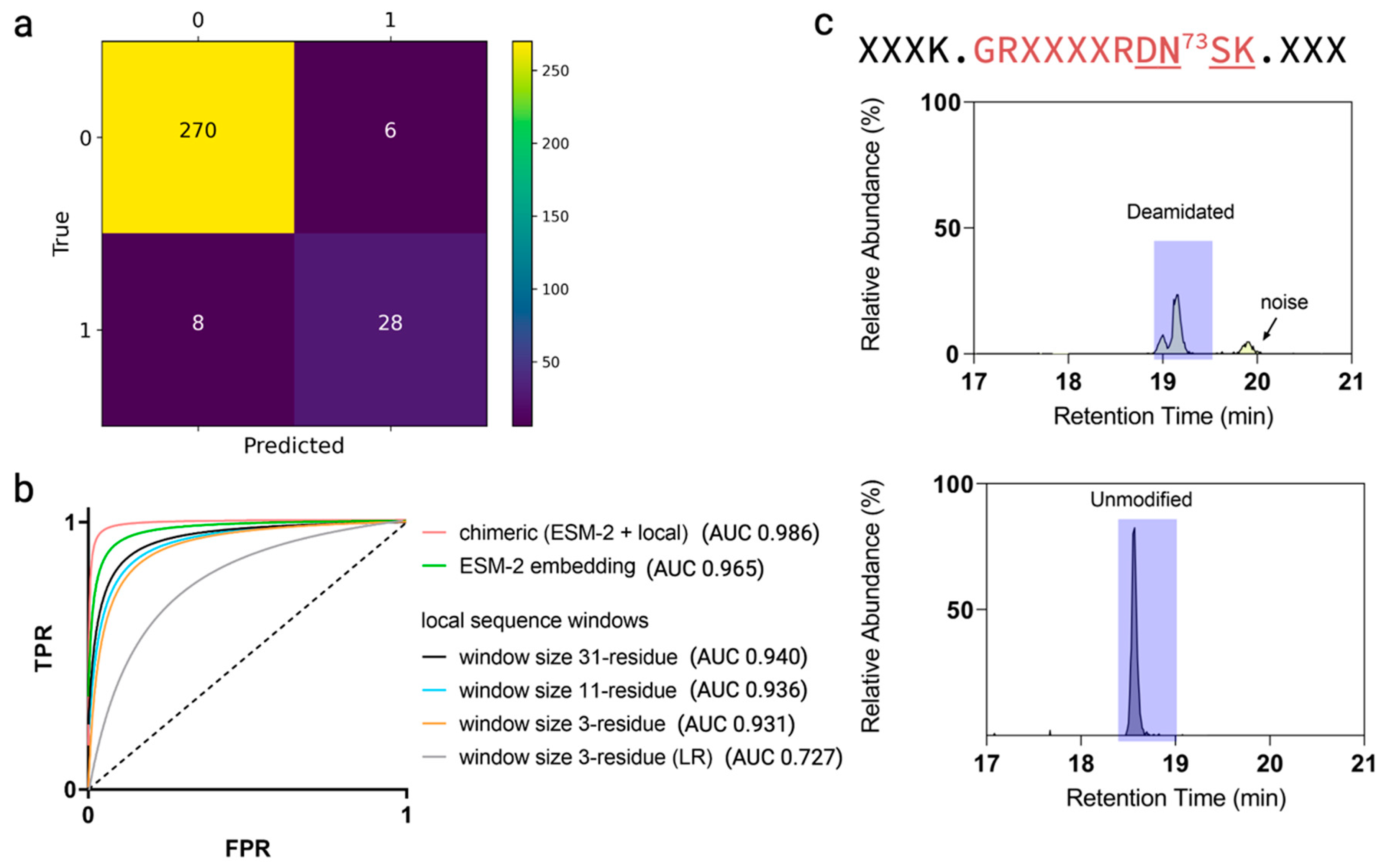
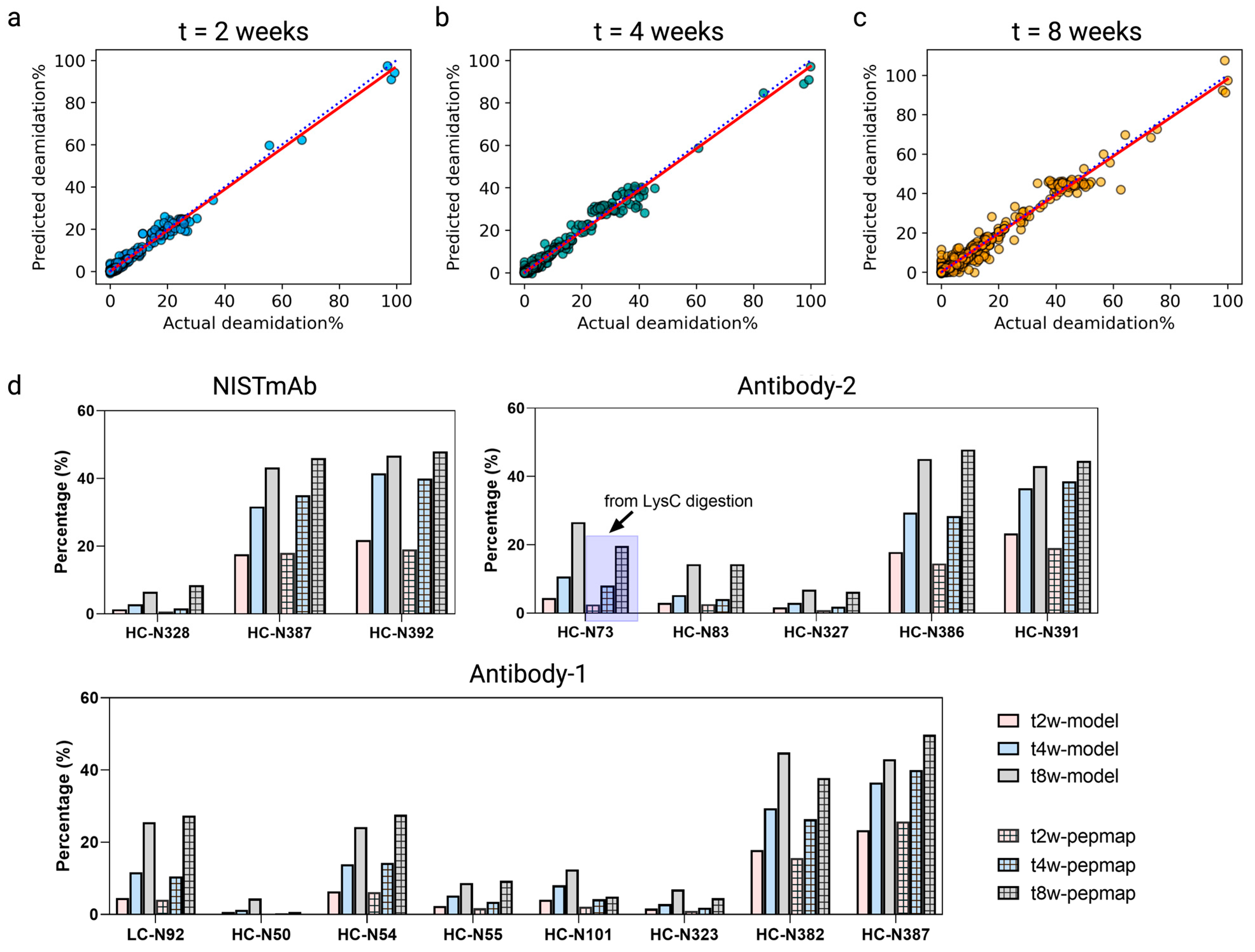
3.6. Quantitative Deamidation Extents Prediction
3.7. Model Implementation for High-Throughput Screening Drug Candidates
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Ab | Antibody |
| ANN | Artificial neural network |
| Asn | Asparagine |
| Asp | Aspartic acid |
| AUC | Area under the curve |
| CDR | Complementarity-determining region |
| CID | Collision-induced dissociation |
| cIEF | Capillary isoelectric focusing |
| CNN | Convolutional neural network |
| Cys | Cysteine |
| DNN | Deep neural network |
| DTT | Dithiothreitol |
| EDTA | Ethylenediaminetetraacetic acid |
| ESM | Evolutionary scale modeling |
| ETD | Electron-transfer dissociation |
| FN | False negative |
| FP | False positive |
| FPR | False positive rate |
| FTE | Full-time employee |
| GdnHCl | Guanidine hydrochloride |
| HCD | Higher energy collision dissociation |
| HTP | High-throughput |
| IAM | Iodoacetamide |
| LC-MS | Liquid chromatography–mass spectrometry |
| LSTM | Long short-term memory |
| Lys | Lysine |
| LysC | Endoproteinase Lys-C enzyme |
| mAb | Monoclonal antibody |
| MCC | Matthews correlation coefficient |
| Met | Methionine |
| MS/MS | Tandem mass spectrometry |
| pCQA | Potential critical quality attributes |
| pLM | Protein language model |
| PTM | Post-translational modification |
| QSAR | Quantitative structure–activity relationship |
| ReLU | Rectified linear unit |
| RNN | Recurrent neural network |
| ROC | Receiver operating characteristics |
| SASA | Solvent-accessible surface area |
| SD | Standard deviation |
| TFA | Trifluoroacetic acid |
| TN | True negative |
| TP | True positive |
| TPR | True positive rate |
| Trp | Tryptophan |
| Tyr | Tyrosine |
| UV | Ultraviolet |
| XIC | Extracted ion chromatogram |
References
- Beck, A.; Reichert, J.M. Therapeutic Fc-fusion proteins and peptides as successful alternatives to antibodies. MAbs 2011, 3, 415–416. [Google Scholar] [CrossRef] [PubMed]
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. MAbs 2015, 7, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.; Meksiriporn, B.; Tan, J.; Spangler, J.B. Mechanism-Driven Design of Multispecific Antibodies for Targeted Disease Treatment. Annu. Rev. Chem. Biomol. Eng. 2024, 15, 415–416. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Jiskoot, W.; Schoneich, C.; Rathore, A.S. Oxidation and Deamidation of Monoclonal Antibody Products: Potential Impact on Stability, Biological Activity, and Efficacy. J. Pharm. Sci. 2022, 111, 903–918. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, A.A.R.; D’Angelo, S.; Erasmus, M.F.; Leal-Lopes, C.; Ferrara, F.; Spector, L.P.; Naranjo, L.; Molina, E.; Max, T.; DeAguero, A.; et al. Simultaneous affinity maturation and developability enhancement using natural liability-free CDRs. MAbs 2022, 14, 2115200. [Google Scholar] [CrossRef] [PubMed]
- Gervais, D. Protein deamidation in biopharmaceutical manufacture: Understanding, control and impact. J. Chem. Technol. Biot. 2016, 91, 569–575. [Google Scholar] [CrossRef]
- Lu, X.; Machiesky, L.A.; De Mel, N.; Du, Q.; Xu, W.; Washabaugh, M.; Jiang, X.R.; Wang, J. Characterization of IgG1 Fc Deamidation at Asparagine 325 and Its Impact on Antibody-dependent Cell-mediated Cytotoxicity and FcgammaRIIIa Binding. Sci. Rep. 2020, 10, 383. [Google Scholar] [CrossRef] [PubMed]
- Nowak, C.; Cheung, J.K.; Dellatore, S.M.; Katiyar, A.; Bhat, R.; Sun, J.; Ponniah, G.; Neill, A.; Mason, B.; Beck, A.; et al. Forced degradation of recombinant monoclonal antibodies: A practical guide. MAbs 2017, 9, 1217–1230. [Google Scholar] [CrossRef] [PubMed]
- Federici, M.; Lubiniecki, A.; Manikwar, P.; Volkin, D.B. Analytical lessons learned from selected therapeutic protein drug comparability studies. Biologicals 2013, 41, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Sandra, K.; Vandenheede, I.; Sandra, P. Modern chromatographic and mass spectrometric techniques for protein biopharmaceutical characterization. J. Chromatogr. A 2014, 1335, 81–103. [Google Scholar] [CrossRef] [PubMed]
- Carillo, S.; Criscuolo, A.; Fussl, F.; Cook, K.; Bones, J. Intact multi-attribute method (iMAM): A flexible tool for the analysis of monoclonal antibodies. Eur. J. Pharm. Biopharm. 2022, 177, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Mouchahoir, T.; Schiel, J.E.; Rogers, R.; Heckert, A.; Place, B.J.; Ammerman, A.; Li, X.; Robinson, T.; Schmidt, B.; Chumsae, C.M.; et al. Attribute Analytics Performance Metrics from the MAM Consortium Interlaboratory Study. J. Am. Soc. Mass. Spectrom. 2022, 33, 1659–1677. [Google Scholar] [CrossRef] [PubMed]
- Pohl, T.; Gervais, A.; Dirksen, E.H.C.; D’Alessio, V.; Bechtold-Peters, K.; Burkitt, W.; Cao, L.; Greven, S.; Lennard, A.; Li, X.; et al. Technical considerations for the implementation of the multi-attribute-method by mass spectrometry in a quality control laboratory. Eur. J. Pharm. Biopharm. 2023, 188, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Plotnikov, N.V.; Rouse, J.C.; Singh, S.K. Biopharmaceutical Informatics: Supporting biologic drug development via molecular modelling and informatics. J. Pharm. Pharmacol. 2018, 70, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, N.V.; Singh, S.K.; Rouse, J.C.; Kumar, S. Quantifying the Risks of Asparagine Deamidation and Aspartate Isomerization in Biopharmaceuticals by Computing Reaction Free-Energy Surfaces. J. Phys. Chem. B 2017, 121, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.E.; Robinson, A.B. Prediction of protein deamidation rates from primary and three-dimensional structure. Proc. Natl. Acad. Sci. USA 2001, 98, 4367–4372. [Google Scholar] [CrossRef] [PubMed]
- Vatsa, S. In silico prediction of post-translational modifications in therapeutic antibodies. MAbs 2022, 14, 2023938. [Google Scholar] [CrossRef] [PubMed]
- Delmar, J.A.; Wang, J.; Choi, S.W.; Martins, J.A.; Mikhail, J.P. Machine Learning Enables Accurate Prediction of Asparagine Deamidation Probability and Rate. Mol. Ther. Methods Clin. Dev. 2019, 15, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, D.; Bauer, J.; Kossner, M.; Henry, A.; Karow-Zwick, A.R.; Licari, G. Predicting deamidation and isomerization sites in therapeutic antibodies using structure-based in silico approaches. MAbs 2024, 16, 2333436. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Sun, Y. Protein asparagine deamidation prediction based on structures with machine learning methods. PLoS ONE 2017, 12, e0181347. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, J.R.; Alonso, L.G.; Sanchez, I.E. Prediction of Spontaneous Protein Deamidation from Sequence-Derived Secondary Structure and Intrinsic Disorder. PLoS ONE 2015, 10, e0145186. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, J.R.; Leonetti, C.O.; Alonso, L.G.; Sanchez, I.E. NGOME-Lite: Proteome-wide prediction of spontaneous protein deamidation highlights differences between taxa. Methods 2022, 200, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Sydow, J.F.; Lipsmeier, F.; Larraillet, V.; Hilger, M.; Mautz, B.; Molhoj, M.; Kuentzer, J.; Klostermann, S.; Schoch, J.; Voelger, H.R.; et al. Structure-based prediction of asparagine and aspartate degradation sites in antibody variable regions. PLoS ONE 2014, 9, e100736. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Huang, M.; Lewis, M.J.; Hu, P. Structure Based Prediction of Asparagine Deamidation Propensity in Monoclonal Antibodies. MAbs 2018, 10, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.E.; Robinson, Z.W.; Robinson, B.R.; Robinson, A.L.; Robinson, J.A.; Robinson, M.L.; Robinson, A.B. Structure-dependent nonenzymatic deamidation of glutaminyl and asparaginyl pentapeptides. J. Pept. Res. 2004, 63, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Tunnermann, L.; Lofstedt, T.; Gratz, R. Transformer-based deep learning for predicting protein properties in the life sciences. Elife 2023, 12, e82819. [Google Scholar] [CrossRef] [PubMed]
- Brandes, N.; Ofer, D.; Peleg, Y.; Rappoport, N.; Linial, M. ProteinBERT: A universal deep-learning model of protein sequence and function. Bioinformatics 2022, 38, 2102–2110. [Google Scholar] [CrossRef]
- Rives, A.; Meier, J.; Sercu, T.; Goyal, S.; Lin, Z.; Liu, J.; Guo, D.; Ott, M.; Zitnick, C.L.; Ma, J.; et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. USA 2021, 118, e2016239118. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rehawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M.; et al. ProtTrans: Toward Understanding the Language of Life Through Self-Supervised Learning. IEEE Trans. Pattern. Anal. Mach. Intell. 2022, 44, 7112–7127. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Su, H.; Wang, W.; Ye, L.; Wei, H.; Peng, Z.; Anishchenko, I.; Baker, D.; Yang, J. The trRosetta server for fast and accurate protein structure prediction. Nat. Protoc. 2021, 16, 5634–5651. [Google Scholar] [CrossRef] [PubMed]
- Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical assessment of methods of protein structure prediction (CASP)-Round XIII. Proteins 2019, 87, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Wang, R.; Sun, Y.; Liu, J.; Chen, Z.; Zhang, Y.J. Interpretable feature extraction and dimensionality reduction in ESM2 for protein localization prediction. Brief Bioinform. 2024, 25, bbad534. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Jiang, L.; Chen, Y.; Zhang, Y.; Li, X.; Ma, Z.; Fu, Z.; He, F.; Sun, P.; Ren, Z.; et al. THPLM: A sequence-based deep learning framework for protein stability changes prediction upon point variations using pretrained protein language model. Bioinformatics 2023, 39, btad646. [Google Scholar] [CrossRef] [PubMed]
- Pakhrin, S.C.; Pokharel, S.; Pratyush, P.; Chaudhari, M.; Ismail, H.D.; Kc, D.B. LMPhosSite: A Deep Learning-Based Approach for General Protein Phosphorylation Site Prediction Using Embeddings from the Local Window Sequence and Pretrained Protein Language Model. J. Proteome Res. 2023, 22, 2548–2557. [Google Scholar] [CrossRef] [PubMed]
- Pokharel, S.; Pratyush, P.; Heinzinger, M.; Newman, R.H.; Kc, D.B. Improving protein succinylation sites prediction using embeddings from protein language model. Sci. Rep. 2022, 12, 16933. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.E.; Dubois, H.; Hoffmann, M.; D’Eri, S.; Fromentin, Y.; Wiesner, J.; Pfenninger, A.; Clavier, S.; Pieper, A.; Duhau, L.; et al. Automated mass spectrometry multi-attribute method analyses for process development and characterization of mAbs. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2021, 1166, 122540. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhang, J.; Buettner, A.; Vosika, E.; Sadek, M.; Hao, Z.; Reusch, D.; Koenig, M.; Chan, W.; Bathke, A.; et al. Mass spectrometry-based multi-attribute method in protein therapeutics product quality monitoring and quality control. MAbs 2023, 15, 2197668. [Google Scholar] [CrossRef] [PubMed]
- Harding-Larsen, D.; Funk, J.; Madsen, N.G.; Gharabli, H.; Acevedo-Rocha, C.G.; Mazurenko, S.; Welner, D.H. Protein Representations: Encoding Biological Information for Machine Learning in Biocatalysis. ChemRxiv 2024. [Google Scholar]
- Suzek, B.E.; Huang, H.; McGarvey, P.; Mazumder, R.; Wu, C.H. UniRef: Comprehensive and non-redundant UniProt reference clusters. Bioinformatics 2007, 23, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Iman, M.; Arabnia, H.R.; Rasheed, K. A Review of Deep Transfer Learning and Recent Advancements. Technologies 2023, 11, 40. [Google Scholar] [CrossRef]
- Villegas-Morcillo, A.; Makrodimitris, S.; van Ham, R.; Gomez, A.M.; Sanchez, V.; Reinders, M.J.T. Unsupervised protein embeddings outperform hand-crafted sequence and structure features at predicting molecular function. Bioinformatics 2021, 37, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Weissenow, K.; Heinzinger, M.; Rost, B. Protein language-model embeddings for fast, accurate, and alignment-free protein structure prediction. Structure 2022, 30, 1169–1177e1164. [Google Scholar] [CrossRef] [PubMed]
- Jha, K.; Saha, S.; Singh, H. Prediction of protein-protein interaction using graph neural networks. Sci. Rep. 2022, 12, 8360. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.T.; Winter, R.; Clevert, D.A. Unsupervised Representation Learning for Proteochemometric Modeling. Int. J. Mol. Sci. 2021, 22, 2882. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tian, B. Protein–DNA binding sites prediction based on pre-trained protein language model and contrastive learning. Brief. Bioinform. 2024, 25, bbad488. [Google Scholar] [CrossRef]
- Lu, X.; Nobrega, R.P.; Lynaugh, H.; Jain, T.; Barlow, K.; Boland, T.; Sivasubramanian, A.; Vasquez, M.; Xu, Y. Deamidation and isomerization liability analysis of 131 clinical-stage antibodies. MAbs 2019, 11, 45–57. [Google Scholar] [CrossRef] [PubMed]
- 16—Development issues: Antibody stability, developability, immunogenicity, and comparability. In Therapeutic Antibody Engineering; Strohl, W.R., Strohl, L.M., Eds.; Woodhead Publishing: Sawston, UK, 2012; pp. 377–595. [Google Scholar] [CrossRef]
- Chelius, D.; Rehder, D.S.; Bondarenko, P.V. Identification and characterization of deamidation sites in the conserved regions of human immunoglobulin gamma antibodies. Anal. Chem. 2005, 77, 6004–6011. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, J.L.; Inglese, J.; Walters, M.A. Mitigating risk in academic preclinical drug discovery. Nat. Rev. Drug Discov. 2015, 14, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Abel, R.; Mondal, S.; Masse, C.; Greenwood, J.; Harriman, G.; Ashwell, M.A.; Bhat, S.; Wester, R.; Frye, L.; Kapeller, R.; et al. Accelerating drug discovery through tight integration of expert molecular design and predictive scoring. Curr. Opin. Struct. Biol. 2017, 43, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Khetan, R.; Curtis, R.; Deane, C.M.; Hadsund, J.T.; Kar, U.; Krawczyk, K.; Kuroda, D.; Robinson, S.A.; Sormanni, P.; Tsumoto, K.; et al. Current advances in biopharmaceutical informatics: Guidelines, impact and challenges in the computational developability assessment of antibody therapeutics. MAbs 2022, 14, 2020082. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Niu, B.; Jimenez, R.B.; Wang, J.; Albarghouthi, M. Fully automated peptide mapping multi-attribute method by liquid chromatography-mass spectrometry with robotic liquid handling system. J. Pharm. Biomed. Anal. 2021, 198, 113988. [Google Scholar] [CrossRef] [PubMed]
- Hosna, A.; Merry, E.; Gyalmo, J.; Alom, Z.; Aung, Z.; Azim, M.A. Transfer learning: A friendly introduction. J. Big. Data 2022, 9, 102. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.E.; Robinson, A.B. Prediction of primary structure deamidation rates of asparaginyl and glutaminyl peptides through steric and catalytic effects. J. Pept. Res. 2004, 63, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Sun, J.; Sun, H.; Wang, Y.; Hou, S.; Jiang, B.; Xie, Y.; Ji, R.; Xue, L.; Ding, X.; et al. Identification and characterization of an unexpected isomerization motif in CDRH2 that affects antibody activity. MAbs 2023, 15, 2215364. [Google Scholar] [CrossRef] [PubMed]
- Dick, L.W., Jr.; Qiu, D.; Wong, R.B.; Cheng, K.-C. Isomerization in the CDR2 of a monoclonal antibody: Binding analysis and factors that influence the isomerization rate. Biotechnol. Bioeng. 2010, 105, 515–523. [Google Scholar] [CrossRef]
- Kim, M.S.; Pandey, A. Electron transfer dissociation mass spectrometry in proteomics. Proteomics 2012, 12, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Schmirler, R.; Heinzinger, M.; Rost, B. Fine-tuning protein language models boosts predictions across diverse tasks. bioRxiv 2023, 15, 7407. [Google Scholar] [CrossRef]
- Sledzieski, S.; Kshirsagar, M.; Baek, M.; Berger, B.; Dodhia, R.; Ferres, J.L. Democratizing Protein Language Models with Parameter-Efficient Fine-Tuning. bioRxiv 2023, 121, e2405840121. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Descriptors | Accuracy | Precision | Recall | Specificity | F1-Score | MCC |
|---|---|---|---|---|---|---|
| Local sequence only | 0.932 ± 0.014 | 0.745 ± 0.044 | 0.679 ± 0.039 | 0.967 ± 0.019 | 0.710 ± 0.035 | 0.673 ± 0.030 |
| Global embeddings only | 0.944 ± 0.012 | 0.798 ± 0.049 | 0.728 ± 0.043 | 0.975 ± 0.016 | 0.761 ± 0.046 | 0.731 ± 0.027 |
| Local + global embeddings | 0.956 ± 0.014 | 0.835 ± 0.059 | 0.790 ± 0.036 | 0.979 ± 0.016 | 0.812 ± 0.031 | 0.787 ± 0.038 |
| Classifier | Accuracy | Precision | Recall | MCC | Specificity |
|---|---|---|---|---|---|
| Decision tree model [25] | 0.949 | 0.833 | 0.694 | 0.733 | 0.982 |
| Random forest model [21] | 0.952 | 0.818 | 0.750 | 0.757 | 0.978 |
| NGOME [23] | 0.942 | 0.781 | 0.694 | 0.705 | 0.975 |
| NG, NS, and NN motifs | 0.917 | 0.586 | 0.944 | 0.704 | 0.913 |
| Chimeric model | 0.955 | 0.823 | 0.778 | 0.775 | 0.978 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, B.; Lee, B.; Wang, L.; Chen, W.; Johnson, J. The Accurate Prediction of Antibody Deamidations by Combining High-Throughput Automated Peptide Mapping and Protein Language Model-Based Deep Learning. Antibodies 2024, 13, 74. https://doi.org/10.3390/antib13030074
Niu B, Lee B, Wang L, Chen W, Johnson J. The Accurate Prediction of Antibody Deamidations by Combining High-Throughput Automated Peptide Mapping and Protein Language Model-Based Deep Learning. Antibodies. 2024; 13(3):74. https://doi.org/10.3390/antib13030074
Chicago/Turabian StyleNiu, Ben, Benjamin Lee, Lili Wang, Wen Chen, and Jeffrey Johnson. 2024. "The Accurate Prediction of Antibody Deamidations by Combining High-Throughput Automated Peptide Mapping and Protein Language Model-Based Deep Learning" Antibodies 13, no. 3: 74. https://doi.org/10.3390/antib13030074
APA StyleNiu, B., Lee, B., Wang, L., Chen, W., & Johnson, J. (2024). The Accurate Prediction of Antibody Deamidations by Combining High-Throughput Automated Peptide Mapping and Protein Language Model-Based Deep Learning. Antibodies, 13(3), 74. https://doi.org/10.3390/antib13030074