The Endothelium as a Driver of Liver Fibrosis and Regeneration


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
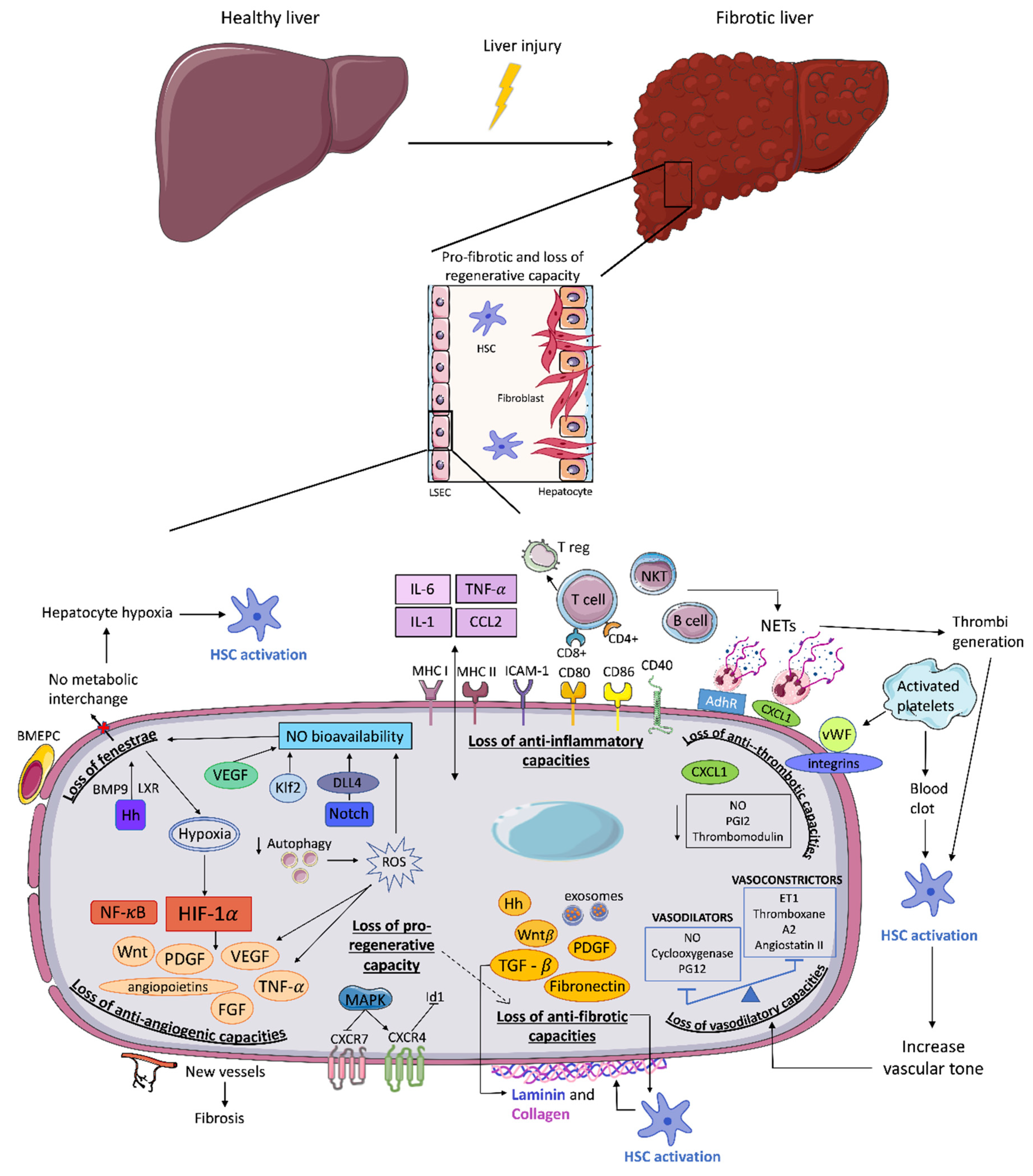
2. Triggers for LSEC Dysfunction
3. Endothelial Dysfunction and Fibrosis Progression
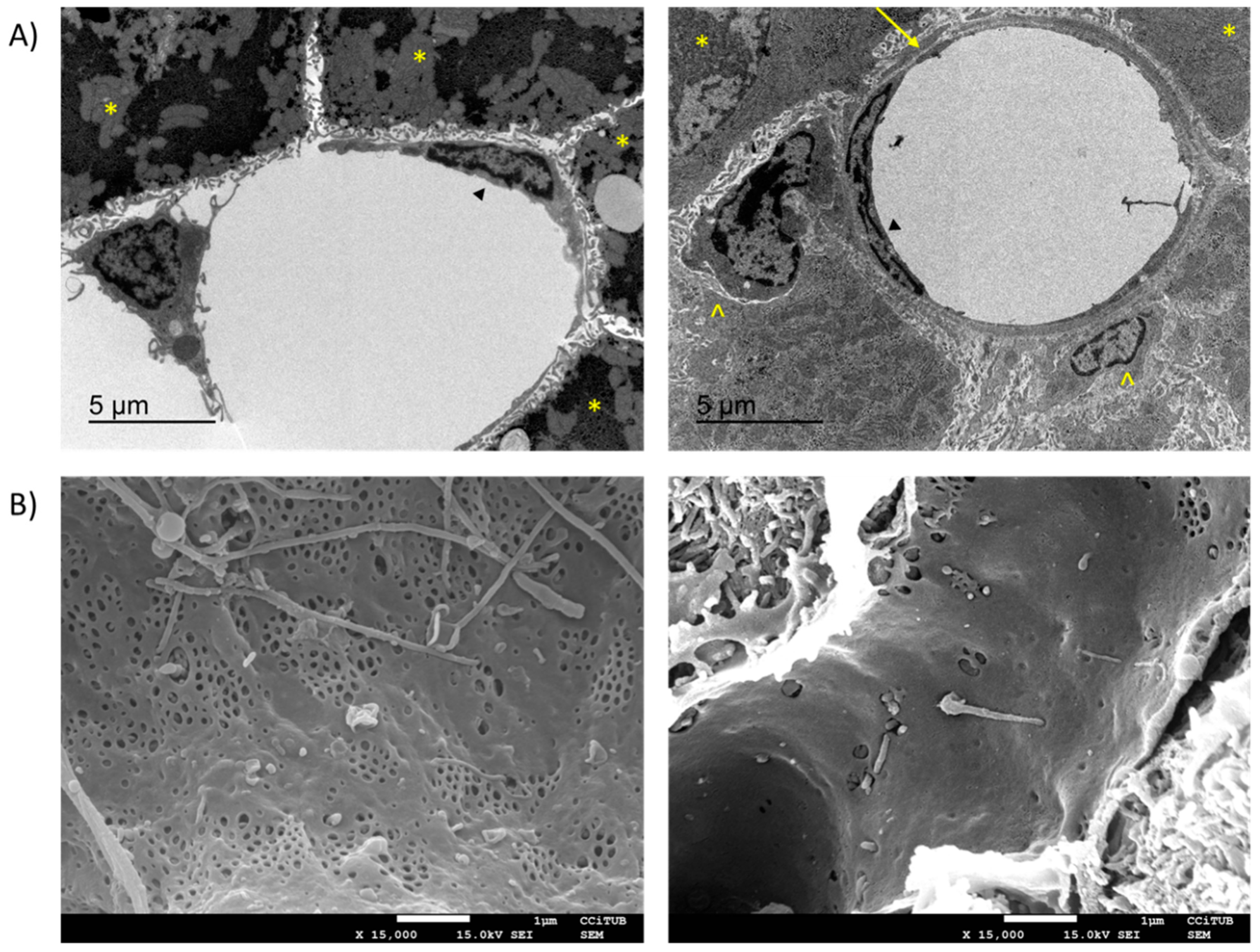
3.1. Loss of LSEC Fenestrae
3.2. Loss of Vasodilatory Capacities
3.3. Loss of Anti-Inflammatory Capacities
3.4. Loss of Anti-Thrombotic Capacities
3.5. Loss of Anti-Angiogenic Capacities
3.6. Loss of Anti-Fibrotic Capacities
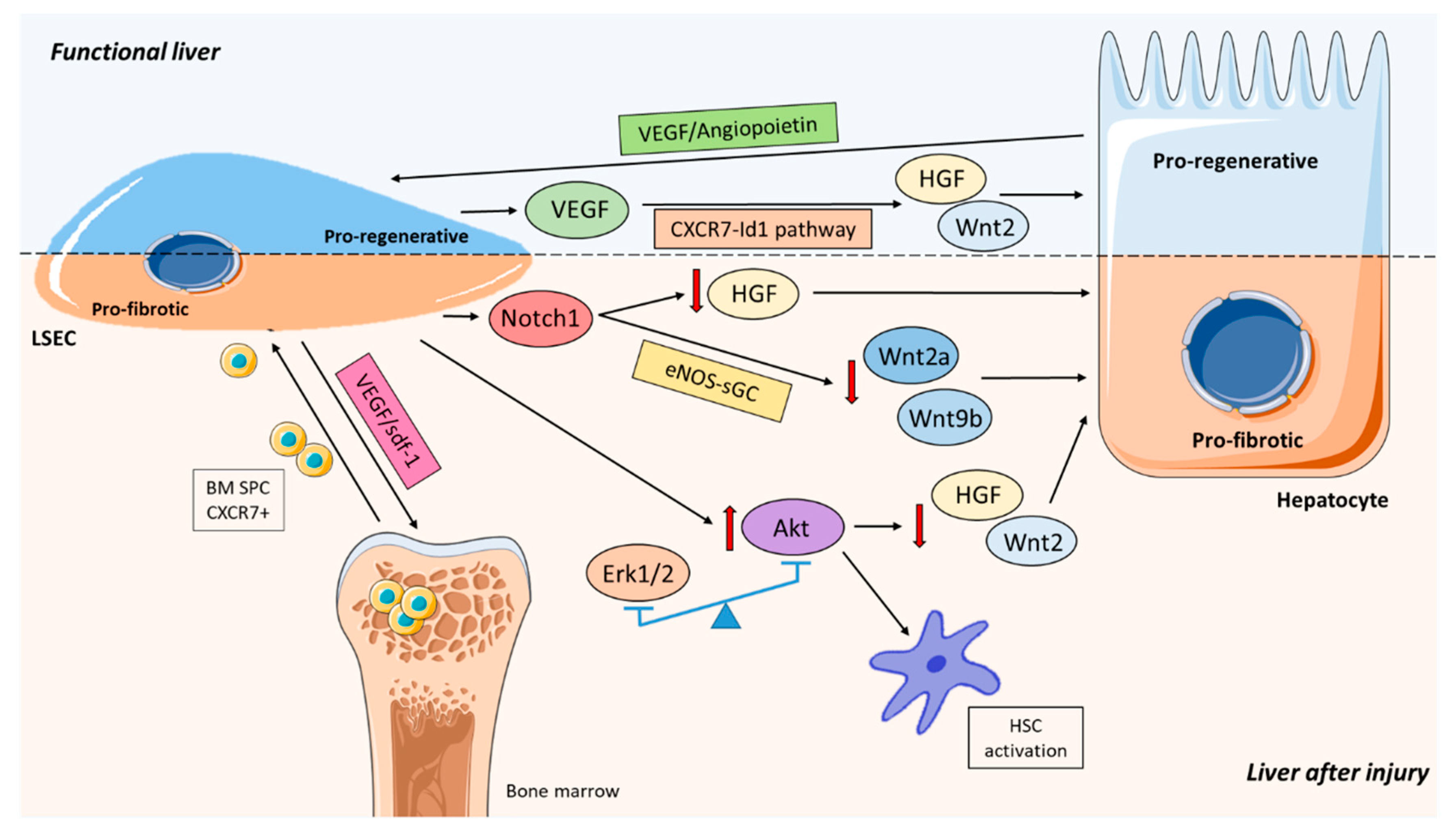
3.7. Loss of Pro-Regenerative Capacity
4. Effect of Mechanical Forces on LSEC Phenotype
4.1. ECM Stiffness
4.2. Shear Stress
5. Role of LSEC Balancing Liver Regeneration and Fibrosis
6. Fibrosis Resolution
7. LSEC Targeting: Potential for Therapy
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APCs | Antigen presenting cells |
| BMEPC | Bone marrow endothelial progenitor cells |
| BM SPC | Bone marrow sinusoidal progenitor cells |
| CD44 | Cluster determinant 44 |
| DAMPs | Danger-associated molecular patterns |
| DLL4 | Delta-like ligand 4 |
| ED | Endothelial dysfunction |
| ET-1 | Endothelin-1 |
| ECM | Extracellular matrix |
| FFAs | Free fatty acids |
| Hh | Hedgehog |
| HSC | Hepatic stellate cells |
| HCV | Hepatitis C virus |
| HGF | Hepatocyte growth factor |
| RHAMM | Hyaluronate-mediated motility |
| HA | Hyaluronic acid |
| HARE | Hyaluronic acid receptor for endocytosis |
| HIF | Hypoxia-inducible transcription factors |
| ICAM-1 | Intercellular adhesion molecule-1 |
| Klf2 | Krüppel-like transcription factor 2 |
| KC | Kupffer cells |
| LSEC | Liver sinusoidal endothelial cells |
| LXR | Liver X receptor |
| MMPs | Matrix metalloproteases |
| NETs | Neutrophil extracellular traps |
| NO | Nitric oxide |
| NAFLD | Non-alcoholic fatty liver disease |
| NS5A | Non-structural protein 5A |
| PAMPs | Pathogen-associated molecular patterns |
| PGI2 | Prostaglandin I2 |
| PARs | Protease-activated receptors |
| QDs | Quantum dots |
| ROS | Reactive oxygen species |
| Tregs | Regulatory T cells |
| SEM | Scanning electron microscopy |
| SK1 | Sphingosine kinase 1 |
| TIMPs | Tissue inhibitors of matrix metalloproteases |
| TEM | Transmission electron microscopy |
| VAP-1 | Vascular adhesion protein-1 |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| VEGF | Vascular endothelial growth factor |
| VEGFR-2 | Vascular endothelial growth factor receptor-2 |
References
- Wisse, E.; de Zanger, R.B.; Charels, K.; van der Smissen, P.; McCuskey, R.S. The Liver Sieve: Considerations Concerning the Structure and Function of Endothelial Fenestrae, the Sinusoidal Wall and the Space of Disse. Hepatology 1985, 5, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.-E. Liver Sinusoidal Endothelial Cells: Physiology and Role in Liver Diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Couvelard, A.; Scoazec, J.Y.; Dauge, M.C.; Bringuier, A.F.; Potet, F.; Feldmann, G. Structural and Functional Differentiation of Sinusoidal Endothelial Cells during Liver Organogenesis in Humans. Blood 1996, 87, 4568–4580. [Google Scholar] [CrossRef] [PubMed]
- McCuskey, R.S. Morphological Mechanisms for Regulating Blood Flow through Hepatic Sinusoids. Liver Int. 2000, 20, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Tanoi, T.; Tamura, T.; Sano, N.; Nakayama, K.; Fukunaga, K.; Zheng, Y.-W.; Akhter, A.; Sakurai, Y.; Hayashi, Y.; Harashima, H.; et al. Protecting Liver Sinusoidal Endothelial Cells Suppresses Apoptosis in Acute Liver Damage. Hepatol. Res. 2016, 46, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Golse, N.; Bucur, P.O.; Adam, R.; Castaing, D.; Sa Cunha, A.; Vibert, E. New Paradigms in Post-Hepatectomy Liver Failure. J. Gastrointest. Surg. 2013, 17, 593–605. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver Fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver Fibrosis and Repair: Immune Regulation of Wound Healing in a Solid Organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef]
- Povero, D.; Busletta, C.; Novo, E.; Di Bonzo, L.V.; Cannito, S.; Paternostro, C.; Parola, M. Liver Fibrosis: A Dynamic and Potentially Reversible Process. Histol. Histopathol. 2010. [Google Scholar] [CrossRef]
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of Liver Fibrosis. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 425–456. [Google Scholar] [CrossRef]
- DeLeve, L.D.; Wang, X.; Guo, Y. Sinusoidal Endothelial Cells Prevent Rat Stellate Cell Activation and Promote Reversion to Quiescence. Hepatology 2008, 48, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Wang, X.; Wang, L.L.; Wang, L.L.; Atkinson, R.D.; Kanel, G.C.; Gaarde, W.A.; Deleve, L.D. Role of Differentiation of Liver Sinusoidal Endothelial Cells in Progression and Regression of Hepatic Fibrosis in Rats. Gastroenterology 2012, 142, 918–927.e6. [Google Scholar] [CrossRef] [PubMed]
- Smedsrød, B.; Pertoft, H.; Gustafson, S.; Laurent, T.C. Scavenger Functions of the Liver Endothelial Cell. Biochem. J. 1990, 266, 313. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.-S.; Cao, Z.; Lis, R.; Nolan, D.J.; Guo, P.; Simons, M.; Penfold, M.E.; Shido, K.; Rabbany, S.Y.; Rafii, S. Divergent Angiocrine Signals from Vascular Niche Balance Liver Regeneration and Fibrosis. Nature 2014, 505, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Arteel, G.E. Oxidants and Antioxidants in Alcohol-Induced Liver Disease. Gastroenterology 2003, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D.; Horton, J.D. Molecular Mediators of Hepatic Steatosis and Liver Injury. J. Clin. Investig. 2004, 147–152. [Google Scholar] [CrossRef]
- Okuda, M.; Li, K.; Beard, M.R.; Showalter, L.A.; Scholle, F.; Lemon, S.M.; Weinman, S.A. Mitochondrial Injury, Oxidative Stress, and Antioxidant Gene Expression Are Induced by Hepatitis C Virus Core Protein. Gastroenterology 2002, 122, 366–375. [Google Scholar] [CrossRef]
- Gong, G.; Waris, G.; Tanveer, R.; Siddiqui, A. Human Hepatitis C Virus NS5A Protein Alters Intracellular Calcium Levels, Induces Oxidative Stress, and Activates STAT-3 and NF-ΚB. Proc. Natl. Acad. Sci. USA 2001, 98, 9599–9604. [Google Scholar] [CrossRef]
- Guillaume, M.; Rodriguez-Vilarrupla, A.; Gracia-Sancho, J.; Rosado, E.; Mancini, A.; Bosch, J.; Garcia-Pagán, J.C. Recombinant Human Manganese Superoxide Dismutase Reduces Liver Fibrosis and Portal Pressure in CCl4-Cirrhotic Rats. J. Hepatol. 2013, 58, 240–246. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Laviña, B.; Rodríguez-Vilarrupla, A.; García-Calderó, H.; Fernández, M.; Bosch, J.; García-Pagán, J.-C. Increased Oxidative Stress in Cirrhotic Rat Livers: A Potential Mechanism Contributing to Reduced Nitric Oxide Bioavailability. Hepatology 2008, 47, 1248–1256. [Google Scholar] [CrossRef]
- Di Pascoli, M.; Diví, M.; Rodríguez-Vilarrupla, A.; Rosado, E.; Gracia-Sancho, J.; Vilaseca, M.; Bosch, J.; García-Pagán, J.C. Resveratrol Improves Intrahepatic Endothelial Dysfunction and Reduces Hepatic Fibrosis and Portal Pressure in Cirrhotic Rats. J. Hepatol. 2013, 58, 904–910. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014. [Google Scholar] [CrossRef]
- Del Campo, J.A.; Gallego, P.; Grande, L. Role of Inflammatory Response in Liver Diseases: Therapeutic Strategies. World J. Hepatol. 2018. [Google Scholar] [CrossRef]
- Klatt, E.C.; Kumar, V. Tissue Renewal and Repair: Regeneration, Healing, and Fibrosis. In Robbins and Cotran Review of Pathology; Elsevier: Philadelphia, PA, USA, 2010; pp. 22–26. [Google Scholar] [CrossRef]
- Bingol, B.; Sheng, M. Mechanisms of Mitophagy: PINK1, Parkin, USP30 and Beyond. Free Radic. Biol. Med. 2016, 100, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Wang, H.; Li, S.; Liu, Q.; Sha, H. The Anti-Inflammatory and Anti-Oxidant Mechanisms of the Keap1/Nrf2/ARE Signaling Pathway in Chronic Diseases. Aging Dis. 2019, 10, 637. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Vilarrupla, A.; Bosch, J.; García-Pagán, J.-C. Potential Role of Antioxidants in the Treatment of Portal Hypertension. J. Hepatol. 2007, 46, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Novo, E.; Busletta, C.; Di Bonzo, L.V.; Povero, D.; Paternostro, C.; Mareschi, K.; Ferrero, I.; David, E.; Bertolani, C.; Caligiuri, A.; et al. Intracellular Reactive Oxygen Species Are Required for Directional Migration of Resident and Bone Marrow-Derived Hepatic pro-Fibrogenic Cells. J. Hepatol. 2011, 54, 964–974. [Google Scholar] [CrossRef] [PubMed]
- Cogger, V.C.; Muller, M.; Fraser, R.; McLean, A.J.; Khan, J.; Le Couteur, D.G. The Effects of Oxidative Stress on the Liver Sieve. J. Hepatol. 2004, 41, 370–376. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Maeso-Díaz, R.; Fernández-Iglesias, A.; Navarro-Zornoza, M.; Bosch, J. New Cellular and Molecular Targets for the Treatment of Portal Hypertension. Hepatol. Int. 2015, 9, 183–191. [Google Scholar] [CrossRef]
- Marrone, G.; Shah, V.H.; Gracia-Sancho, J. Sinusoidal Communication in Liver Fibrosis and Regeneration. J. Hepatol. 2016, 65, 608–617. [Google Scholar] [CrossRef]
- Sørensen, K.K.; Simon-Santamaria, J.; McCuskey, R.S.; Smedsrød, B. Liver Sinusoidal Endothelial Cells. In Comprehensive Physiology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; Volume 5, pp. 1751–1774. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of Hepatic Stellate Cell Activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Hamer, I.; Wattiaux, R.; Coninck, S.W.-D. Deleterious Effects of Xanthine Oxidase on Rat Liver Endothelial Cells after Ischemia/Reperfusion. Biochim. Biophys. Acta - Mol. Cell Res. 1995, 1269, 145–152. [Google Scholar] [CrossRef][Green Version]
- Deleve, L. Glutathione Defense in Non-Parenchymal Cells. Semin. Liver Dis. 1998, 18, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Mandili, G.; Alchera, E.; Merlin, S.; Imarisio, C.; Chandrashekar, B.R.; Riganti, C.; Bianchi, A.; Novelli, F.; Follenzi, A.; Carini, R. Mouse Hepatocytes and LSEC Proteome Reveal Novel Mechanisms of Ischemia/Reperfusion Damage and Protection by A2aR Stimulation. J. Hepatol. 2015, 62, 573–580. [Google Scholar] [CrossRef]
- Ruart, M.; Chavarria, L.; Campreciós, G.; Suárez-Herrera, N.; Montironi, C.; Guixé-Muntet, S.; Bosch, J.; Friedman, S.L.; Garcia-Pagán, J.C.; Hernández-Gea, V. Impaired Endothelial Autophagy Promotes Liver Fibrosis by Aggravating the Oxidative Stress Response during Acute Liver Injury. J. Hepatol. 2019, 70, 458–469. [Google Scholar] [CrossRef]
- Temkin, V.; Karin, M. From Death Receptor to Reactive Oxygen Species and C-Jun N-Terminal Protein Kinase: The Receptor-Interacting Protein 1 Odyssey. Immunol. Rev. 2007, 8–21. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat. Med. 2018. [Google Scholar] [CrossRef]
- Tanaka, M.; Miyajima, A. Liver Regeneration and Fibrosis after Inflammation. Inflamm. Regen. 2016, 36. [Google Scholar] [CrossRef]
- Wynn, T.A. Common and Unique Mechanisms Regulate Fibrosis in Various Fibroproliferative Diseases. J. Clin. Investig. 2007, 524–529. [Google Scholar] [CrossRef]
- Heymann, F.; Tacke, F. Immunology in the Liver-from Homeostasis to Disease. Nat. Rev. Gastroenterol. Hepatol. 2016. [Google Scholar] [CrossRef]
- Crispe, I.N. The Liver as a Lymphoid Organ. Annu. Rev. Immunol. 2009, 27, 147–163. [Google Scholar] [CrossRef] [PubMed]
- DeLeve, L.; Maretti-Mira, A. Liver Sinusoidal Endothelial Cell: An Update. Semin. Liver Dis. 2017, 37, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Boaru, S.G.; Borkham-Kamphorst, E.; Tihaa, L.; Haas, U.; Weiskirchen, R. Expression Analysis of Inflammasomes in Experimental Models of Inflammatory and Fibrotic Liver Disease. J. Inflamm. (United Kingd.) 2012. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.K.; Roth, J.; Oppenheim, J.J.; Tracey, K.J.; Vogl, T.; Feldmann, M.; Horwood, N.; Nanchahal, J. Alarmins: Awaiting a Clinical Response. J. Clin. Investig. 2012. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Nuñez, G. Sterile Inflammation: Sensing and Reacting to Damage. Nat. Rev. Immunol. 2010. [Google Scholar] [CrossRef]
- Sprague, A.H.; Khalil, R.A. Inflammatory Cytokines in Vascular Dysfunction and Vascular Disease. Biochem. Pharmacol. 2009, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Hickey, P.L.; Angus, P.W.; Mclean, A.J.; Morgan, D.J. Oxygen Supplementation Restores Theophylline Clearance to Normal in Cirrhotic Rats. Gastroenterology 1995, 108, 1504–1509. [Google Scholar] [CrossRef]
- Morgan, D.J.; McLean, A.J. Therapeutic Implications of Impaired Hepatic Oxygen Diffusion in Chronic Liver Disease. Hepatology 1991, 14, 1280–1282. [Google Scholar] [CrossRef]
- Vanheule, E.; Geerts, A.M.; Van Huysse, J.; Schelfhout, D.; Praet, M.; Van Vlierberghe, H.; De Vos, M.; Colle, I. An Intravital Microscopic Study of the Hepatic Microcirculation in Cirrhotic Mice Models: Relationship between Fibrosis and Angiogenesis. Int. J. Exp. Pathol. 2008, 89, 419–432. [Google Scholar] [CrossRef]
- Hickey, P.L.; McLean, A.J.; Angus, P.W.; Choo, E.F.; Morgan, D.J. Increased Sensitivity of Propranolol Clearance to Reduced Oxygen Delivery in the Isolated Perfused Cirrhotic Rat Liver. Gastroenterology 1996, 111, 1039–1048. [Google Scholar] [CrossRef]
- Braet, F.; Wisse, E. Structural and Functional Aspects of Liver Sinusoidal Endothelial Cell Fenestrae: A Review. Comp. Hepatol. 2002, 1, 1. [Google Scholar] [CrossRef] [PubMed]
- Babbs, C.; Haboubi, N.Y.; Mellor, J.M.; Smith, A.; Rowan, B.P.; Warnes, T.W. Endothelial Cell Transformation in Primary Biliary Cirrhosis: A Morphological and Biochemical Study. Hepatology 1990, 11, 723–729. [Google Scholar] [CrossRef] [PubMed]
- DeLeve, L.D.; Wang, X.; Kanel, G.C.; Atkinson, R.D.; McCuskey, R.S. Prevention of Hepatic Fibrosis in a Murine Model of Metabolic Syndrome with Nonalcoholic Steatohepatitis. Am. J. Pathol. 2008, 173, 993–1001. [Google Scholar] [CrossRef]
- Horn, T.; Christoffersen, P.; Henriksen, J.H. Alcoholic Liver Injury: Defenestration in Noncirrhotic Livers—A Scanning Electron Microscopic Study. Hepatology 1987, 7, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Fraser, R.; Rogers, G.W.T.; Bowler, L.M.; Day, W.A.; Dobbs, B. Defenestration and Vitamin A Status in a Rat Model of Cirrhosis. Cells Hepatic Sinusoid 1991, 3, 195–198. [Google Scholar]
- Francque, S.; Laleman, W.; Verbeke, L.; Van Steenkiste, C.; Casteleyn, C.; Kwanten, W.; Van Dyck, C.; D’Hondt, M.; Ramon, A.; Vermeulen, W.; et al. Increased Intrahepatic Resistance in Severe Steatosis: Endothelial Dysfunction, Vasoconstrictor Overproduction and Altered Microvascular Architecture. Lab. Investig. 2012. [Google Scholar] [CrossRef] [PubMed]
- Pasarín, M.; La Mura, V.; Gracia-Sancho, J.; García-Calderó, H.; Rodríguez-Vilarrupla, A.; García-Pagán, J.C.; Bosch, J.; Abraldes, J.G. Sinusoidal Endothelial Dysfunction Precedes Inflammation and Fibrosis in a Model of NAFLD. PLoS ONE 2012, 7, e32785. [Google Scholar] [CrossRef]
- Miyao, M.; Kotani, H.; Ishida, T.; Kawai, C.; Manabe, S.; Abiru, H.; Tamaki, K. Pivotal Role of Liver Sinusoidal Endothelial Cells in NAFLD/NASH Progression. Lab. Investig. 2015. [Google Scholar] [CrossRef]
- García-Pagán, J.-C.; Gracia-Sancho, J.; Bosch, J. Functional Aspects on the Pathophysiology of Portal Hypertension in Cirrhosis. J. Hepatol. 2012, 57, 458–461. [Google Scholar] [CrossRef]
- Tateya, S.; Rizzo, N.O.; Handa, P.; Cheng, A.M.; Morgan-Stevenson, V.; Daum, G.; Clowes, A.W.; Morton, G.J.; Schwartz, M.W.; Kim, F. Endothelial NO/CGMP/VASP Signaling Attenuates Kupffer Cell Activation and Hepatic Insulin Resistance Induced by High-Fat Feeding. Diabetes 2011, 60, 2792–2801. [Google Scholar] [CrossRef]
- Braet, F.; de Zanger, R.; Baekeland, M.; Crabbé, E.; van der Smissen, P.; Wisse, E. Structure and Dynamics of the Fenestrae-Associated Cytoskeleton of Rat Liver Sinusoidal Endothelial Cells. Hepatology 1995. [Google Scholar] [CrossRef]
- Braet, F.; Spector, I.; De Zanger, R.; Wisse, E. A Novel Structure Involved in the Formation of Liver Endothelial Cell Fenestrae Revealed by Using the Actin Inhibitor Misakinolide. Proc. Natl. Acad. Sci. USA 1998. [Google Scholar] [CrossRef] [PubMed]
- Fraser, R.; Dobbs, B.R.; Rogers, G.W.T. Lipoproteins and the Liver Sieve: The Role of the Fenestrated Sinusoidal Endothelium in Lipoprotein Metabolism, Atherosclerosis, and Cirrhosis. Hepatology 1995. [Google Scholar] [CrossRef]
- McGuire, R.F.; Bissell, D.M.; Boyles, J.; Roll, F.J. Role of Extracellular Matrix in Regulating Fenestrations of Sinusoidal Endothelial Cells Isolated from Normal Rat Liver. Hepatology 1992. [Google Scholar] [CrossRef] [PubMed]
- Yamane, A.; Seetharam, L.; Yamaguchi, S.; Gotoh, N.; Takahashi, T.; Neufeld, G.; Shibuya, M. A New Communication System between Hepatocytes and Sinusoidal Endothelial Cells in Liver through Vascular Endothelial Growth Factor and Flt Tyrosine Kinase Receptor Family (Flt-1 and KDR/Flk-1). Oncogene 1994, 9, 2683–2690. [Google Scholar] [PubMed]
- DeLeve, L.D.; Wang, X.; Hu, L.; McCuskey, M.K.; McCuskey, R.S. Rat Liver Sinusoidal Endothelial Cell Phenotype Is Maintained by Paracrine and Autocrine Regulation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G757–G763. [Google Scholar] [CrossRef] [PubMed]
- Marrone, G.; Maeso-Díaz, R.; García-Cardena, G.; Abraldes, J.G.; García-Pagán, J.C.; Bosch, J.; Gracia-Sancho, J. KLF2 Exerts Antifibrotic and Vasoprotective Effects in Cirrhotic Rat Livers: Behind the Molecular Mechanisms of Statins. Gut 2015, 64, 1434–1443. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; García-Calderó, H.; Hide, D.; Marrone, G.; Guixé-Muntet, S.; Peralta, C.; García-Pagán, J.C.; Abraldes, J.G.; Bosch, J. Simvastatin Maintains Function and Viability of Steatotic Rat Livers Procured for Transplantation. J. Hepatol. 2013, 58, 1140–1146. [Google Scholar] [CrossRef]
- Guixé-Muntet, S.; de Mesquita, F.C.; Vila, S.; Hernández-Gea, V.; Peralta, C.; García-Pagán, J.C.; Bosch, J.; Gracia-Sancho, J. Cross-Talk between Autophagy and KLF2 Determines Endothelial Cell Phenotype and Microvascular Function in Acute Liver Injury. J. Hepatol. 2017, 66, 86–94. [Google Scholar] [CrossRef]
- Duan, J.-L.L.; Ruan, B.; Yan, X.-C.C.; Liang, L.; Song, P.; Yang, Z.-Y.Y.; Liu, Y.; Dou, K.-F.F.; Han, H.; Wang, L. Endothelial Notch Activation Reshapes the Angiocrine of Sinusoidal Endothelia to Aggravate Liver Fibrosis and Blunt Regeneration in Mice. Hepatology 2018. [Google Scholar] [CrossRef]
- Chen, L.; Gu, T.; Li, B.; Li, F.; Ma, Z.; Zhang, Q.; Cai, X.; Lu, L. Delta-like Ligand 4/DLL4 Regulates the Capillarization of Liver Sinusoidal Endothelial Cell and Liver Fibrogenesis. Biochim. Biophys. Acta Mol. Cell Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Matz-Soja, M.; Gebhardt, R. The Many Faces of Hedgehog Signalling in the Liver: Recent Progress Reveals Striking Cellular Diversity and the Importance of Microenvironments. J. Hepatol. 2014, 61, 1449–1450. [Google Scholar] [CrossRef] [PubMed]
- Witek, R.P.; Yang, L.; Liu, R.; Jung, Y.; Omenetti, A.; Syn, W.; Choi, S.S.; Cheong, Y.; Fearing, C.M.; Agboola, K.M.; et al. Liver Cell–Derived Microparticles Activate Hedgehog Signaling and Alter Gene Expression in Hepatic Endothelial Cells. Gastroenterology 2009, 136, 320–330.e2. [Google Scholar] [CrossRef]
- Xie, G.; Choi, S.S.; Syn, W.-K.; Michelotti, G.A.; Swiderska, M.; Karaca, G.; Chan, I.S.; Chen, Y.; Diehl, A.M. Hedgehog Signalling Regulates Liver Sinusoidal Endothelial Cell Capillarisation. Gut 2013, 62, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Zhao, T.; Gao, X.; Wu, Y. Liver X Receptor α Is Essential for the Capillarization of Liver Sinusoidal Endothelial Cells in Liver Injury. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Desroches-Castan, A.; Tillet, E.; Ricard, N.; Ouarné, M.; Mallet, C.; Belmudes, L.; Couté, Y.; Boillot, O.; Scoazec, J.Y.; Bailly, S.; et al. Bone Morphogenetic Protein 9 Is a Paracrine Factor Controlling Liver Sinusoidal Endothelial Cell Fenestration and Protecting Against Hepatic Fibrosis. Hepatology 2019. [Google Scholar] [CrossRef]
- Desroches-Castan, A.; Tillet, E.; Ricard, N.; Ouarné, M.; Mallet, C.; Feige, J.-J.; Bailly, S. Differential Consequences of Bmp9 Deletion on Sinusoidal Endothelial Cell Differentiation and Liver Fibrosis in 129/Ola and C57BL/6 Mice. Cells 2019, 8, 1079. [Google Scholar] [CrossRef]
- Yokomori, H.; Oda, M.; Ogi, M.; Kamegaya, Y.; Tsukada, N.; Ishii, H. Endothelial Nitric Oxide Synthase and Caveolin-1 Are Co-Localized in Sinusoidal Endothelial Fenestrae. Liver 2001. [Google Scholar] [CrossRef]
- Warren, A.; Cogger, V.C.; Arias, I.M.; McCuskey, R.S.; Le Couteur, D.G. Liver Sinusoidal Endothelial Fenestrations in Caveolin-1 Knockout Mice. Microcirculation 2010, 17, 32–38. [Google Scholar] [CrossRef]
- Venkatraman, L.; Tucker-Kellogg, L. The CD47-Binding Peptide of Thrombospondin-1 Induces Defenestration of Liver Sinusoidal Endothelial Cells. Liver Int. 2013, 33, 1386–1397. [Google Scholar] [CrossRef]
- Maretti-Mira, A.C.; Wang, X.; Wang, L.; DeLeve, L.D. Incomplete Differentiation of Engrafted Bone Marrow Endothelial Progenitor Cells Initiates Hepatic Fibrosis in the Rat. Hepatology 2019. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.; Haddad, F.G.; Garcia-Cardena, G.; Frangos, J.A.; Mennone, A.; Groszmann, R.J.; Sessa, W.C. Liver Sinusoidal Endothelial Cells Are Responsible for Nitric Oxide Modulation of Resistance in the Hepatic Sinusoids. J. Clin. Investig. 1997, 100, 2923–2930. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Chung, J.J. Reduced Nitric Oxide Production by Endothelial Cells in Cirrhotic Rat Liver: Endothelial Dysfunction in Portal Hypertension. Gastroenterology 1998, 114, 344–351. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Laviña, B.; Rodríguez-Vilarrupla, A.; García-Calderó, H.; Bosch, J.; García-Pagán, J.C. Enhanced Vasoconstrictor Prostanoid Production by Sinusoidal Endothelial Cells Increases Portal Perfusion Pressure in Cirrhotic Rat Livers. J. Hepatol. 2007, 47, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, K.K.; McCourt, P.; Berg, T.; Crossley, C.; Le Couteur, D.; Wake, K.; Smedsrød, B. The Scavenger Endothelial Cell: A New Player in Homeostasis and Immunity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R1217–R1230. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, M.; Kurihara, H.; Kimura, S.; Tomobe, Y.; Kobayashi, M.; Mitsui, Y.; Yazaki, Y.; Goto, K.; Masaki, T. A Novel Potent Vasoconstrictor Peptide Produced by Vascular Endothelial Cells. Nature 1988, 332, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Sancho-Bru, P.; Ginês, P.; Brenner, D.A. Liver Fibrogenesis: A New Role for the Renin–Angiotensin System. Antioxid. Redox Signal. 2005, 7, 1346–1355. [Google Scholar] [CrossRef]
- Rockey, D.C. Hepatic Blood Flow Regulation by Stellate Cells in Normal and Injured Liver. Semin. Liver Dis. 2001, 21, 337–350. [Google Scholar] [CrossRef]
- Rockey, D. Vascular Mediators in the Injured Liver. Hepatology 2003, 37, 4–12. [Google Scholar] [CrossRef]
- Rockey, D.C.; Weisiger, R.A. Endothelin Induced Contractility of Stellate Cells from Normal and Cirrhotic Rat Liver: Implications for Regulation of Portal Pressure and Resistance. Hepatology 1996, 24, 233–240. [Google Scholar] [CrossRef]
- Feder, L.S.; Todaro, J.A.; Laskin, D.L. Characterization of Interleukin-1 and Interleukin-6 Production by Hepatic Endothelial Cells and Macrophages. J. Leukoc. Biol. 1993, 53, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Tacke, F. Roles for Chemokines in Liver Disease. Gastroenterology 2014. [Google Scholar] [CrossRef] [PubMed]
- Connolly, M.K.; Bedrosian, A.S.; Malhotra, A.; Henning, J.R.; Ibrahim, J.; Vera, V.; Cieza-Rubio, N.E.; Hassan, B.U.; Pachter, H.L.; Cohen, S.; et al. In Hepatic Fibrosis, Liver Sinusoidal Endothelial Cells Acquire Enhanced Immunogenicity. J. Immunol. 2010, 185, 2200–2208. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.J.; Jain, G.; Rajagopalan, P. Designing a Fibrotic Microenvironment to Investigate Changes in Human Liver Sinusoidal Endothelial Cell Function. Acta Biomater. 2015. [Google Scholar] [CrossRef] [PubMed]
- Miyachi, Y.; Tsuchiya, K.; Komiya, C.; Shiba, K.; Shimazu, N.; Yamaguchi, S.; Deushi, M.; Osaka, M.; Inoue, K.; Sato, Y.; et al. Roles for Cell-Cell Adhesion and Contact in Obesity-Induced Hepatic Myeloid Cell Accumulation and Glucose Intolerance. Cell Rep. 2017, 18, 2766–2779. [Google Scholar] [CrossRef]
- Tomita, K.; Tamiya, G.; Ando, S.; Ohsumi, K.; Chiyo, T.; Mizutani, A.; Kitamura, N.; Toda, K.; Kaneko, T.; Horie, Y.; et al. Tumour Necrosis Factor Signalling through Activation of Kupffer Cells Plays an Essential Role in Liver Fibrosis of Non-Alcoholic Steatohepatitis in Mice. Gut 2006, 55, 415–424. [Google Scholar] [CrossRef]
- Wu, J.; Meng, Z.; Jiang, M.; Zhang, E.; Trippler, M.; Broering, R.; Bucchi, A.; Krux, F.; Dittmer, U.; Yang, D.; et al. Toll-like Receptor-Induced Innate Immune Responses in Non-Parenchymal Liver Cells Are Cell Type-Specific. Immunology 2010, 129, 363–374. [Google Scholar] [CrossRef]
- Weston, C.J.; Shepherd, E.L.; Claridge, L.C.; Rantakari, P.; Curbishley, S.M.; Tomlinson, J.W.; Hubscher, S.G.; Reynolds, G.M.; Aalto, K.; Anstee, Q.M.; et al. Vascular Adhesion Protein-1 Promotes Liver Inflammation and Drives Hepatic Fibrosis. J. Clin. Investig. 2015, 125, 501–520. [Google Scholar] [CrossRef]
- Shetty, S.; Lalor, P.F.; Adams, D.H. Lymphocyte Recruitment to the Liver: Molecular Insights into the Pathogenesis of Liver Injury and Hepatitis. Toxicology 2008. [Google Scholar] [CrossRef]
- Lalor, P.F.; Hields, P.S.; Grant, A.J.; Adams, D.H. Recruitment of Lymphocytes to the Human Liver. Immunol. Cell Biol. 2002. [Google Scholar] [CrossRef]
- Oteiza, A.; Li, R.; McCuskey, R.S.; Smedsrød, B.; Sørensen, K.K. Effects of Oxidized Low-Density Lipoproteins on the Hepatic Microvasculature. Am. J. Physiol. Liver Physiol. 2011, 301, G684–G693. [Google Scholar] [CrossRef] [PubMed]
- van Oosten, M.; van de Bilt, E.; de Vries, H.E.; van Berkel, T.J.C.; Kuiper, J. Vascular Adhesion Molecule–1 and Intercellular Adhesion Molecule–1 Expression on Rat Liver Cells after Lipopolysaccharide Administrationin Vivo. Hepatology 1995, 22, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Lefere, S.; Van de Velde, F.; Hoorens, A.; Raevens, S.; Van Campenhout, S.; Vandierendonck, A.; Neyt, S.; Vandeghinste, B.; Vanhove, C.; Debbaut, C.; et al. Angiopoietin-2 Promotes Pathological Angiogenesis and Is a Therapeutic Target in Murine Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 1087–1104. [Google Scholar] [CrossRef]
- Ip, E.; Farrell, G.; Hall, P.; Robertson, G.; Leclercq, I. Administration of the Potent PPAR? Agonist, Wy-14,643, Reverses Nutritional Fibrosis and Steatohepatitis in Mice. Hepatology 2004, 39, 1286–1296. [Google Scholar] [CrossRef] [PubMed]
- dela Peña, A.; Leclercq, I.; Field, J.; George, J.; Jones, B.; Farrell, G. NF-ΚB Activation, Rather Than TNF, Mediates Hepatic Inflammation in a Murine Dietary Model of Steatohepatitis. Gastroenterology 2005, 129, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Limmer, A.; Ohl, J.; Kurts, C.; Ljunggren, H.G.; Reiss, Y.; Groettrup, M.; Momburg, F.; Arnold, B.; Knolle, P.A. Efficient Presentation of Exogenous Antigen by Liver Endothelial Cells to CD8+ T Cells Results in Antigen-Specific T-Cell Tolerance. Nat. Med. 2000, 6, 1348–1354. [Google Scholar] [CrossRef]
- Knolle, P.A.; Schmitt, E.; Jin, S.; Germann, T.; Duchmann, R.; Hegenbarth, S.; Gerken, G.; Lohse, A.W. Induction of Cytokine Production in Naive CD4+ T Cells by Antigen- Presenting Murine Liver Sinusoidal Endothelial Cells but Failure to Induce Differentiation toward T(H1) Cells. Gastroenterology 1999, 116, 1428–1440. [Google Scholar] [CrossRef]
- Carambia, A.; Freund, B.; Schwinge, D.; Heine, M.; Laschtowitz, A.; Huber, S.; Wraith, D.C.; Korn, T.; Schramm, C.; Lohse, A.W.; et al. TGF-β-Dependent Induction of CD4+CD25+Foxp3+ Tregs by Liver Sinusoidal Endothelial Cells. J. Hepatol. 2014, 61, 594–599. [Google Scholar] [CrossRef]
- Chakraborty, K.; Chatterjee, S.; Bhattacharyya, A. Impact of Treg on Other T Cell Subsets in Progression of Fibrosis in Experimental Lung Fibrosis. Tissue Cell 2018, 53, 87–92. [Google Scholar] [CrossRef]
- Langhans, B.; Krämer, B.; Louis, M.; Nischalke, H.D.; Hüneburg, R.; Staratschek-Jox, A.; Odenthal, M.; Manekeller, S.; Schepke, M.; Kalff, J.; et al. Intrahepatic IL-8 Producing Foxp3+CD4+ Regulatory T Cells and Fibrogenesis in Chronic Hepatitis C. J. Hepatol. 2013, 59, 229–235. [Google Scholar] [CrossRef]
- Diehl, L.; Schurich, A.; Grochtmann, R.; Hegenbarth, S.; Chen, L.; Knolle, P.A. Tolerogenic Maturation of Liver Sinusoidal Endothelial Cells Promotes B7-Homolog 1-Dependent CD8+ T Cell Tolerance. Hepatology 2008, 47, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Limmer, A.; Ohl, J.; Wingender, G.; Berg, M.; Jüngerkes, F.; Schumak, B.; Djandji, D.; Scholz, K.; Klevenz, A.; Hegenbarth, S.; et al. Cross-Presentation of Oral Antigens by Liver Sinusoidal Endothelial Cells Leads to CD8 T Cell Tolerance. Eur. J. Immunol. 2005, 35, 2970–2981. [Google Scholar] [CrossRef] [PubMed]
- Berg, M.; Wingender, G.; Djandji, D.; Hegenbarth, S.; Momburg, F.; Hämmerling, G.; Limmer, A.; Knolle, P. Cross-Presentation of Antigens from Apoptotic Tumor Cells by Liver Sinusoidal Endothelial Cells Leads to Tumor-Specific CD8+ T Cell Tolerance. Eur. J. Immunol. 2006, 36, 2960–2970. [Google Scholar] [CrossRef] [PubMed]
- Schurich, A.; Berg, M.; Stabenow, D.; Böttcher, J.; Kern, M.; Schild, H.-J.; Kurts, C.; Schuette, V.; Burgdorf, S.; Diehl, L.; et al. Dynamic Regulation of CD8 T Cell Tolerance Induction by Liver Sinusoidal Endothelial Cells. J. Immunol. 2010, 184, 4107–4114. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.; Lalor, P.F.; Adams, D.H. Liver Sinusoidal Endothelial Cells — Gatekeepers of Hepatic Immunity. Nat. Rev. Gastroenterol. Hepatol. 2018, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Safadi, R.; Ohta, M.; Alvarez, C.E.; Fiel, M.I.; Bansal, M.; Mehal, W.Z.; Friedman, S.L. Immune Stimulation of Hepatic Fibrogenesis by CD8 Cells and Attenuation by Transgenic Interleukin-10 from Hepatocytes. Gastroenterology 2004, 127, 870–882. [Google Scholar] [CrossRef]
- Shetty, S.; Bruns, T.; Weston, C.J.; Stamataki, Z.; Oo, Y.H.; Long, H.M.; Reynolds, G.M.; Pratt, G.; Moss, P.; Jalkanen, S.; et al. Recruitment Mechanisms of Primary and Malignant B Cells to the Human Liver. Hepatology 2012, 56, 1521–1531. [Google Scholar] [CrossRef]
- Wehr, A.; Baeck, C.; Heymann, F.; Niemietz, P.M.; Hammerich, L.; Martin, C.; Zimmermann, H.W.; Pack, O.; Gassler, N.; Hittatiya, K.; et al. Chemokine Receptor CXCR6-Dependent Hepatic NK T Cell Accumulation Promotes Inflammation and Liver Fibrosis. J. Immunol. 2013, 190, 5226–5236. [Google Scholar] [CrossRef]
- Novobrantseva, T.I.; Majeau, G.R.; Amatucci, A.; Kogan, S.; Brenner, I.; Casola, S.; Shlomchik, M.J.; Koteliansky, V.; Hochman, P.S.; Ibraghimov, A. Attenuated Liver Fibrosis in the Absence of B Cells. J. Clin. Investig. 2005, 115, 3072–3082. [Google Scholar] [CrossRef]
- Thapa, M.; Chinnadurai, R.; Velazquez, V.M.; Tedesco, D.; Elrod, E.; Han, J.H.; Sharma, P.; Ibegbu, C.; Gewirtz, A.; Anania, F.; et al. Liver Fibrosis Occurs through Dysregulation of MyD88-Dependent Innate B-Cell Activity. Hepatology 2015, 61, 2067–2079. [Google Scholar] [CrossRef]
- Ju, C.; Tacke, F. Hepatic Macrophages in Homeostasis and Liver Diseases: From Pathogenesis to Novel Therapeutic Strategies. Cell. Mol. Immunol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F. Targeting Hepatic Macrophages to Treat Liver Diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef]
- Parola, M.; Pinzani, M. Liver Fibrosis: Pathophysiology, Pathogenetic Targets and Clinical Issues. Mol. Asp. Med. 2019, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Colburn, P.; Buonassisi, V. Anti-Clotting Activity of Endothelial Cell Cultures and Heparan Sulfate Proteoglycans. Biochem. Biophys. Res. Commun. 1982, 104, 220–227. [Google Scholar] [CrossRef]
- Moncada, S.; Gryglewski, R.; Bunting, S.; Vane, J.R. An Enzyme Isolated from Arteries Transforms Prostaglandin Endoperoxides to an Unstable Substance That Inhibits Platelet Aggregation. Nature 1976, 263, 663–665. [Google Scholar] [CrossRef] [PubMed]
- Mellion, B.T.; Ignarro, L.J.; Ohlstein, E.H.; Pontecorvo, E.G.; Hyman, A.L.; Kadowitz, P.J. Evidence for the Inhibitory Role of Guanosine 3’,5’-Monophosphate in ADP-Induced Human Platelet Aggregation in the Presence of Nitric Oxide and Related Vasodilators. Blood 1981, 57, 946–955. [Google Scholar] [CrossRef]
- Radomski, M.W.; Palmer, R.M.J.; Moncada, S. ENDOGENOUS NITRIC OXIDE INHIBITS HUMAN PLATELET ADHESION TO VASCULAR ENDOTHELIUM. Lancet 1987, 330, 1057–1058. [Google Scholar] [CrossRef]
- Bochenek, M.L.; Schäfer, K. Role of Endothelial Cells in Acute and Chronic Thrombosis. Hamostaseologie 2019, 128–139. [Google Scholar] [CrossRef]
- Esmon, C.T. Basic Mechanisms and Pathogenesis of Venous Thrombosis. Blood Rev. 2009, 23, 225–229. [Google Scholar] [CrossRef]
- Bevilacqua, M.P.; Pober, J.S.; Majeau, G.R.; Cotran, R.S.; Gimbrone, M.A. Interleukin 1 (IL-1) Induces Biosynthesis and Cell Surface Expression of Procoagulant Activity in Human Vascular Endothelial Cells. J. Exp. Med. 1984, 160, 618–623. [Google Scholar] [CrossRef]
- Myers, D.; Farris, D.; Hawley, A.; Wrobleski, S.; Chapman, A.; Stoolman, L.; Knibbs, R.; Strieter, R.; Wakefield, T. Selectins Influence Thrombosis in a Mouse Model of Experimental Deep Venous Thrombosis. J. Surg. Res. 2002, 108, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Wanless, I.R.; Liu, J.J.; Butany, J. Role of Thrombosis in the Pathogenesis of Congestive Hepatic Fibrosis (Cardiac Cirrhosis). Hepatology 1995, 21, 1232–1237. [Google Scholar] [CrossRef] [PubMed]
- Wanless, I.R.; Wong, F.; Blendis, L.M.; Greig, P.; Heathcote, E.J.; Levy, G. Hepatic and Portal Vein Thrombosis in Cirrhosis: Possible Role in Development of Parenchymal Extinction and Portal Hypertension. Hepatology 1995, 21, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Wright, M.; Goldin, R.; Thursz, M.R. Parenchymal Extinction: Coagulation and Hepatic Fibrogenesis. Clin. Liver Disease 2009, 117–126. [Google Scholar] [CrossRef]
- Cerini, F.; Vilaseca, M.; Lafoz, E.; García-Irigoyen, O.; García-Calderó, H.; Tripathi, D.M.; Avila, M.; Reverter, J.C.; Bosch, J.; Gracia-Sancho, J.; et al. Enoxaparin Reduces Hepatic Vascular Resistance and Portal Pressure in Cirrhotic Rats. J. Hepatol. 2016, 64. [Google Scholar] [CrossRef]
- Chambers, R.C.; Dabbagh, K.; McAnulty, R.J.; Gray, A.J.; Blanc-Brude, O.P.; Laurent, G.J. Thrombin Stimulates Fibroblast Procollagen Production via Proteolytic Activation of Protease-Activated Receptor 1. Biochem. J. 1998, 333, 121–127. [Google Scholar] [CrossRef]
- Gaça, M.D.A.; Zhou, X.; Benyon, R.C. Regulation of Hepatic Stellate Cell Proliferation and Collagen Synthesis by Proteinase-Activated Receptors. J. Hepatol. 2002, 36, 362–369. [Google Scholar] [CrossRef]
- Fiorucci, S.; Antonelli, E.; Distrutti, E.; Severino, B.; Fiorentina, R.; Baldoni, M.; Caliendo, G.; Santagada, V.; Morelli, A.; Cirino, G. PAR1 Antagonism Protects Against Experimental Liver Fibrosis. Role of Proteinase Receptors in Stellate Cell Activation. Hepatology 2004, 39, 365–375. [Google Scholar] [CrossRef]
- Hilscher, M.B.; Sehrawat, T.; Arab, J.P.; Zeng, Z.; Gao, J.; Liu, M.; Kostallari, E.; Gao, Y.; Simonetto, D.A.; Yaqoob, U.; et al. Mechanical Stretch Increases Expression of CXCL1 in Liver Sinusoidal Endothelial Cells to Recruit Neutrophils, Generate Sinusoidal Microthombi, and Promote Portal Hypertension. Gastroenterology 2019, 157, 193–209.e9. [Google Scholar] [CrossRef]
- Coulon, S.; Heindryckx, F.; Geerts, A.; Van Steenkiste, C.; Colle, I.; Van Vlierberghe, H. Angiogenesis in Chronic Liver Disease and Its Complications. Liver Int. 2011, 146–162. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular Mechanisms and Clinical Applications of Angiogenesis. Nature 2011, 298–307. [Google Scholar] [CrossRef]
- Dewhirst, M.W.; Cao, Y.; Moeller, B. Cycling Hypoxia and Free Radicals Regulate Angiogenesis and Radiotherapy Response. Nat. Rev. Cancer 2008, 425–437. [Google Scholar] [CrossRef]
- Semela, D.; Das, A.; Langer, D.; Kang, N.; Leof, E.; Shah, V. PDGF Signaling through Ephrin-B2 Regulates Hepatic Vascular Structure and Function. Gastroenterology 2009, 135, 671–679. [Google Scholar] [CrossRef]
- Augustin, H.G.; Young Koh, G.; Thurston, G.; Alitalo, K. Control of Vascular Morphogenesis and Homeostasis through the Angiopoietin - Tie System. Nat. Rev. Mol. Cell Biol. 2009, 165–177. [Google Scholar] [CrossRef]
- Phng, L.K.; Potente, M.; Leslie, J.D.; Babbage, J.; Nyqvist, D.; Lobov, I.; Ondr, J.K.; Rao, S.; Lang, R.A.; Thurston, G.; et al. Nrarp Coordinates Endothelial Notch and Wnt Signaling to Control Vessel Density in Angiogenesis. Dev. Cell 2009, 16, 70–82. [Google Scholar] [CrossRef]
- Povero, D.; Eguchi, A.; Niesman, I.R.; Andronikou, N.; Du Jeu, X.D.M.; Mulya, A.; Berk, M.; Lazic, M.; Thapaliya, S.; Parola, M.; et al. Lipid-Induced Toxicity Stimulates Hepatocytes to Release Angiogenic Microparticles That Require Vanin-1 for Uptake by Endothelial Cells. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef]
- Povero, D.; Panera, N.; Eguchi, A.; Johnson, C.D.; Papouchado, B.G.; de Araujo Horcel, L.; Pinatel, E.M.; Alisi, A.; Nobili, V.; Feldstein, A.E. Lipid-Induced Hepatocyte-Derived Extracellular Vesicles Regulate Hepatic Stellate Cells via MicroRNA Targeting Peroxisome Proliferator-Activated Receptor-γ. CMGH 2015, 1, 646–663.e4. [Google Scholar] [CrossRef]
- Lemoinne, S.; Cadoret, A.; Rautou, P.E.; El Mourabit, H.; Ratziu, V.; Corpechot, C.; Rey, C.; Bosselut, N.; Barbu, V.; Wendum, D.; et al. Portal Myofibroblasts Promote Vascular Remodeling Underlying Cirrhosis Formation through the Release of Microparticles. Hepatology 2015, 61, 1041–1055. [Google Scholar] [CrossRef]
- Deregibus, M.C.; Cantaluppi, V.; Calogero, R.; Lo Iacono, M.; Tetta, C.; Biancone, L.; Bruno, S.; Bussolati, B.; Camussi, G. Endothelial Progenitor Cell - Derived Microvesicles Activate an Angiogenic Program in Endothelial Cells by a Horizontal Transfer of MRNA. Blood 2007, 110, 2440–2448. [Google Scholar] [CrossRef]
- Mause, S.F.; Ritzel, E.; Liehn, E.A.; Hristov, M.; Bidzhekov, K.; Müller-Newen, G.; Soehnlein, O.; Weber, C. Platelet Microparticles Enhance the Vasoregenerative Potential of Angiogenic Early Outgrowth Cells after Vascular Injury. Circulation 2010, 122, 495–506. [Google Scholar] [CrossRef]
- Kitade, M.; Yoshiji, H.; Kojima, H.; Ikenaka, Y.; Noguchi, R.; Kaji, K.; Yoshii, J.; Yanase, K.; Namisaki, T.; Asada, K.; et al. Leptin-Mediated Neovascularization Is a Prerequisite for Progression of Nonalcoholic Steatohepatitis in Rats. Hepatology 2006, 44, 983–991. [Google Scholar] [CrossRef]
- Ehling, J.; Bartneck, M.; Wei, X.; Gremse, F.; Fech, V.; Möckel, D.; Baeck, C.; Hittatiya, K.; Eulberg, D.; Luedde, T.; et al. CCL2-Dependent Infiltrating Macrophages Promote Angiogenesis in Progressive Liver Fibrosis. Gut 2014, 63, 1960–1971. [Google Scholar] [CrossRef]
- Zhou, L.Y.; Zeng, H.; Wang, S.; Chen, J.X. Regulatory Role of Endothelial PHD2 in the Hepatic Steatosis. Cell. Physiol. Biochem. 2018, 48, 1003–1011. [Google Scholar] [CrossRef]
- Patsenker, E.; Popov, Y.; Stickel, F.; Schneider, V.; Ledermann, M.; Sägesser, H.; Niedobitek, G.; Goodman, S.L.; Schuppan, D. Pharmacological Inhibition of Integrin Avβ3 Aggravates Experimental Liver Fibrosis and Suppresses Hepatic Angiogenesis. Hepatology 2009, 50, 1501–1511. [Google Scholar] [CrossRef]
- Yang, L.; Kwon, J.; Popov, Y.; Gajdos, G.B.; Ordog, T.; Brekken, R.A.; Mukhopadhyay, D.; Schuppan, D.; Bi, Y.; Simonetto, D.; et al. Vascular Endothelial Growth Factor Promotes Fibrosis Resolution and Repair in Mice. Gastroenterology 2014, 146. [Google Scholar] [CrossRef]
- Kantari-Mimoun, C.; Castells, M.; Klose, R.; Meinecke, A.K.; Lemberger, U.J.; Rautou, P.E.; Pinot-Roussel, H.; Badoual, C.; Schrödter, K.; Österreicher, C.H.; et al. Resolution of Liver Fibrosis Requires Myeloid Cell-Driven Sinusoidal Angiogenesis. Hepatology 2015, 61, 2042–2055. [Google Scholar] [CrossRef]
- Neubauer, K.; Krüger, M.; Quondamatteo, F.; Knittel, T.; Saile, B.; Ramadori, G. Transforming Growth Factor-Β1 Stimulates the Synthesis of Basement Membrane Proteins Laminin, Collagen Type IV and Entactin in Rat Liver Sinusoidal Endothelial Cells. J. Hepatol. 1999, 31, 692–702. [Google Scholar] [CrossRef]
- Ribera, J.; Pauta, M.; Melgar-Lesmes, P.; Córdoba, B.; Bosch, A.; Calvo, M.; Rodrigo-Torres, D.; Sancho-Bru, P.; Mira, A.; Jiménez, W.; et al. A Small Population of Liver Endothelial Cells Undergoes Endothelial-Tomesenchymal Transition in Response to Chronic Liver Injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2017. [Google Scholar] [CrossRef]
- Jarnagin, W.R.; Rockey, D.C.; Koteliansky, V.E.; Wang, S.S.; Bissell, D.M. Expression of Variant Fibronectins in Wound Healing: Cellular Source and Biological Activity of the EIIIA Segment in Rat Hepatic Fibrogenesis. J. Cell Biol. 1994, 127, 2037–2048. [Google Scholar] [CrossRef]
- Friedman, S.L. Liver Fibrosis - From Bench to Bedside. J. Hepatol. 2003. [Google Scholar] [CrossRef]
- Wang, B.; Zhao, L.; Fish, M.; Logan, C.Y.; Nusse, R. Self-Renewing Diploid Axin2+ Cells Fuel Homeostatic Renewal of the Liver. Nature 2015, 524, 180. [Google Scholar] [CrossRef]
- Sakata, K.; Eda, S.; Lee, E.-S.; Hara, M.; Imoto, M.; Kojima, S. Neovessel Formation Promotes Liver Fibrosis via Providing Latent Transforming Growth Factor-β. Biochem. Biophys. Res. Commun. 2014, 443, 950–956. [Google Scholar] [CrossRef]
- Ding, B.-S.; Nolan, D.J.; Butler, J.M.; James, D.; Babazadeh, A.O.; Rosenwaks, Z.; Mittal, V.; Kobayashi, H.; Shido, K.; Lyden, D.; et al. Inductive Angiocrine Signals from Sinusoidal Endothelium Are Required for Liver Regeneration. Nature 2010, 468, 310–315. [Google Scholar] [CrossRef]
- Glińska-Suchocka, K.; Orłowska, A.; Spużak, J.; Jankowski, M.; Kubiak, K. Suitability of Using Serum Hialuronic Acid Concentrations in the Diagnosis of Canine Liver Fibrosis. Pol. J. Vet. Sci. 2015, 18, 873–878. [Google Scholar] [CrossRef][Green Version]
- Baranova, A.; Lal, P.; Birerdinc, A.; Younossi, Z.M. Non-Invasive Markers for Hepatic Fibrosis. BMC Gastroenterol. 2011, 11, 91. [Google Scholar] [CrossRef]
- Wong, L.; Yamasaki, G.; Johnson, R.J.; Friedman, S.L. Induction of Beta-Platelet-Derived Growth Factor Receptor in Rat Hepatic Lipocytes during Cellular Activation in Vivo and in Culture. J. Clin. Investig. 1994, 94, 1563–1569. [Google Scholar] [CrossRef]
- Sato, Y.; Okada, F.; Abe, M.; Seguchi, T.; Kuwano, M.; Sato, S.; Furuya, A.; Hanai, N.; Tamaoki, T. The Mechanism for the Activation of Latent TGF-Beta during Co-Culture of Endothelial Cells and Smooth Muscle Cells: Cell-Type Specific Targeting of Latent TGF-Beta to Smooth Muscle Cells. J. Cell Biol. 1993, 123, 1249–1254. [Google Scholar] [CrossRef]
- Rocha, A.S.; Vidal, V.; Mertz, M.; Kendall, T.J.; Charlet, A.; Okamoto, H.; Schedl, A. The Angiocrine Factor Rspondin3 Is a Key Determinant of Liver Zonation. Cell Rep. 2015, 13, 1757–1764. [Google Scholar] [CrossRef]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and Characterization of Exosomes from Cell Culture Supernatants and Biological Fluids. Curr. Protoc. Cell Biol. 2006. [Google Scholar] [CrossRef]
- Wang, R.; Ding, Q.; Yaqoob, U.; De Assuncao, T.D.M.; Verma, V.K.; Hirsova, P.; Cao, S.; Mukhopadhyay, D.; Huebert, R.C.; Shah, V.H. Exosome Adherence and Internalization by Hepatic Stellate Cells Triggers Sphingosine 1-Phosphate-Dependent Migration. J. Biol. Chem. 2015. [Google Scholar] [CrossRef]
- Kuramitsu, K.; Sverdlov, D.Y.; Liu, S.B.; Csizmadia, E.; Burkly, L.; Schuppan, D.; Hanto, D.W.; Otterbein, L.E.; Popov, Y. Failure of Fibrotic Liver Regeneration in Mice Is Linked to a Severe Fibrogenic Response Driven by Hepatic Progenitor Cell Activation. Am. J. Pathol. 2013. [Google Scholar] [CrossRef]
- Cordero-Espinoza, L.; Huch, M. The Balancing Act of the Liver: Tissue Regeneration versus Fibrosis. J. Clin. Investig. 2018. [CrossRef] [PubMed]
- Greene, A.K.; Wiener, S.; Puder, M.; Yoshida, A.; Shi, B.; Perez-Atayde, A.R.; Efstathiou, J.A.; Holmgren, L.; Adamis, A.P.; Rupnick, M.; et al. Endothelial-Directed Hepatic Regeneration after Partial Hepatectomy. Ann. Surg. 2003. [Google Scholar] [CrossRef] [PubMed]
- Foucher, J.; Chanteloup, E.; Vergniol, J.; Castéra, L.; Le Bail, B.; Adhoute, X.; Bertet, J.; Couzigou, P.; De Lédinghen, V. Diagnosis of Cirrhosis by Transient Elastography (FibroScan): A Prospective Study. Gut 2006, 55, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.G. The Role of Matrix Stiffness in Regulating Cell Behavior. Hepatology 2008, 1394–1400. [Google Scholar] [CrossRef]
- Olsen, A.L.; Bloomer, S.A.; Chan, E.P.; Gaça, M.D.A.; Georges, P.C.; Sackey, B.; Uemura, M.; Janmey, P.A.; Wells, R.G. Hepatic Stellate Cells Require a Stiff Environment for Myofibroblastic Differentiation. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301. [Google Scholar] [CrossRef]
- Wells, R.G. The Role of Matrix Stiffness in Hepatic Stellate Cell Activation and Liver Fibrosis. J. Clin. Gastroenterol. 2005. [Google Scholar] [CrossRef]
- Natarajan, V.; Berglund, E.J.; Chen, D.X.; Kidambi, S. Substrate Stiffness Regulates Primary Hepatocyte Functions. RSC Adv. 2015, 5, 80956–80966. [Google Scholar] [CrossRef]
- Desai, S.S.; Tung, J.C.; Zhou, V.X.; Grenert, J.P.; Malato, Y.; Rezvani, M.; Español-Suñer, R.; Willenbring, H.; Weaver, V.M.; Chang, T.T. Physiological Ranges of Matrix Rigidity Modulate Primary Mouse Hepatocyte Function in Part through Hepatocyte Nuclear Factor 4 Alpha. Hepatology 2016, 64, 261–275. [Google Scholar] [CrossRef]
- Matrix Stiffness Regulate Liver Sinusoidal Endothelial Cells (LSECs) Function: Importance for Liver Fibrosis Progression. Available online: https://www.fasebj.org/doi/abs/10.1096/fasebj.2019.33.1_supplement.496.39 (accessed on 24 October 2019).
- Juin, A.; Planus, E.; Guillemot, F.; Horakova, P.; Albiges-Rizo, C.; Génot, E.; Rosenbaum, J.; Moreau, V.; Saltel, F. Extracellular Matrix Rigidity Controls Podosome Induction in Microvascular Endothelial Cells. Biol. Cell 2013, 105, 46–57. [Google Scholar] [CrossRef]
- Kilarski, W.W.; Samolov, B.; Petersson, L.; Kvanta, A.; Gerwins, P. Biomechanical Regulation of Blood Vessel Growth during Tissue Vascularization. Nat. Med. 2009, 15, 657–664. [Google Scholar] [CrossRef]
- Korff, T.; Augustin, H.G. Tensional Forces in Fibrillar Extracellular Matrices Control Directional Capillary Sprouting. J. Cell Sci. 1999, 112, 3249–3258. [Google Scholar] [PubMed]
- Bishop, P.N. The Role of Extracellular Matrix in Retinal Vascular Development and Preretinal Neovascularization. Exp. Eye Res. 2015, 133, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; You, Z.; Yu, H.; Zhou, L.; Zhao, H.; Yan, X.; Li, D.; Wang, B.; Zhu, L.; Xu, Y.; et al. Mechanotransduction-Modulated Fibrotic Microniches Reveal the Contribution of Angiogenesis in Liver Fibrosis. Nat. Mater. 2017, 16, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.F. Flow-Mediated Endothelial Mechanotransduction. Physiol. Rev. 1995, 519–560. [Google Scholar] [CrossRef] [PubMed]
- Ayajiki, K.; Kindermann, M.; Hecker, M.; Fleming, I.; Busse, R. Intracellular PH and Tyrosine Phosphorylation but Not Calcium Determine Shear Stress-Induced Nitric Oxide Production in Native Endothelial Cells. Circ. Res. 1996, 78, 750–758. [Google Scholar] [CrossRef]
- Hwa, A.J.; Fry, R.C.; Sivaraman, A.; So, P.T.; Samson, L.D.; Stolz, D.B.; Griffith, L.G. Rat Liver Sinusoidal Endothelial Cells Survive without Exogenous VEGF in 3D Perfused Co-Cultures with Hepatocytes. FASEB J. 2007, 21, 2564–2579. [Google Scholar] [CrossRef]
- Domansky, K.; Inman, W.; Serdy, J.; Dash, A.; Lim, M.H.M.; Griffith, L.G. Perfused Multiwell Plate for 3D Liver Tissue Engineering. Lab Chip 2010, 10, 51–58. [Google Scholar] [CrossRef]
- Ortega-Ribera, M.; Fernández-Iglesias, A.; Illa, X.; Moya, A.; Molina, V.; Maeso-Díaz, R.; Fondevila, C.; Peralta, C.; Bosch, J.; Villa, R.; et al. Resemblance of the Human Liver Sinusoid in a Fluidic Device with Biomedical and Pharmaceutical Applications. Biotechnol. Bioeng. 2018, 115, 2585–2594. [Google Scholar] [CrossRef]
- Parmar, K.M.; Larman, H.B.; Dai, G.; Zhang, Y.; Wang, E.T.; Moorthy, S.N.; Kratz, J.R.; Lin, Z.; Jain, M.K.; Gimbrone, M.A.; et al. Integration of Flow-Dependent Endothelial Phenotypes by Kruppel-like Factor 2. J. Clin. Investig. 2006, 116, 49–58. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Villarreal, G.; Zhang, Y.; Yu, J.X.; Liu, Y.; Tullius, S.G.; García-Cardeña, G. Flow Cessation Triggers Endothelial Dysfunction during Organ Cold Storage Conditions: Strategies for Pharmacologic Intervention. Transplantation 2010, 90, 142–149. [Google Scholar] [CrossRef]
- Marrone, G.; Russo, L.; Rosado, E.; Hide, D.; García-Cardeña, G.; García-Pagán, J.C.; Bosch, J.; Gracia-Sancho, J. The Transcription Factor KLF2 Mediates Hepatic Endothelial Protection and Paracrine Endothelial-Stellate Cell Deactivation Induced by Statins. J. Hepatol. 2013. [Google Scholar] [CrossRef]
- Shay-Salit, A.; Shushy, M.; Wolfovitz, E.; Yahav, H.; Breviario, F.; Dejana, E.; Resnick, N. VEGF Receptor 2 and the Adherens Junction as a Mechanical Transducer in Vascular Endothelial Cells. Proc. Natl. Acad. Sci. USA 2002, 99, 9462–9467. [Google Scholar] [CrossRef]
- Braet, F.; Shleper, M.; Paizi, M.; Brodsky, S.; Kopeiko, N.; Resnick, N.; Spira, G. Liver Sinusoidal Endothelial Cell Modulation upon Resection and Shear Stress in Vitro. Comp. Hepatol. 2004, 3. [Google Scholar] [CrossRef]
- Roskams, T. Relationships Among Stellate Cell Activation, Progenitor Cells, and Hepatic Regeneration. Clin. Liver Dis. 2008. [Google Scholar] [CrossRef]
- You, Q.; Holt, M.; Yin, H.; Li, G.; Hu, C.J.; Ju, C. Role of Hepatic Resident and Infiltrating Macrophages in Liver Repair after Acute Injury. Biochem. Pharmacol. 2013. [Google Scholar] [CrossRef]
- Widmann, J.J.; Fahimi, H.D. Proliferation of Mononuclear Phagocytes (Kupffer Cells) and Endothelial Cells in Regenerating Rat Liver. A Light and Electron Microscopic Cytochemical Study. Am. J. Pathol. 1975, 80, 349. [Google Scholar]
- Abshagen, K.; Eipel, C.; Kalff, J.C.; Menger, M.D.; Vollmar, B. Loss of NF-ΚB Activation in Kupffer Cell-Depleted Mice Impairs Liver Regeneration after Partial Hepatectomy. Am. J. Physiol. Gastrointest. Liver Physiol. 2007. [Google Scholar] [CrossRef]
- Xu, C.S.; Jiang, Y.; Zhang, L.X.; Chang, C.F.; Wang, G.P.; Shi, R.J.; Yang, Y.J. The Role of Kupffer Cells in Rat Liver Regeneration Revealed by Cell-Specific Microarray Analysis. J. Cell. Biochem. 2012. [Google Scholar] [CrossRef]
- Mabuchi, A.; Mullaney, I.; Sheard, P.W.; Hessian, P.A.; Mallard, B.L.; Tawadrous, M.N.; Zimmermann, A.; Senoo, H.; Wheatley, A.M. Role of Hepatic Stellate Cell/Hepatocyte Interaction and Activation of Hepatic Stellate Cells in the Early Phase of Liver Regeneration in the Rat. J. Hepatol. 2004. [Google Scholar] [CrossRef]
- Manavski, Y.; Abel, T.; Hu, J.; Kleinlützum, D.; Buchholz, C.J.; Belz, C.; Augustin, H.G.; Boon, R.A.; Dimmeler, S. Endothelial Transcription Factor KLF2 Negatively Regulates Liver Regeneration via Induction of Activin A. Proc. Natl. Acad. Sci. USA 2017. [Google Scholar] [CrossRef]
- Sackey-Aboagye, B.; Olsen, A.L.; Mukherjee, S.M.; Ventriglia, A.; Yokosaki, Y.; Greenbaum, L.E.; Lee, G.Y.; Naga, H.; Wells, R.G. Fibronectin Extra Domain a Promotes Liver Sinusoid Repair Following Hepatectomy. PLoS ONE 2016, 11, e0163737. [Google Scholar] [CrossRef]
- DeLeve, L.D.; Wang, X.; Wang, L. VEGF-Sdf1 Recruitment of CXCR7+ Bone Marrow Progenitors of Liver Sinusoidal Endothelial Cells Promotes Rat Liver Regeneration. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G739–G746. [Google Scholar] [CrossRef]
- Maher, J.J. Cell-Specific Expression of Hepatocyte Growth Factor in Liver: Upregulation in Sinusoidal Endothelial Cells after Carbon Tetrachloride. J. Clin. Investig. 1993, 91, 2244–2252. [Google Scholar] [CrossRef]
- Nanji, A.A.; Tahan, S.R.; Wei, Y.; Sadrzadeh, S.M.H. Hepatic Sinusoidal Endothelial Cell G1/S Arrest Correlates with Severity of Alcoholic Liver Injury in the Rat. Gastroenterology 1994. [Google Scholar] [CrossRef]
- LeCouter, J.; Moritz, D.R.; Li, B.; Phillips, G.L.; Liang, X.H.; Gerber, H.P.; Hillan, K.J.; Ferraral, N. Angiogenesis-Independent Endothelial Protection of Liver: Role of VEGFR-1. Science 2003. [Google Scholar] [CrossRef]
- Wang, L.L.; Wang, X.; Wang, L.L.; Chiu, J.D.; Van De Ven, G.; Gaarde, W.A.; Deleve, L.D. Hepatic Vascular Endothelial Growth Factor Regulates Recruitment of Rat Liver Sinusoidal Endothelial Cell Progenitor Cells. Gastroenterology 2012. [Google Scholar] [CrossRef]
- Wang, L.; Wang, X.; Xie, G.; Wang, L.; Hill, C.K.; DeLeve, L.D. Liver Sinusoidal Endothelial Cell Progenitor Cells Promote Liver Regeneration in Rats. J. Clin. Investig. 2012, 122, 1567–1573. [Google Scholar] [CrossRef]
- Steffan, A.-M.; Gendrault, J.-L.; McCuskey, R.S.; McCuskey, P.A.; Kirn, A. Phagocytosis, an Unrecognized Property of Murine Endothelial Liver Cells. Hepatology 1986. [Google Scholar] [CrossRef]
- Taniguchi, E.; Sakisaka, S.; Matsuo, K.; Tanikawa, K.; Sata, M. Expression and Role of Vascular Endothelial Growth Factor in Liver Regeneration after Partial Hepatectomy in Rats. J. Histochem. Cytochem. 2001. [Google Scholar] [CrossRef]
- Xu, C.S.; Chen, X.G.; Chang, C.F.; Wang, G.P.; Wang, W.B.; Zhang, L.X.; Zhu, Q.S.; Wang, L. Analysis of Time-Course Gene Expression Profiles of Sinusoidal Endothelial Cells during Liver Regeneration in Rats. Mol. Cell. Biochem. 2011. [Google Scholar] [CrossRef]
- Sato, T.; El-Assal, O.N.; Ono, T.; Yamanoi, A.; Kumar Dhar, D.; Nagasue, N. Sinusoidal Endothelial Cell Proliferation and Expression of Angiopoietin/Tie Family in Regenerating Rat Liver. J. Hepatol. 2001. [Google Scholar] [CrossRef]
- Mochida, S.; Ishikawa, K.; Inao, M.; Shibuya, M.; Fujiwara, K. Increased Expressions of Vascular Endothelial Growth Factor and Its Receptors, Flt-1 and KDR/Flk-1, in Regenerating Rat Liver. Biochem. Biophys. Res. Commun. 1996. [Google Scholar] [CrossRef] [PubMed]
- Shubham, S.; Kumar, D.; Rooge, S.; Maras, J.S.; Maheshwari, D.; Nautiyal, N.; Kumari, R.; Bhat, A.; Kumar, G.; Rastogi, A.; et al. Cellular and Functional Loss of Liver Endothelial Cells Correlates with Poor Hepatocyte Regeneration in Acute-on-Chronic Liver Failure. Hepatol. Int. 2019. [Google Scholar] [CrossRef] [PubMed]
- Lao, Y.; Li, Y.; Zhang, P.; Shao, Q.; Lin, W.; Qiu, B.; Lv, Y.; Tang, L.; Su, S.; Zhang, H.; et al. Targeting Endothelial Erk1/2-Akt Axis as a Regeneration Strategy to Bypass Fibrosis during Chronic Liver Injury in Mice. Mol. Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.S.; Ferrara, N. Developmental and Pathological Angiogenesis. Annu. Rev. Cell Dev. Biol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; Van Der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of Putative Progenitor Endothelial Cells for Angiogenesis. Science 1997. [Google Scholar] [CrossRef]
- Shi, Q.; Rafii, S.; Wu Hong-De, M.; Wijelath, E.S.; Yu, C.; Ishida, A.; Fujita, Y.; Kothari, S.; Mohle, R.; Sauvage, L.R.; et al. Evidence for Circulating Bone Marrow-Derived Endothelial Cells. Blood 1998, 92, 362–367. [Google Scholar] [CrossRef]
- Peichev, M.; Naiyer, A.J.; Pereira, D.; Zhu, Z.; Lane, W.J.; Williams, M.; Oz, M.C.; Hicklin, D.J.; Witte, L.; Moore, M.A.S.; et al. Expression of VEGFR-2 and AC133 by Circulating Human CD34 + Cells Identifies a Population of Functional Endothelial Precursors. Blood 2000, 95, 952–958. [Google Scholar] [CrossRef]
- Katagiri, H.; Kushida, Y.; Nojima, M.; Kuroda, Y.; Wakao, S.; Ishida, K.; Endo, F.; Kume, K.; Takahara, T.; Nitta, H.; et al. A Distinct Subpopulation of Bone Marrow Mesenchymal Stem Cells, Muse Cells, Directly Commit to the Replacement of Liver Components. Am. J. Transplant. 2016. [Google Scholar] [CrossRef]
- Yu, Y.; Liang, Y.; Liu, X.; Yang, H.; Su, Y.; Xia, X.; Wang, H. Id1 Modulates Endothelial Progenitor Cells Function through Relieving the E2-2-Mediated Repression of FGFR1 and VEGFR2 in Vitro. Mol. Cell. Biochem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liang, Y.; Yin, C.; Liu, X.; Su, Y.; Zhang, L.; Wang, H. Inhibitor of DNA-Binding 1 Promotes Endothelial Progenitor Cell Proliferation and Migration by Suppressing E2-2 through the Helix-Loop-Helix Domain. Int. J. Mol. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Yu, Y.; Zhang, L.; Ma, Y.; Wang, H. Inhibitor of DNA Binding 1 Regulates Cell Cycle Progression of Endothelial Progenitor Cells through Induction of Wnt2 Expression. Mol. Med. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Harb, R.; Xie, G.; Lutzko, C.; Guo, Y.; Wang, X.; Hill, C.K.; Kanel, G.C.; DeLeve, L.D. Bone Marrow Progenitor Cells Repair Rat Hepatic Sinusoidal Endothelial Cells After Liver Injury. Gastroenterology 2009. [Google Scholar] [CrossRef]
- Wang, X.; Maretti-Mira, A.C.; Wang, L.; DeLeve, L.D. Liver-Selective MMP-9 Inhibition in the Rat Eliminates Ischemia-Reperfusion Injury and Accelerates Liver Regeneration. Hepatology 2019. [Google Scholar] [CrossRef]
- Bautch, V.L. Stem Cells and the Vasculature. Nat. Med. 2011. [Google Scholar] [CrossRef]
- Medina, R.J.; Barber, C.L.; Sabatier, F.; Dignat-George, F.; Melero-Martin, J.M.; Khosrotehrani, K.; Ohneda, O.; Randi, A.M.; Chan, J.K.Y.; Yamaguchi, T.; et al. Endothelial Progenitors: A Consensus Statement on Nomenclature. Stem Cells Transl. Med. 2017. [Google Scholar] [CrossRef]
- Dickson, I. Stem Cell Therapy for Liver Cirrhosis UnREALISTIC? Nat. Rev. Gastroenterol. Hepatol. 2018. [Google Scholar] [CrossRef]
- Singhal, M.; Liu, X.; Inverso, D.; Jiang, K.; Dai, J.; He, H.; Bartels, S.; Li, W.; Abdul Pari, A.A.; Gengenbacher, N.; et al. Endothelial Cell Fitness Dictates the Source of Regenerating Liver Vasculature. J. Exp. Med. 2018. [Google Scholar] [CrossRef]
- Sun, M.; Kisseleva, T. Reversibility of Liver Fibrosis. Clin. Res. Hepatol. Gastroenterol. 2015, S60–S63. [Google Scholar] [CrossRef]
- Fernández Carrillo, C.; Lens, S.; Llop, E.; Pascasio, J.M.; Crespo, J.; Arenas, J.; Fernández, I.; Baliellas, C.; Carrión, J.A.; de la Mata, M.; et al. Treatment of Hepatitis C Virus Infection in Patients with Cirrhosis and Predictive Value of Model for End-Stage Liver Disease: Analysis of Data from the Hepa-C Registry. Hepatology 2017. [Google Scholar] [CrossRef] [PubMed]
- Lens, S.; Alvarado, E.; Mariño, Z.; Martinez, J.; Londoño, M.; Llop, E.; Panero, C.; Torras, X.; Baiges, A.; Hernandez-gea, V.; et al. PS-150 Long-Term Impact of HCV Eradication after All-Oral Therapy in Patients with Clinical Significant Portal Hypertension. J. Hepatol. 2018, 68, S84. [Google Scholar] [CrossRef]
- Mauro, E.; Crespo, G.; Montironi, C.; Londoño, M.-C.; Hernandez-Gea, V.; Ruiz, P.; Sastre, L.; Lombardo, J.; Mariño, Z.; Diaz, A.; et al. Portal Pressure and Liver Stiffness Measurements in the Prediction of Fibrosis Regression after SVR in Recurrent Hepatitis C Ezequiel. Hepatology 2017. [Google Scholar] [CrossRef]
- Iredale, J.P.; Benyon, R.C.; Pickering, J.; McCullen, M.; Northrop, M.; Pawley, S.; Hovell, C.; Arthur, M.J. Mechanisms of Spontaneous Resolution of Rat Liver Fibrosis. Hepatic Stellate Cell Apoptosis and Reduced Hepatic Expression of Metalloproteinase Inhibitors. J. Clin. Investig. 1998. [Google Scholar] [CrossRef]
- Troeger, J.S.; Mederacke, I.; Gwak, G.Y.; Dapito, D.H.; Mu, X.; Hsu, C.C.; Pradere, J.P.; Friedman, R.A.; Schwabe, R.F. Deactivation of Hepatic Stellate Cells during Liver Fibrosis Resolution in Mice. Gastroenterology 2012, 143. [Google Scholar] [CrossRef]
- Issa, R.; Williams, E.; Trim, N.; Kendall, T.; Arthur, M.J.P.; Reichen, J.; Benyon, R.C.; Iredale, J.P. Apoptosis of Hepatic Stellate Cells: Involvement in Resolution of Biliary Fibrosis and Regulation by Soluble Growth Factors. Gut 2001. [Google Scholar] [CrossRef] [PubMed]
- Iredale, J.P.; Bataller, R. Identifying Molecular Factors That Contribute to Resolution of Liver Fibrosis. Gastroenterology 2014, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Iredale, J.P. Models of Liver Fibrosis: Exploring the Dynamic Nature of Inflammation and Repair in a Solid Organ. J. Clin. Investig. 2007, 539–548. [Google Scholar] [CrossRef]
- Iredale, J.P.; Thompson, A.; Henderson, N.C. Extracellular Matrix Degradation in Liver Fibrosis: Biochemistry and Regulation. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 876–883. [Google Scholar] [CrossRef]
- Campana, L.; Iredale, J.P. Regression of Liver Fibrosis. Semin. Liver Dis. 2017, 37, 1–10. [Google Scholar] [CrossRef]
- Issa, R.; Zhou, X.; Constandinou, C.M.; Fallowfield, J.; Millward-Sadler, H.; Gaca, M.D.A.; Sands, E.; Suliman, I.; Trim, N.; Knorr, A.; et al. Spontaneous Recovery from Micronodular Cirrhosis: Evidence for Incomplete Resolution Associated with Matrix Cross-Linking. Gastroenterology 2004. [Google Scholar] [CrossRef] [PubMed]
- Iredale, J.P. Tissue Inhibitors of Metalloproteinases in Liver Fibrosis. Int. J. Biochem. Cell Biol. 1997, 43–54. [Google Scholar] [CrossRef]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P.; Leibovich, S.; et al. Selective Depletion of Macrophages Reveals Distinct, Opposing Roles during Liver Injury and Repair. J. Clin. Investig. 2005. [Google Scholar] [CrossRef]
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T.; et al. Differential Ly-6C Expression Identifies the Recruited Macrophage Phenotype, Which Orchestrates the Regression of Murine Liver Fibrosis. Proc. Natl. Acad. Sci. USA 2012. [Google Scholar] [CrossRef]
- Thomas, J.A.; Pope, C.; Wojtacha, D.; Robson, A.J.; Gordon-Walker, T.T.; Hartland, S.; Ramachandran, P.; Van Deemter, M.; Hume, D.A.; Iredale, J.P.; et al. Macrophage Therapy for Murine Liver Fibrosis Recruits Host Effector Cells Improving Fibrosis, Regeneration, and Function. Hepatology 2011, 53, 2003–2015. [Google Scholar] [CrossRef]
- Kantari-Mimoun, C.; Krzywinska, E.; Castells, M.; Milien, C.; Klose, R.; Meinecke, A.K.; Lemberger, U.; Mathivet, T.; Gojkovic, M.; Schrödter, K.; et al. Boosting the Hypoxic Response in Myeloid Cells Accelerates Resolution of Fibrosis and Regeneration of the Liver in Mice. Oncotarget 2017, 8, 15085–15100. [Google Scholar] [CrossRef]
- DeLeve, L.D. Liver Sinusoidal Endothelial Cells in Hepatic Fibrosis. Hepatology 2015, 61, 1740–1746. [Google Scholar] [CrossRef]
- Malovic, I.; Sørensen, K.K.; Elvevold, K.H.; Nedredal, G.I.; Paulsen, S.; Erofeev, A.V.; Smedsrød, B.H.; McCourt, P.A.G. The Mannose Receptor on Murine Liver Sinusoidal Endothelial Cells Is the Main Denatured Collagen Clearance Receptor. Hepatology 2007, 45, 1454–1461. [Google Scholar] [CrossRef]
- Hellevik, T.; Bondevik, A.; Smedsrød, B. Intracellular Fate of Endocytosed Collagen in Rat Liver Endothelial Cells. Exp. Cell Res. 1996, 223, 39–49. [Google Scholar] [CrossRef]
- Smedsrod, B.; Johansson, S.; Pertoft, H. Studies in Vivo and in Vitro on the Uptake and Degradation of Soluble Collagen A1(I) Chains in Rat Liver Endothelial and Kupffer Cells. Biochem. J. 1985, 228, 415–424. [Google Scholar] [CrossRef]
- Smedsrod, B. Receptor-Mediated Endocytosis of Connective Tissue Macromolecules in Liver Endothelial Cells. Scand. J. Clin. Lab. Investig. Suppl. 1990, 50, 148–151. [Google Scholar]
- Smedsrod, B.; Melkko, J.; Risteli, L.; Risteli, J. Circulating C-Terminal Propeptide of Type I Procollagen Is Cleared Mainly via the Mannose Receptor in Liver Endothelial Cells. Biochem. J. 1990, 271, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Choi, J.; Son, T.; Wee, H.J.; Bae, S.J.; Seo, J.H.; Park, J.H.; Ryu, S.H.; Lee, D.; Jang, M.K.; et al. Altered AKAP12 Expression in Portal Fibroblasts and Liver Sinusoids Mediates Transition from Hepatic Fibrogenesis to Fibrosis Resolution. Exp. Mol. Med. 2018, 50. [Google Scholar] [CrossRef] [PubMed]
- Falkowski, M.; Schledzewski, K.; Hansen, B.; Goerdt, S. Expression of Stabilin-2, a Novel Fasciclin-like Hyaluronan Receptor Protein, in Murine Sinusoidal Endothelia, Avascular Tissues, and at Solid/ Liquid Interfaces. Histochem. Cell Biol. 2003, 120, 361–369. [Google Scholar] [CrossRef]
- Zhou, B.; Weigel, J.A.; Fauss, L.; Weigel, P.H. Identification of the Hyaluronan Receptor for Endocytosis (HARE). J. Biol. Chem. 2000, 275, 37733–37741. [Google Scholar] [CrossRef]
- Toriyabe, N.; Hayashi, Y.; Hyodo, M.; Harashima, H. Synthesis and Evaluation of Stearylated Hyaluronic Acid for the Active Delivery of Liposomes to Liver Endothelial Cells. Biol. Pharm. Bull. 2011, 34, 1084–1089. [Google Scholar] [CrossRef][Green Version]
- Takei, Y.; Maruyama, A.; Ferdous, A.; Nishimura, Y.; Kawano, S.; Ikejima, K.; Okumura, S.; Asayama, S.; Nogawa, M.; Hashimoto, M.; et al. Targeted Gene Delivery to Sinusoidal Endothelial Cells: DNA Nanoassociate Bearing Hyaluronan-Glycocalyx. FASEB J. 2004, 18, 699–701. [Google Scholar] [CrossRef]
- Szafraniec, J.; Błazejczyk, A.; Kus, E.; Janik, M.; Zajac, G.; Wietrzyk, J.; Chlopicki, S.; Zapotoczny, S. Robust Oil-Core Nanocapsules with Hyaluronate-Based Shells as Promising Nanovehicles for Lipophilic Compounds. Nanoscale 2017, 9, 18867–18880. [Google Scholar] [CrossRef]
- Kren, B.T.; Unger, G.M.; Sjeklocha, L.; Trossen, A.A.; Korman, V.; Diethelm-Okita, B.M.; Reding, M.T.; Steer, C.J. Nanocapsule-Delivered Sleeping Beauty Mediates Therapeutic Factor VIII Expression in Liver Sinusoidal Endothelial Cells of Hemophilia A Mice. J. Clin. Investig. 2009, 119, 2086–2099. [Google Scholar] [CrossRef]
- Lee, H.; Mok, H.; Lee, S.; Oh, Y.K.; Park, T.G. Target-Specific Intracellular Delivery of SiRNA Using Degradable Hyaluronic Acid Nanogels. J. Control. Release 2007, 119, 245–252. [Google Scholar] [CrossRef]
- Marquez, J.; Fernandez-Piñeiro, I.; Araúzo-Bravo, M.J.; Poschmann, G.; Stühler, K.; Khatib, A.M.; Sanchez, A.; Unda, F.; Ibarretxe, G.; Bernales, I.; et al. Targeting Liver Sinusoidal Endothelial Cells with MiR-20a-Loaded Nanoparticles Reduces Murine Colon Cancer Metastasis to the Liver. Int. J. Cancer 2018, 143, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Hunt, N.J.; McCourt, P.A.G.; Le Couteur, D.G.; Cogger, V.C. Novel Targets for Delaying Aging: The Importance of the Liver and Advances in Drug Delivery. Adv. Drug Deliv. Rev. 2018, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, M.; Weeke-Klimp, A.H.; Hoenselaar, E.P.D.; Stuart, M.C.A.; Meijer, D.K.F.; Scherphof, G.L.; Kamps, J.A.A.M. Stabilized Lipid Coated Lipoplexes for the Delivery of Antisense Oligonucleotides to Liver Endothelial Cells in Vitro and in Vivo. J. Drug Target. 2004, 12, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, M.; Weeke-Klimp, A.H.; Morselt, H.W.M.; Kimpfler, A.; Ásgeirsdóttir, S.A.; Schubert, R.; Meijer, D.K.F.; Scherphof, G.L.; Kamps, J.A.A.M. Optimized Targeting of Polyethylene Glycol-Stabilized Anti-Intercellular Adhesion Molecule 1 Oligonucleotide/Lipid Particles to Liver Sinusoidal Endothelial Cells. Mol. Pharmacol. 2005, 67, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Hunt, N.J.; Lockwood, G.P.; Le Couteur, F.H.; McCourt, P.A.G.; Singla, N.; Kang, S.W.S.; Burgess, A.; Kuncic, Z.; Le Couteur, D.G.; Cogger, V.C. Rapid Intestinal Uptake and Targeted Delivery to the Liver Endothelium Using Orally Administered Silver Sulfide Quantum Dots. ACS Nano 2020, 14, 1492–1507. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, X.; Liu, X.; Kumar, S.; Gochman, G.; Ji, Y.; Liao, Y.P.; Chang, C.H.; Situ, W.; Lu, J.; et al. Use of Polymeric Nanoparticle Platform Targeting the Liver To Induce Treg-Mediated Antigen-Specific Immune Tolerance in a Pulmonary Allergen Sensitization Model. ACS Nano 2019, 13, 4778–4794. [Google Scholar] [CrossRef]
- Yu, X.; Chen, L.; Liu, J.; Dai, B.; Xu, G.; Shen, G.; Luo, Q.; Zhang, Z. Immune Modulation of Liver Sinusoidal Endothelial Cells by Melittin Nanoparticles Suppresses Liver Metastasis. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Akhter, A.; Hayashi, Y.; Sakurai, Y.; Ohga, N.; Hida, K.; Harashima, H. A Liposomal Delivery System That Targets Liver Endothelial Cells Based on a New Peptide Motif Present in the ApoB-100 Sequence. Int. J. Pharm. 2013, 456, 195–201. [Google Scholar] [CrossRef]
- Carambia, A.; Freund, B.; Schwinge, D.; Bruns, O.T.; Salmen, S.C.; Ittrich, H.; Reimer, R.; Heine, M.; Huber, S.; Waurisch, C.; et al. Nanoparticle-Based Autoantigen Delivery to Treg-Inducing Liver Sinusoidal Endothelial Cells Enables Control of Autoimmunity in Mice. J. Hepatol. 2015, 62, 1349–1356. [Google Scholar] [CrossRef]
- Liang, X.; Grice, J.E.; Zhu, Y.; Liu, D.; Sanchez, W.Y.; Li, Z.; Crawford, D.H.G.; Le Couteur, D.G.; Cogger, V.C.; Liu, X.; et al. Intravital Multiphoton Imaging of the Selective Uptake of Water-Dispersible Quantum Dots into Sinusoidal Liver Cells. Small 2015, 11, 1711–1720. [Google Scholar] [CrossRef]
- Bargheer, D.; Giemsa, A.; Freund, B.; Heine, M.; Waurisch, C.; Stachowski, G.M.; Hickey, S.G.; Eychmüller, A.; Heeren, J.; Nielsen, P. The Distribution and Degradation of Radiolabeled Superparamagnetic Iron Oxide Nanoparticles and Quantum Dots in Mice. Beilstein J. Nanotechnol. 2015, 6, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Hatakeyama, H.; Hyodo, M.; Harashima, H. Relationship Between the Physicochemical Properties of Lipid Nanoparticles and the Quality of SiRNA Delivery to Liver Cells. Mol. Ther. 2016, 24, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Sano, N.; Tamura, T.; Toriyabe, N.; Nowatari, T.; Nakayama, K.; Tanoi, T.; Murata, S.; Sakurai, Y.; Hyodo, M.; Fukunaga, K.; et al. New Drug Delivery System for Liver Sinusoidal Endothelial Cells for Ischemia-Reperfusion Injury. World J. Gastroenterol. 2015, 21, 12778–12786. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; De Minicis, S.; Österreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 Enhances TGF-β Signaling and Hepatic Fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Chauhan, A.K.; Motto, D.G.; Lamb, C.B.; Bergmeier, W.; Dockal, M.; Plaimauer, B.; Scheiflinger, F.; Ginsburg, D.; Wagner, D.D. Systemic Antithrombotic Effects of ADAMTS13. J. Exp. Med. 2006, 203, 767–776. [Google Scholar] [CrossRef]
- Geys, L.; Bauters, D.; Roose, E.; Tersteeg, C.; Vanhoorelbeke, K.; Hoylaerts, M.F.; Lijnen, R.H.; Scroyen, I. ADAMTS13 Deficiency Promotes Microthrombosis in a Murine Model of Diet-Induced Liver Steatosis. Thromb. Haemost. 2017, 117, 19–26. [Google Scholar] [CrossRef]
- Huang, Y.; Feng, H.; Kan, T.; Huang, B.; Zhang, M.; Li, Y.; Shi, C.; Wu, M.; Luo, Y.; Yang, J.; et al. Bevacizumab Attenuates Hepatic Fibrosis in Rats by Inhibiting Activation of Hepatic Stellate Cells. PLoS ONE 2013, 8, e73492. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lafoz, E.; Ruart, M.; Anton, A.; Oncins, A.; Hernández-Gea, V. The Endothelium as a Driver of Liver Fibrosis and Regeneration. Cells 2020, 9, 929. https://doi.org/10.3390/cells9040929
Lafoz E, Ruart M, Anton A, Oncins A, Hernández-Gea V. The Endothelium as a Driver of Liver Fibrosis and Regeneration. Cells. 2020; 9(4):929. https://doi.org/10.3390/cells9040929
Chicago/Turabian StyleLafoz, Erica, Maria Ruart, Aina Anton, Anna Oncins, and Virginia Hernández-Gea. 2020. "The Endothelium as a Driver of Liver Fibrosis and Regeneration" Cells 9, no. 4: 929. https://doi.org/10.3390/cells9040929
APA StyleLafoz, E., Ruart, M., Anton, A., Oncins, A., & Hernández-Gea, V. (2020). The Endothelium as a Driver of Liver Fibrosis and Regeneration. Cells, 9(4), 929. https://doi.org/10.3390/cells9040929