In Silico Methods in Antibody Design


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
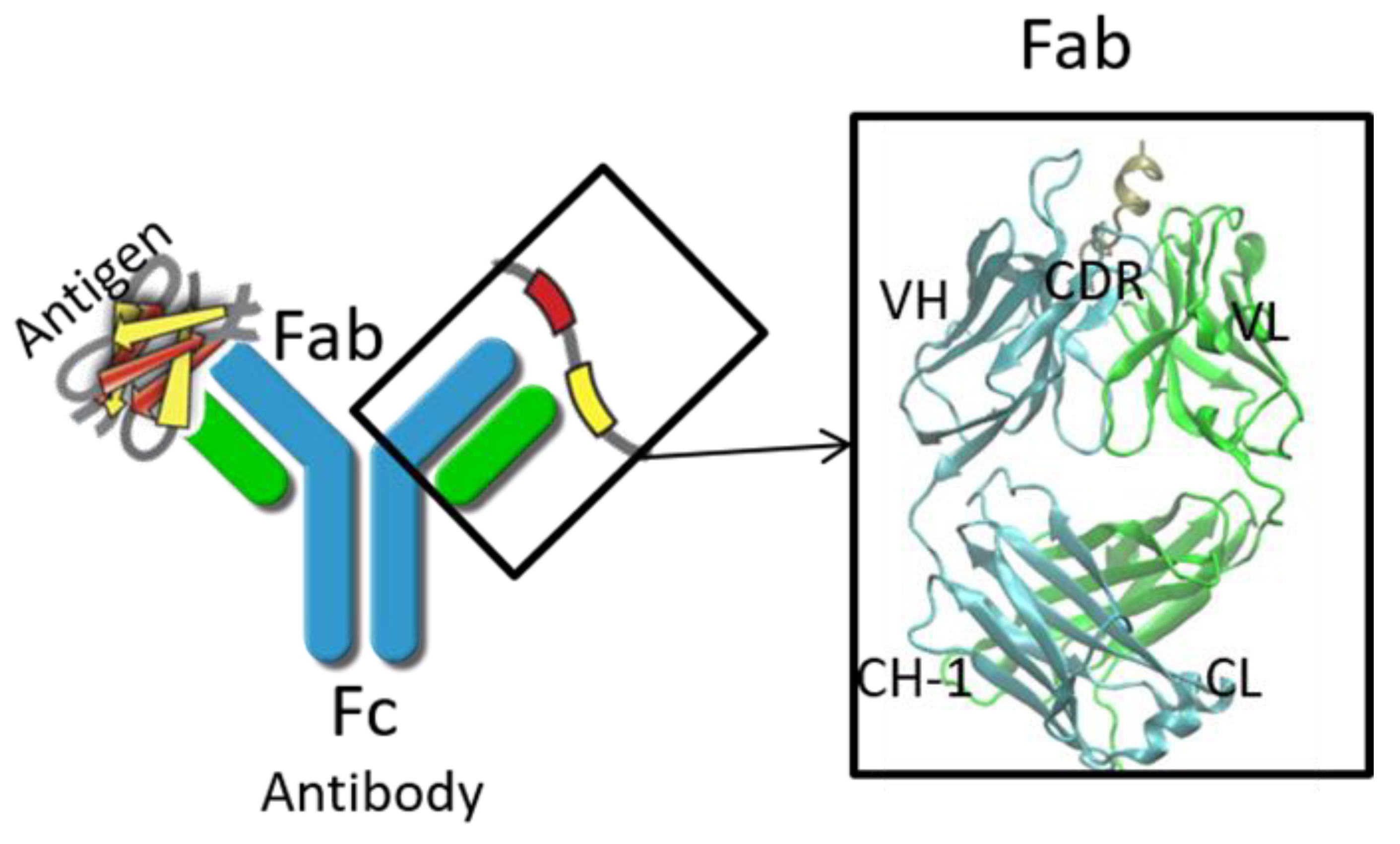
2. Structure Prediction of Variable Domain and Complementarity-Determining Regions
3. Antibody-Antigen Binding Prediction, Epitope Mapping, and Affinity Maturation
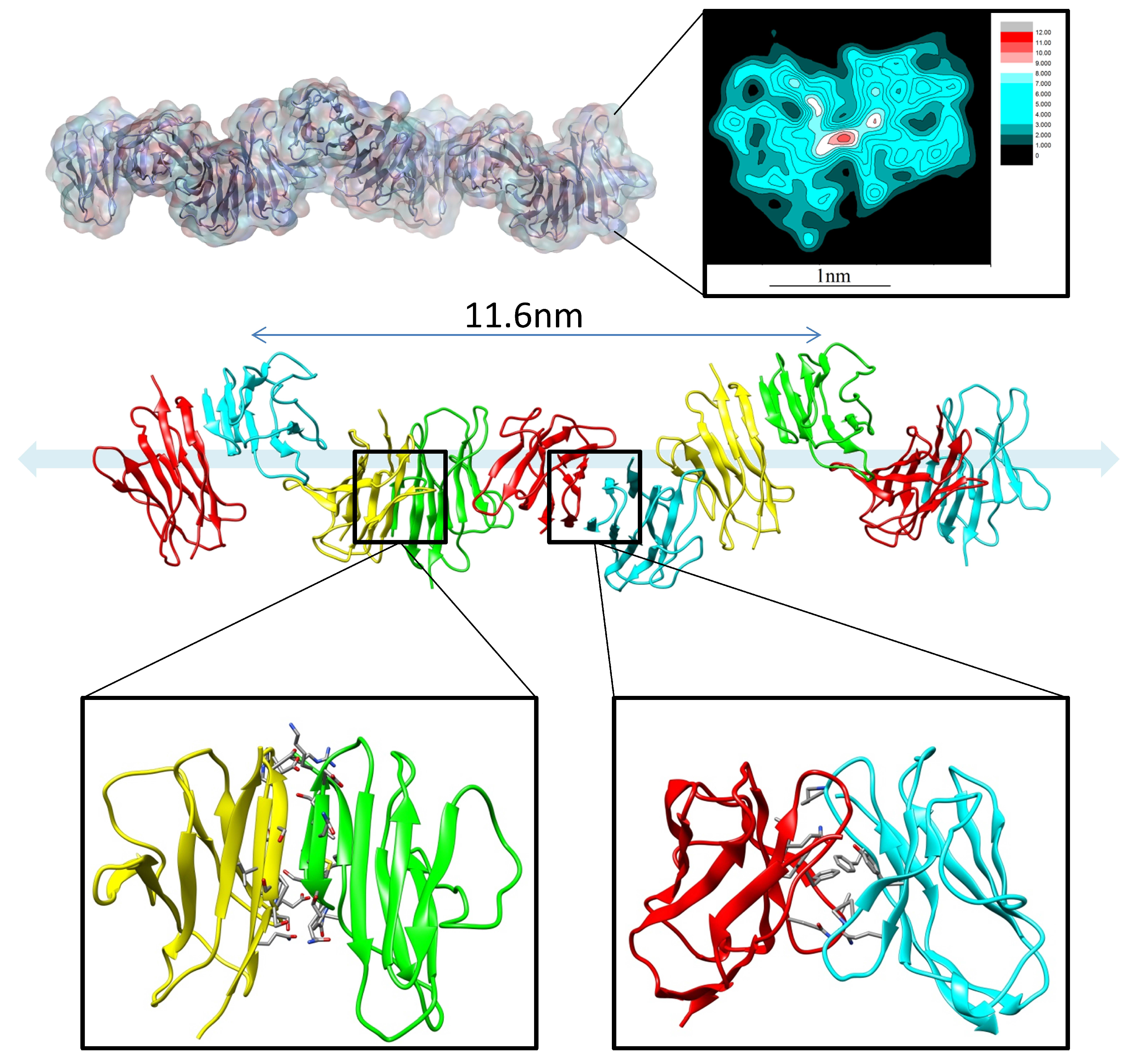
4. Antibody Aggregation, Stability, and Immunogenicity
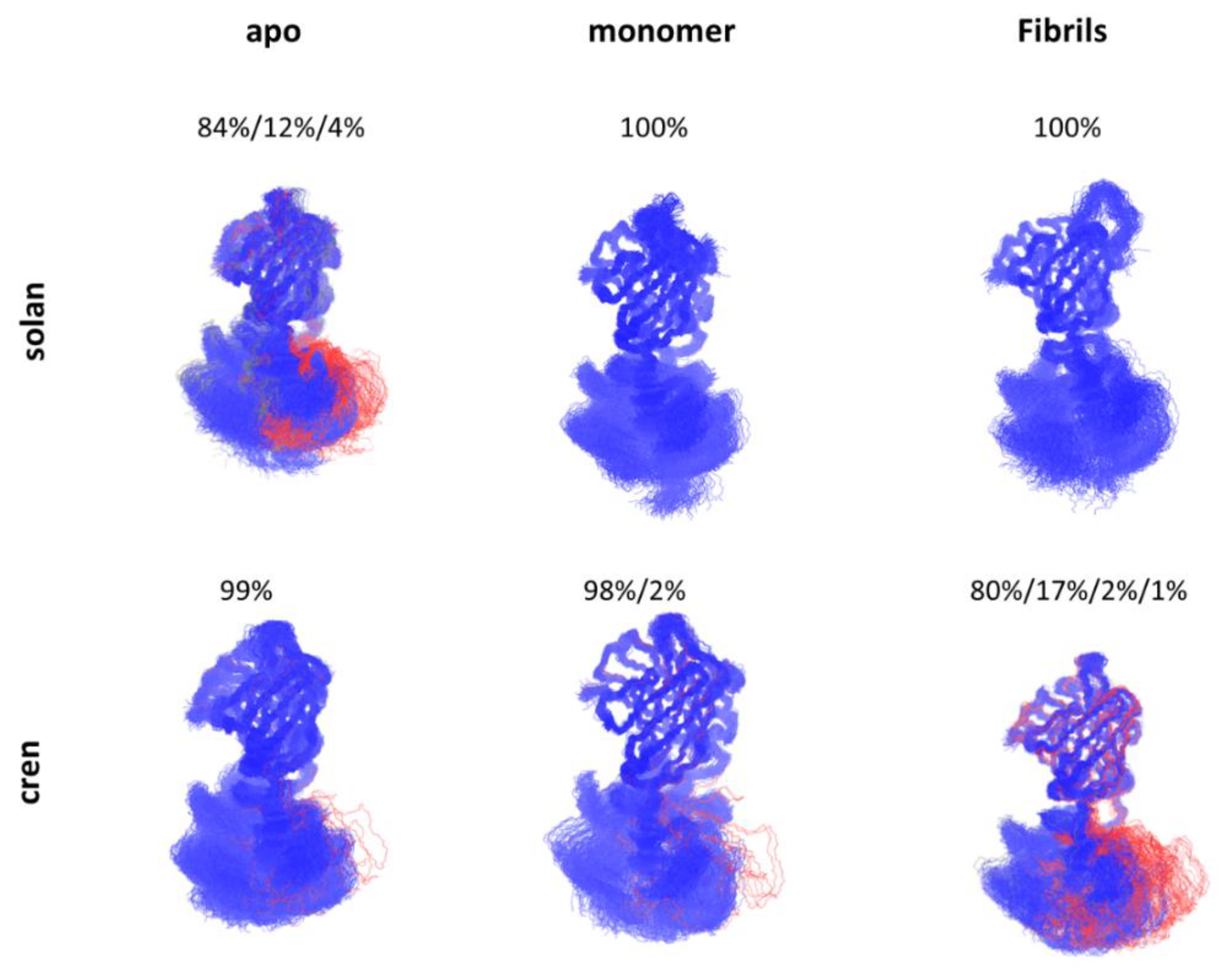
5. Allosteric Effects in Antibodies
6. Modulation of the Effector Functions
7. In Silico Vaccine Design
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pincetic, A.; Bournazos, S.; DiLillo, D.J.; Maamary, J.; Wang, T.T.; Dahan, R.; Fiebiger, B.M.; Ravetch, J.V. Type I and type II Fc receptors regulate innate and adaptive immunity. Nat. Immunol. 2014, 15, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, H.W., Jr.; Cavacini, L. Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 2010, 125, S41–S52. [Google Scholar] [CrossRef] [PubMed]
- Mian, I.S.; Bradwell, A.R.; Olson, A.J. Structure, function and properties of antibody binding sites. J. Mol. Biol. 1991, 217, 133–151. [Google Scholar] [CrossRef]
- Torres, M.; Casadevall, A. The immunoglobulin constant region contributes to affinity and specificity. Trends Immunol. 2008, 29, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Dall’Acqua, W.F.; Cook, K.E.; Damschroder, M.M.; Woods, R.M.; Wu, H. Modulation of the effector functions of a human IgG1 through engineering of its hinge region. J. Immunol. 2006, 177, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Higel, F.; Seidl, A.; Sorgel, F.; Friess, W. N-glycosylation heterogeneity and the influence on structure, function and pharmacokinetics of monoclonal antibodies and Fc fusion proteins. Eur. J. Pharm. Biopharm. 2016, 100, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Flynn, G.C.; Chen, X.; Liu, Y.D.; Shah, B.; Zhang, Z. Naturally occurring glycan forms of human immunoglobulins G1 and G2. Mol. Immunol. 2010, 47, 2074–2082. [Google Scholar] [CrossRef] [PubMed]
- Janda, A.; Bowen, A.; Greenspan, N.S.; Casadevall, A. Ig constant region effects on variable region structure and function. Front. Microbiol. 2016, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Tomaras, G.D.; Ferrari, G.; Shen, X.; Alam, S.M.; Liao, H.X.; Pollara, J.; Bonsignori, M.; Moody, M.A.; Fong, Y.; Chen, X.; et al. Vaccine-induced plasma IgA specific for the C1 region of the HIV-1 envelope blocks binding and effector function of IgG. Proc. Natl. Acad. Sci. USA 2013, 110, 9019–9024. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.J.; Shikhman, A.R.; Glass, D.D.; Kangisser, D.; Cunningham, M.W.; Greenspan, N.S. Role of heavy chain constant domains in antibody-antigen interaction. Apparent specificity differences among streptococcal IgG antibodies expressing identical variable domains. J. Immunol. 1993, 150, 2231–2242. [Google Scholar] [PubMed]
- Kato, K.; Matsunaga, C.; Odaka, A.; Yamato, S.; Takaha, W.; Shimada, I.; Arata, Y. Carbon-13 NMR study of switch variant anti-dansyl antibodies: Antigen binding and domain-domain interactions. Biochemistry 1991, 30, 6604–6610. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.; May, R.; Scharff, M.D.; Casadevall, A. Variable-region-identical antibodies differing in isotype demonstrate differences in fine specificity and idiotype. J. Immunol. 2005, 174, 2132–2142. [Google Scholar] [CrossRef] [PubMed]
- Gilliland, G.L.; Luo, J.; Vafa, O.; Almagro, J.C. Leveraging SBDD in protein therapeutic development: Antibody engineering. Methods Mol. Biol. 2012, 841, 321–349. [Google Scholar] [PubMed]
- Ying, T.; Gong, R.; Ju, T.W.; Prabakaran, P.; Dimitrov, D.S. Engineered fc based antibody domains and fragments as novel scaffolds. Biochim. Biophys. Acta 2014, 1844, 1977–1982. [Google Scholar] [CrossRef] [PubMed]
- Hagihara, Y.; Saerens, D. Engineering disulfide bonds within an antibody. Biochim. Biophys. Acta 2014, 1844, 2016–2023. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.Y.; Gejman, R.S.; Brea, E.J.; Oh, C.Y.; Mathias, M.D.; Pankov, D.; Casey, E.; Dao, T.; Scheinberg, D.A. Opportunities and challenges for TCR mimic antibodies in cancer therapy. Expert Opin. Biol. Ther. 2016, 16, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.K.; Allan, B.W.; Chai, Q.; Atwell, S.; Lu, J. Therapeutic antibody engineering to improve viscosity and phase separation guided by crystal structure. Mol. Pharm. 2016, 13, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Chothia, C.; Lesk, A.M. Canonical structures for the hypervariable regions of immunoglobulins. J. Mol. Biol. 1987, 196, 901–917. [Google Scholar] [CrossRef]
- Chothia, C.; Lesk, A.M.; Tramontano, A.; Levitt, M.; Smith-Gill, S.J.; Air, G.; Sheriff, S.; Padlan, E.A.; Davies, D.; Tulip, W.R.; et al. Conformations of immunoglobulin hypervariable regions. Nature 1989, 342, 877–883. [Google Scholar] [CrossRef] [PubMed]
- North, B.; Lehmann, A.; Dunbrack, R.L., Jr. A new clustering of antibody CDR loop conformations. J. Mol. Biol. 2011, 406, 228–256. [Google Scholar] [CrossRef] [PubMed]
- Al-Lazikani, B.; Lesk, A.M.; Chothia, C. Standard conformations for the canonical structures of immunoglobulins. J. Mol. Biol. 1997, 273, 927–948. [Google Scholar] [CrossRef] [PubMed]
- Chailyan, A.; Marcatili, P.; Cirillo, D.; Tramontano, A. Structural repertoire of immunoglobulin lambda light chains. Proteins 2011, 79, 1513–1524. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, D.; Shirai, H.; Kobori, M.; Nakamura, H. Systematic classification of CDR-L3 in antibodies: Implications of the light chain subtypes and the VL-VH interface. Proteins 2009, 75, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.C.; Thornton, J.M. Structural families in loops of homologous proteins: Automatic classification, modelling and application to antibodies. J. Mol. Biol. 1996, 263, 800–815. [Google Scholar] [CrossRef] [PubMed]
- Sircar, A.; Sanni, K.A.; Shi, J.; Gray, J.J. Analysis and modeling of the variable region of camelid single-domain antibodies. J. Immunol. 2011, 186, 6357–6367. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ekiert, D.C.; Ahmad, I.; Yu, W.; Zhang, Y.; Bazirgan, O.; Torkamani, A.; Raudsepp, T.; Mwangi, W.; Criscitiello, M.F.; et al. Reshaping antibody diversity. Cell 2013, 153, 1379–1393. [Google Scholar] [CrossRef] [PubMed]
- Marcatili, P.; Rosi, A.; Tramontano, A. Pigs: Automatic prediction of antibody structures. Bioinformatics 2008, 24, 1953–1954. [Google Scholar] [CrossRef] [PubMed]
- Whitelegg, N.R.; Rees, A.R. WAM: An improved algorithm for modelling antibodies on the web. Protein Eng. 2000, 13, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Do, R.K.; Sali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Soto, C.S.; Honig, B. Evaluating conformational free energies: The colony energy and its application to the problem of loop prediction. Proc. Natl. Acad. Sci. USA 2002, 99, 7432–7437. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.; Kortemme, T. Improvements to robotics-inspired conformational sampling in rosetta. PLoS ONE 2013, 8, e63090. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Day, T.; Warshaviak, D.; Murrett, C.; Friesner, R.; Pearlman, D. Antibody structure determination using a combination of homology modeling, energy-based refinement, and loop prediction. Proteins 2014, 82, 1646–1655. [Google Scholar] [CrossRef] [PubMed]
- Almagro, J.C.; Teplyakov, A.; Luo, J.; Sweet, R.W.; Kodangattil, S.; Hernandez-Guzman, F.; Gilliland, G.L. Second antibody modeling assessment (AMA-II). Proteins 2014, 82, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Almagro, J.C.; Beavers, M.P.; Hernandez-Guzman, F.; Maier, J.; Shaulsky, J.; Butenhof, K.; Labute, P.; Thorsteinson, N.; Kelly, K.; Teplyakov, A.; et al. Antibody modeling assessment. Proteins 2011, 79, 3050–3066. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.C.; Cheetham, J.C.; Rees, A.R. Modeling antibody hypervariable loops: A combined algorithm. Proc. Natl. Acad. Sci. USA 1989, 86, 9268–9272. [Google Scholar] [CrossRef] [PubMed]
- Sircar, A.; Kim, E.T.; Gray, J.J. Rosettaantibody: Antibody variable region homology modeling server. Nucleic Acids Res. 2009, 37, W474–W479. [Google Scholar] [CrossRef] [PubMed]
- Weitzner, B.D.; Kuroda, D.; Marze, N.; Xu, J.; Gray, J.J. Blind prediction performance of rosettaantibody 3.0: Grafting, relaxation, kinematic loop modeling, and full CDR optimization. Proteins 2014, 82, 1611–1623. [Google Scholar] [CrossRef] [PubMed]
- Kiyoshi, M.; Caaveiro, J.M.; Miura, E.; Nagatoishi, S.; Nakakido, M.; Soga, S.; Shirai, H.; Kawabata, S.; Tsumoto, K. Affinity improvement of a therapeutic antibody by structure-based computational design: Generation of electrostatic interactions in the transition state stabilizes the antibody-antigen complex. PLoS ONE 2014, 9, e87099. [Google Scholar] [CrossRef] [PubMed]
- Lippow, S.M.; Wittrup, K.D.; Tidor, B. Computational design of antibody-affinity improvement beyond in vivo maturation. Nat. Biotechnol. 2007, 25, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Sircar, A.; Gray, J.J. Snugdock: Paratope structural optimization during antibody-antigen docking compensates for errors in antibody homology models. PLoS Comput. Biol. 2010, 6, e1000644. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.J.; Moughon, S.; Wang, C.; Schueler-Furman, O.; Kuhlman, B.; Rohl, C.A.; Baker, D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 2003, 331, 281–299. [Google Scholar] [CrossRef]
- Zhao, J.; Nussinov, R.; Ma, B. Mechanisms of recognition of amyloid-β (Aβ) monomer, oligomer, and fibril by homologous antibodies. J. Biol. Chem. 2017, 292, 18325–18343. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhu, D.; Zhu, J.; Nussinov, R.; Ma, B. Local and global anatomy of antibody-protein antigen recognition. J. Mol. Recognit. 2017, 31, e2693. [Google Scholar] [CrossRef] [PubMed]
- Tharakaraman, K.; Robinson, L.N.; Hatas, A.; Chen, Y.L.; Siyue, L.; Raguram, S.; Sasisekharan, V.; Wogan, G.N.; Sasisekharan, R. Redesign of a cross-reactive antibody to dengue virus with broad-spectrum activity and increased in vivo potency. Proc. Natl. Acad. Sci. USA 2013, 110, E1555–E1564. [Google Scholar] [CrossRef] [PubMed]
- Ventura, S.; Zurdo, J.; Narayanan, S.; Parreno, M.; Mangues, R.; Reif, B.; Chiti, F.; Giannoni, E.; Dobson, C.M.; Aviles, F.X.; et al. Short amino acid stretches can mediate amyloid formation in globular proteins: The Src homology 3 (SH3) case. Proc. Natl. Acad. Sci. USA 2004, 101, 7258–7263. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.S. Effects of protein aggregates: An immunologic perspective. AAPS J. 2006, 8, E501–E507. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Singh, S.K.; Wang, X.; Rup, B.; Gill, D. Coupling of aggregation and immunogenicity in biotherapeutics: T- and b-cell immune epitopes may contain aggregation-prone regions. Pharm. Res. 2011, 28, 949–961. [Google Scholar] [CrossRef] [PubMed]
- Worn, A.; Pluckthun, A. Stability engineering of antibody single-chain Fv fragments. J. Mol. Biol. 2001, 305, 989–1010. [Google Scholar] [CrossRef] [PubMed]
- Bird, R.E.; Hardman, K.D.; Jacobson, J.W.; Johnson, S.; Kaufman, B.M.; Lee, S.M.; Lee, T.; Pope, S.H.; Riordan, G.S.; Whitlow, M. Single-chain antigen-binding proteins. Science 1988, 242, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Glockshuber, R.; Malia, M.; Pfitzinger, I.; Pluckthun, A. A comparison of strategies to stabilize immunoglobulin Fv-fragments. Biochemistry 1990, 29, 1362–1367. [Google Scholar] [CrossRef] [PubMed]
- Worn, A.; Pluckthun, A. Mutual stabilization of VL and VH in single-chain antibody fragments, investigated with mutants engineered for stability. Biochemistry 1998, 37, 13120–13127. [Google Scholar] [CrossRef] [PubMed]
- Worn, A.; Pluckthun, A. Different equilibrium stability behavior of ScFv fragments: Identification, classification, and improvement by protein engineering. Biochemistry 1999, 38, 8739–8750. [Google Scholar] [CrossRef] [PubMed]
- Caflisch, A. Computational models for the prediction of polypeptide aggregation propensity. Curr. Opin. Chem. Biol. 2006, 10, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Conchillo-Sole, O.; de Groot, N.S.; Aviles, F.X.; Vendrell, J.; Daura, X.; Ventura, S. Aggrescan: A server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinform. 2007, 8, 65. [Google Scholar] [CrossRef] [PubMed]
- Trovato, A.; Seno, F.; Tosatto, S.C. The pasta server for protein aggregation prediction. Protein Eng. Des. Sel. 2007, 20, 521–523. [Google Scholar] [CrossRef] [PubMed]
- Garbuzynskiy, S.O.; Lobanov, M.Y.; Galzitskaya, O.V. Foldamyloid: A method of prediction of amyloidogenic regions from protein sequence. Bioinformatics 2010, 26, 326–332. [Google Scholar] [CrossRef] [PubMed]
- DuBay, K.F.; Pawar, A.P.; Chiti, F.; Zurdo, J.; Dobson, C.M.; Vendruscolo, M. Prediction of the absolute aggregation rates of amyloidogenic polypeptide chains. J. Mol. Biol. 2004, 341, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Stefani, M.; Taddei, N.; Ramponi, G.; Dobson, C.M. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature 2003, 424, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Singh, S.K.; Kumar, S. Potential aggregation-prone regions in complementarity-determining regions of antibodies and their contribution towards antigen recognition: A computational analysis. Pharm. Res. 2010, 27, 1512–1529. [Google Scholar] [CrossRef] [PubMed]
- Chennamsetty, N.; Voynov, V.; Kayser, V.; Helk, B.; Trout, B.L. Design of therapeutic proteins with enhanced stability. Proc. Natl. Acad. Sci. USA 2009, 106, 11937–11942. [Google Scholar] [CrossRef] [PubMed]
- Voynov, V.; Chennamsetty, N.; Kayser, V.; Helk, B.; Trout, B.L. Predictive tools for stabilization of therapeutic proteins. MAbs 2009, 1, 580–582. [Google Scholar] [CrossRef] [PubMed]
- Chennamsetty, N.; Helk, B.; Voynov, V.; Kayser, V.; Trout, B.L. Aggregation-prone motifs in human immunoglobulin G. J. Mol. Biol. 2009, 391, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Nussinov, R. Simulations as analytical tools to understand protein aggregation and predict amyloid conformation. Curr. Opin. Chem. Biol. 2006, 10, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Ding, F.; Dokholyan, N.V. Probing protein aggregation using discrete molecular dynamics. Front. Biosci. 2008, 13, 4795–4808. [Google Scholar] [CrossRef] [PubMed]
- Wall, J.; Schell, M.; Murphy, C.; Hrncic, R.; Stevens, F.J.; Solomon, A. Thermodynamic instability of human lambda 6 light chains: Correlation with fibrillogenicity. Biochemistry 1999, 38, 14101–14108. [Google Scholar] [CrossRef] [PubMed]
- Stevens, P.W.; Raffen, R.; Hanson, D.K.; Deng, Y.L.; Berrios-Hammond, M.; Westholm, F.A.; Murphy, C.; Eulitz, M.; Wetzel, R.; Solomon, A.; et al. Recombinant immunoglobulin variable domains generated from synthetic genes provide a system for in vitro characterization of light-chain amyloid proteins. Protein Sci. 1995, 4, 421–432. [Google Scholar] [CrossRef]
- Kim, Y.; Wall, J.S.; Meyer, J.; Murphy, C.; Randolph, T.W.; Manning, M.C.; Solomon, A.; Carpenter, J.F. Thermodynamic modulation of light chain amyloid fibril formation. J. Biol. Chem. 2000, 275, 1570–1574. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Gertz, M.A. Primary systemic amyloidosis: Clinical and laboratory features in 474 cases. Semin. Hematol. 1995, 32, 45–59. [Google Scholar] [PubMed]
- Keskin, O. Binding induced conformational changes of proteins correlate with their intrinsic fluctuations: A case study of antibodies. BMC Struct. Biol. 2007, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Thielges, M.C.; Zimmermann, J.; Yu, W.; Oda, M.; Romesberg, F.E. Exploring the energy landscape of antibody−antigen complexes: Protein dynamics, flexibility, and molecular recognition. Biochemistry 2008, 47, 7237–7247. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Tracka, M.B.; Uddin, S.; Casas-Finet, J.; Jacobs, D.J.; Livesay, D.R. Redistribution of flexibility in stabilizing antibody fragment mutants follows le chatelier’s principle. PLoS ONE 2014, 9, e92870. [Google Scholar]
- Sela-Culang, I.; Kunik, V.; Ofran, Y. The structural basis of antibody-antigen recognition. Immune Syst. Model. Anal. 2015, 8, 302. [Google Scholar] [CrossRef] [PubMed]
- Jay, J.W.; Bray, B.; Qi, Y.; Igbinigie, E.; Wu, H.; Li, J.; Ren, G. IgG antibody 3D structures and dynamics. Antibodies 2018, 7, 18. [Google Scholar] [CrossRef]
- Adachi, M.; Kurihara, Y.; Nojima, H.; Takeda-Shitaka, M.; Kamiya, K.; Umeyama, H. Interaction between the antigen and antibody is controlled by the constant domains: Normal mode dynamics of the hel–hyhel-10 complex. Protein Sci. 2003, 12, 2125–2131. [Google Scholar] [CrossRef] [PubMed]
- Pritsch, O.; Hudry-Clergeon, G.; Buckle, M.; Pétillot, Y.; Bouvet, J.P.; Gagnon, J.; Dighiero, G. Can immunoglobulin C (H) 1 constant region domain modulate antigen binding affinity of antibodies? J. Clin. Investig. 1996, 98, 2235. [Google Scholar] [CrossRef] [PubMed]
- Dam, T.K.; Torres, M.; Brewer, C.F.; Casadevall, A. Isothermal titration calorimetry reveals differential binding thermodynamics of variable region-identical antibodies differing in constant region for a univalent ligand. J. Biol. Chem. 2008, 283, 31366–31370. [Google Scholar] [CrossRef] [PubMed]
- Tudor, D.; Yu, H.; Maupetit, J.; Drillet, A.-S.; Bouceba, T.; Schwartz-Cornil, I.; Lopalco, L.; Tuffery, P.; Bomsel, M. Isotype modulates epitope specificity, affinity, and antiviral activities of anti–HIV-1 human broadly neutralizing 2F5 antibody. Proc. Natl. Acad. Sci. USA 2012, 109, 12680–12685. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Tracka, M.B.; Uddin, S.; Casas-Finet, J.; Jacobs, D.J.; Livesay, D.R. Rigidity emerges during antibody evolution in three distinct antibody systems: Evidence from QSFR analysis of fab fragments. PLoS Comput. Biol. 2015, 11, e1004327. [Google Scholar] [CrossRef] [PubMed]
- Sela-Culang, I.; Alon, S.; Ofran, Y. A systematic comparison of free and bound antibodies reveals binding-related conformational changes. J. Immunol. 2012, 189, 4890–4899. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Nussinov, R.; Ma, B. Allosteric control of antibody-prion recognition through oxidation of a disulfide bond between the CH and CL chains. Protein Eng. Des. Sel. 2017, 30, 67–76. [Google Scholar] [PubMed]
- Lazar, G.A.; Dang, W.; Karki, S.; Vafa, O.; Peng, J.S.; Hyun, L.; Chan, C.; Chung, H.S.; Eivazi, A.; Yoder, S.C.; et al. Engineered antibody Fc variants with enhanced effector function. Proc. Natl. Acad. Sci. USA 2006, 103, 4005–4010. [Google Scholar] [CrossRef] [PubMed]
- Dahiyat, B.I.; Mayo, S.L. Protein design automation. Protein Sci. 1996, 5, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Raha, K.; Wollacott, A.M.; Italia, M.J.; Desjarlais, J.R. Prediction of amino acid sequence from structure. Protein Sci. 2000, 9, 1106–1119. [Google Scholar] [CrossRef] [PubMed]
- Mimura, Y.; Sondermann, P.; Ghirlando, R.; Lund, J.; Young, S.P.; Goodall, M.; Jefferis, R. Role of oligosaccharide residues of IgG 1-Fc in Fc gamma RIIb binding. J. Biol. Chem. 2001, 276, 45539–45547. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Chu, J.; Zou, Z.; Hamacher, N.B.; Rixon, M.W.; Sun, P.D. Structure of FcgammaRI in complex with Fc reveals the importance of glycan recognition for high-affinity IgG binding. Proc. Natl. Acad. Sci. USA 2015, 112, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.; Tanaka, T.; Takahashi, N.; Sarmay, G.; Arata, Y.; Jefferis, R. A protein structural change in aglycosylated IgG3 correlates with loss of huFc gamma R1 and huFc gamma R111 binding and/or activation. Mol. Immunol. 1990, 27, 1145–1153. [Google Scholar] [CrossRef]
- Walker, M.R.; Lund, J.; Thompson, K.M.; Jefferis, R. Aglycosylation of human IgG1 and IgG3 monoclonal antibodies can eliminate recognition by human cells expressing Fc gamma Ri and/or Fc gamma RII receptors. Biochem. J. 1989, 259, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Jefferis, R. The glycosylation of antibody molecules: Functional significance. Glycoconj. J. 1993, 10, 358–361. [Google Scholar] [PubMed]
- Okbazghi, S.Z.; More, A.S.; White, D.R.; Duan, S.; Shah, I.S.; Joshi, S.B.; Middaugh, C.R.; Volkin, D.B.; Tolbert, T.J. Production, characterization, and biological evaluation of well-defined IgG1 Fc glycoforms as a model system for biosimilarity analysis. J. Pharm. Sci. 2016, 105, 559–574. [Google Scholar] [CrossRef] [PubMed]
- Subedi, G.P.; Barb, A.W. The immunoglobulin G1 N-glycan composition affects binding to each low affinity Fc gamma receptor. MAbs 2016, 8, 1512–1524. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Nishimura, M.; Nagano, M.; Yagi, H.; Sasakawa, H.; Uchida, K.; Shitara, K.; Kato, K. Glycoform-dependent conformational alteration of the Fc region of human immunoglobulin G1 as revealed by NMR spectroscopy. Biochim. Biophys. Acta 2006, 1760, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Shields, R.L.; Lai, J.; Keck, R.; O’Connell, L.Y.; Hong, K.; Meng, Y.G.; Weikert, S.H.; Presta, L.G. Lack of fucose on human IgG1 n-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J. Biol. Chem. 2002, 277, 26733–26740. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Im, W. Effects of n-glycan composition on structure and dynamics of IgG1 Fc and their implications for antibody engineering. Sci. Rep. 2017, 7, 12659. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.; Takahashi, N.; Pound, J.D.; Goodall, M.; Jefferis, R. Multiple interactions of IgG with its core oligosaccharide can modulate recognition by complement and human Fc gamma receptor I and influence the synthesis of its oligosaccharide chains. J. Immunol. 1996, 157, 4963–4969. [Google Scholar] [PubMed]
- Yu, X.; Baruah, K.; Harvey, D.J.; Vasiljevic, S.; Alonzi, D.S.; Song, B.D.; Higgins, M.K.; Bowden, T.A.; Scanlan, C.N.; Crispin, M. Engineering hydrophobic protein-carbohydrate interactions to fine-tune monoclonal antibodies. J. Am. Chem. Soc. 2013, 135, 9723–9732. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.A.; Giddens, J.; Pincetic, A.; Lomino, J.V.; Ravetch, J.V.; Wang, L.X.; Bjorkman, P.J. Structural characterization of anti-inflammatory immunoglobulin g fc proteins. J. Mol. Biol. 2014, 426, 3166–3179. [Google Scholar] [CrossRef] [PubMed]
- Deisenhofer, J. Crystallographic refinement and atomic models of a human fc fragment and its complex with fragment B of protein a from staphylococcus aureus at 2.9- and 2.8-a resolution. Biochemistry 1981, 20, 2361–2370. [Google Scholar] [CrossRef] [PubMed]
- Barb, A.W.; Prestegard, J.H. NMR analysis demonstrates immunoglobulin G N-glycans are accessible and dynamic. Nat. Chem. Biol. 2011, 7, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Subedi, G.P.; Barb, A.W. The structural role of antibody N-glycosylation in receptor interactions. Structure 2015, 23, 1573–1583. [Google Scholar] [CrossRef] [PubMed]
- De Groot, A.S.; Moise, L. New tools, new approaches and new ideas for vaccine development. Expert Rev. Vaccines 2007, 6, 125–127. [Google Scholar] [CrossRef] [PubMed]
- DeLisi, C.; Berzofsky, J.A. T-cell antigenic sites tend to be amphipathic structures. Proc. Natl. Acad. Sci. USA 1985, 82, 7048–7052. [Google Scholar] [CrossRef] [PubMed]
- McMurry, J.; Sbai, H.; Gennaro, M.L.; Carter, E.J.; Martin, W.; De Groot, A.S. Analyzing mycobacterium tuberculosis proteomes for candidate vaccine epitopes. Tuberculosis 2005, 85, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Ju, H.; Liu, Z.; Ning, Q.; Zhang, J.; Zhao, X.; Huang, Y.; Ma, Z.; Li, Y. Bioinformatics resources and tools for conformational b-cell epitope prediction. Comput. Math. Methods Med. 2013, 2013, 943636. [Google Scholar] [CrossRef] [PubMed]
- Sok, D.; Laserson, U.; Laserson, J.; Liu, Y.; Vigneault, F.; Julien, J.P.; Briney, B.; Ramos, A.; Saye, K.F.; Le, K.; et al. The effects of somatic hypermutation on neutralization and binding in the PGT121 family of broadly neutralizing HIV antibodies. PLoS Pathog. 2013, 9, e1003754. [Google Scholar] [CrossRef]
- Scharf, L.; Scheid, J.F.; Lee, J.H.; West, A.P., Jr.; Chen, C.; Gao, H.; Gnanapragasam, P.N.; Mares, R.; Seaman, M.S.; Ward, A.B.; et al. Antibody 8ANC195 reveals a site of broad vulnerability on the HIV-1 envelope spike. Cell Rep. 2014, 7, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Pejchal, R.; Doores, K.J.; Walker, L.M.; Khayat, R.; Huang, P.S.; Wang, S.K.; Stanfield, R.L.; Julien, J.P.; Ramos, A.; Crispin, M.; et al. A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science 2011, 334, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.; Sbai, H.; De Groot, A.S. Bioinformatics tools for identifying class I-restricted epitopes. Methods 2003, 29, 289–298. [Google Scholar] [CrossRef]
- Brusic, V.; Petrovsky, N. Immunoinformatics and its relevance to understanding human immune disease. Expert Rev. Clin. Immunol. 2005, 1, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; LaBute, M.; Yusim, K. Immunoinformatics comes of age. PLoS Comput. Biol. 2006, 2, e71. [Google Scholar] [CrossRef] [PubMed]
- Kringelum, J.V.; Lundegaard, C.; Lund, O.; Nielsen, M. Reliable b cell epitope predictions: Impacts of method development and improved benchmarking. PLoS Comput. Biol. 2012, 8, e1002829. [Google Scholar] [CrossRef] [PubMed]
- Sweredoski, M.J.; Baldi, P. Pepito: Improved discontinuous b-cell epitope prediction using multiple distance thresholds and half sphere exposure. Bioinformatics 2008, 24, 1459–1460. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wu, D.; Xu, T.; Wang, X.; Xu, X.; Tao, L.; Li, Y.X.; Cao, Z.W. SEPPA: A computational server for spatial epitope prediction of protein antigens. Nucleic Acids Res. 2009, 37, W612–W616. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.R. Scaffolding to build a rational vaccine design strategy. Proc. Natl. Acad. Sci. USA 2010, 107, 17859–17860. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Zhu, J. Computational tools for epitope vaccine design and evaluation. Curr. Opin. Virol. 2015, 11, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Zhu, J.; Wu, X.; Moquin, S.; Zhang, B.; Acharya, P.; Georgiev, I.S.; Altae-Tran, H.R.; Chuang, G.Y.; Joyce, M.G.; et al. Multidonor analysis reveals structural elements, genetic determinants, and maturation pathway for HIV-1 neutralization by VRC01-class antibodies. Immunity 2013, 39, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Correia, B.E.; Ban, Y.E.; Friend, D.J.; Ellingson, K.; Xu, H.; Boni, E.; Bradley-Hewitt, T.; Bruhn-Johannsen, J.F.; Stamatatos, L.; Strong, R.K.; et al. Computational protein design using flexible backbone remodeling and resurfacing: Case studies in structure-based antigen design. J. Mol. Biol. 2011, 405, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Correia, B.E.; Bates, J.T.; Loomis, R.J.; Baneyx, G.; Carrico, C.; Jardine, J.G.; Rupert, P.; Correnti, C.; Kalyuzhniy, O.; Vittal, V.; et al. Proof of principle for epitope-focused vaccine design. Nature 2014, 507, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.J. Potent antibody therapeutics by design. Nat. Rev. Immunol. 2006, 6, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Reichert, J.M. Monoclonal antibodies as innovative therapeutics. Curr. Pharm. Biotechnol. 2008, 9, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.L.; Reichert, J.M. Development trends for therapeutic antibody fragments. Nat. Biotechnol. 2009, 27, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Fischman, S.; Ofran, Y. Computational design of antibodies. Curr. Opin. Struct. Biol. 2018, 51, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Lensink, M.F.; Velankar, S.; Wodak, S.J. Modeling protein-protein and protein-peptide complexes: Capri 6th edition. Proteins 2017, 85, 359–377. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.J.; Gindin, T.; Zhang, B.; Wang, L.; Abel, R.; Murret, C.S.; Xu, F.; Bao, A.; Lu, N.J.; Zhou, T.; et al. Free energy perturbation calculation of relative binding free energy between broadly neutralizing antibodies and the GP120 glycoprotein of HIV-1. J. Mol. Biol. 2017, 429, 930–947. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.M.; Murphy, K.P. Prediction of binding energetics from structure using empirical parameterization. Methods Enzymol. 1998, 295, 294–315. [Google Scholar] [PubMed]
- Audie, J.; Scarlata, S. A novel empirical free energy function that explains and predicts protein-protein binding affinities. Biophys. Chem. 2007, 129, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.J.; Vajda, S. Protein docking along smooth association pathways. Proc. Natl. Acad. Sci. USA 2001, 98, 10636–10641. [Google Scholar] [CrossRef] [PubMed]
- Dell’Orco, D.; De Benedetti, P.G.; Fanelli, F. In silico screening of mutational effects on enzyme-proteic inhibitor affinity: A docking-based approach. BMC Struct. Biol. 2007, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, S.; Zhu, Q.; Zhou, Y. A knowledge-based energy function for protein-ligand, protein-protein, and protein-DNA complexes. J. Med. Chem. 2005, 48, 2325–2335. [Google Scholar] [CrossRef] [PubMed]
- Sirin, S.; Apgar, J.R.; Bennett, E.M.; Keating, A.E. Ab-bind: Antibody binding mutational database for computational affinity predictions. Protein Sci. 2016, 25, 393–409. [Google Scholar] [CrossRef] [PubMed]
- Piekarska, B.; Drozd, A.; Konieczny, L.; Krol, M.; Jurkowski, W.; Roterman, I.; Spolnik, P.; Stopa, B.; Rybarska, J. The indirect generation of long-distance structural changes in antibodies upon their binding to antigen. Chem. Biol. Drug Des. 2006, 68, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Bowen, A.; Casadevall, A. Revisiting the immunoglobulin intramolecular signaling hypothesis. Trends Immunol. 2016, 37, 721–723. [Google Scholar] [CrossRef] [PubMed]
- Oda, M.; Kozono, H.; Morii, H.; Azuma, T. Evidence of allosteric conformational changes in the antibody constant region upon antigen binding. Int. Immunol. 2003, 15, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Lane, T.J.; Bowman, G.R.; Beauchamp, K.; Voelz, V.A.; Pande, V.S. Markov state model reveals folding and functional dynamics in ultra-long md trajectories. J. Am. Chem. Soc. 2011, 133, 18413–18419. [Google Scholar] [CrossRef] [PubMed]
- Husic, B.E.; Pande, V.S. Markov state models: From an art to a science. J. Am. Chem. Soc. 2018, 140, 2386–2396. [Google Scholar] [CrossRef]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, J.; Nussinov, R.; Wu, W.-J.; Ma, B. In Silico Methods in Antibody Design. Antibodies 2018, 7, 22. https://doi.org/10.3390/antib7030022
Zhao J, Nussinov R, Wu W-J, Ma B. In Silico Methods in Antibody Design. Antibodies. 2018; 7(3):22. https://doi.org/10.3390/antib7030022
Chicago/Turabian StyleZhao, Jun, Ruth Nussinov, Wen-Jin Wu, and Buyong Ma. 2018. "In Silico Methods in Antibody Design" Antibodies 7, no. 3: 22. https://doi.org/10.3390/antib7030022
APA StyleZhao, J., Nussinov, R., Wu, W.-J., & Ma, B. (2018). In Silico Methods in Antibody Design. Antibodies, 7(3), 22. https://doi.org/10.3390/antib7030022