Potential Therapies Using Myogenic Stem Cells Combined with Bio-Engineering Approaches for Treatment of Muscular Dystrophies


{kind=link}
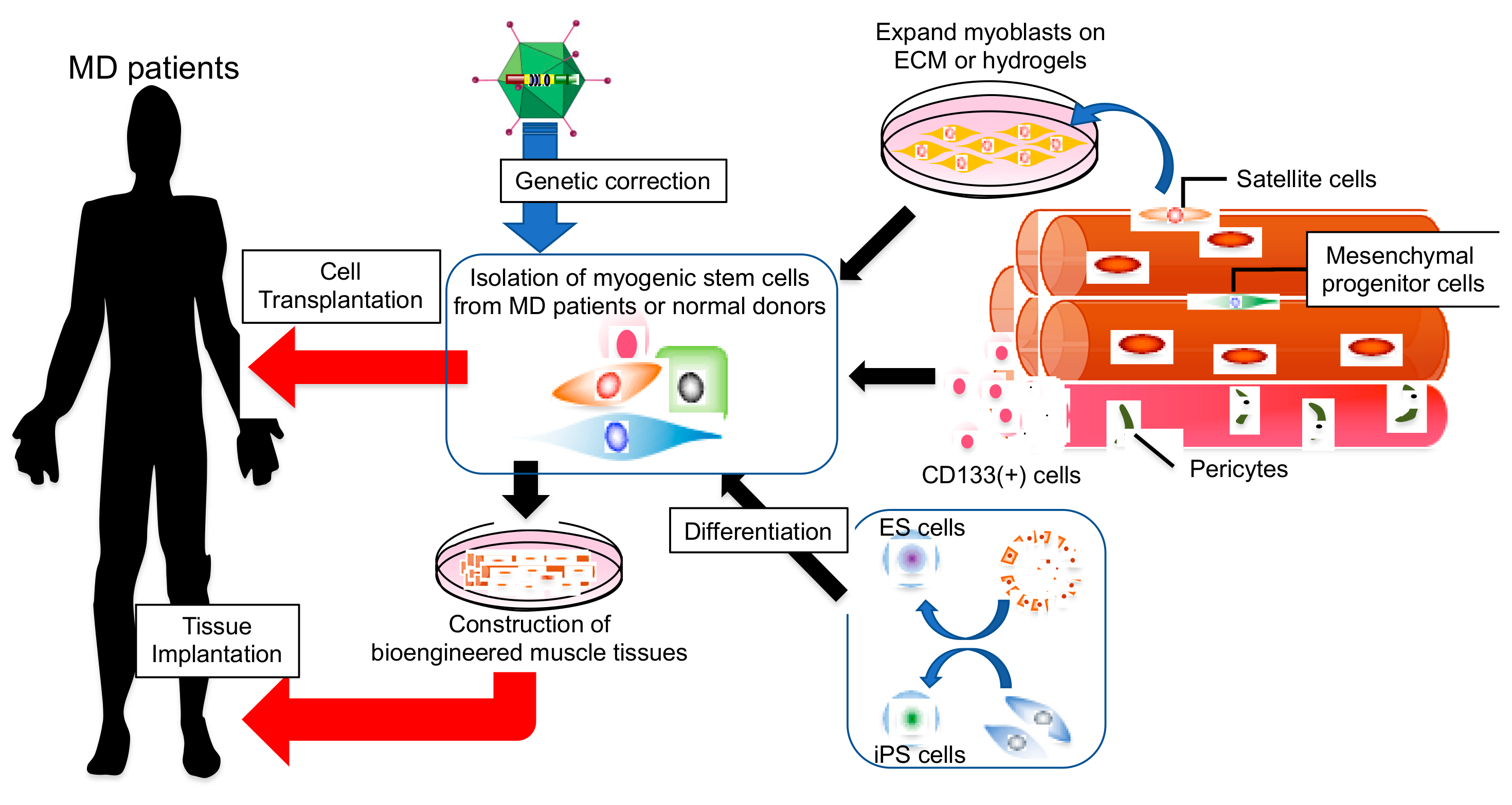
Abstract
1. Introduction
2. Current Status of Myogenic Cell Therapy
3. Cell Sources for Transplantation Therapy
4. Factors That Affect Cell Potential
4.1. Notch Ligands
4.2. Microvascular Environment
4.3. Extracellular Matrix
4.4. Rejuvenation
5. Strategies to Improve or Maintain Myogenic Potential
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Emery, A.E. The muscular dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef]
- Stark, A.E. Determinants of the incidence of Duchenne muscular dystrophy. Ann. Transl. Med. 2015, 3, 287. [Google Scholar] [PubMed]
- Mercuri, E.; Muntoni, F. Muscular dystrophies. Lancet 2013, 381, 845–860. [Google Scholar] [CrossRef]
- Monaco, A.P.; Bertelson, C.J.; Middlesworth, W.; Colletti, C.A.; Aldridge, J.; Fischbeck, K.H.; Bartlett, R.; Pericak-Vance, M.A.; Roses, A.D.; Kunkel, L.M. Detection of deletions spanning the Duchenne muscular dystrophy locus using a tightly linked DNA segment. Nature 1985, 316, 842–845. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Brown, R.H.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.; Beggs, A.H.; Moyer, M.; Scherpf, S.; Heindrich, K.; Bettecken, T.; Meng, G.; Muller, C.R.; Lindlof, M.; Kaariainen, H.; et al. The molecular basis for Duchenne versus Becker muscular dystrophy: Correlation of severity with type of deletion. Am. J. Hum. Genet. 1989, 45, 498–506. [Google Scholar] [PubMed]
- Rahimov, F.; Kunkel, L.M. The cell biology of disease: Cellular and molecular mechanisms underlying muscular dystrophy. J. Cell Biol. 2013, 201, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Walton, J.N.; Nattrass, F.J. On the classification, natural history and treatment of the myopathies. Brain 1954, 77, 169–231. [Google Scholar] [CrossRef] [PubMed]
- Bonne, G.; Di Barletta, M.R.; Varnous, S.; Becane, H.M.; Hammouda, E.H.; Merlini, L.; Muntoni, F.; Greenberg, C.R.; Gary, F.; Urtizberea, J.A.; et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet. 1999, 21, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Sanna, T.; Dello Russo, A.; Toniolo, D.; Vytopil, M.; Pelargonio, G.; De Martino, G.; Ricci, E.; Silvestri, G.; Giglio, V.; Messano, L.; et al. Cardiac features of Emery-Dreifuss muscular dystrophy caused by lamin A/C gene mutations. Eur. Hear. J. 2003, 24, 2227–2236. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, Y.; Kawazura, M.; Haruna, H. A peculiar form of congenital progressive muscular dystrophy. Report of fifteen cases. Paediatr Univ. Tokyo (Tokyo) 1960, 4, 5–8. [Google Scholar]
- McDonald, C.M.; Henricson, E.K.; Abresch, R.T.; Duong, T.; Joyce, N.C.; Hu, F.; Clemens, P.R.; Hoffman, E.P.; Cnaan, A.; Gordish-Dressman, H. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: A prospective cohort study. Lancet 2018, 391, 451–461. [Google Scholar] [CrossRef]
- Shimizu-Motohashi, Y.; Komaki, H.; Motohashi, N.; Takeda, S.; Yokota, T.; Aoki, Y. Restoring Dystrophin Expression in Duchenne Muscular Dystrophy: Current Status of Therapeutic Approaches. J. Pers. Med. 2019, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Verhaart, I.E.C.; Aartsma-Rus, A. Therapeutic developments for Duchenne muscular dystrophy. Nat. Rev. Neurol. 2019, 15, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A. Satellite cell of skeletal muscle fibers. J. Cell Boil. 1961, 9, 493–495. [Google Scholar] [CrossRef]
- Tedesco, F.S.; Dellavalle, A.; Diaz-Manera, J.; Messina, G.; Cossu, G. Repairing skeletal muscle: Regenerative potential of skeletal muscle stem cells. J. Clin. Investig. 2010, 120, 11–19. [Google Scholar] [CrossRef]
- Partridge, T.A.; Morgan, J.E.; Coulton, G.R.; Hoffman, E.P.; Kunkel, L.M. Conversion of mdx myofibres from dystrophin-negative to -positive by injection of normal myoblasts. Nature 1989, 337, 176–179. [Google Scholar] [CrossRef]
- Karpati, G.; Pouliot, Y.; Zubrzycka-Gaarn, E.; Carpenter, S.; Ray, P.N.; Worton, R.G.; Holland, P. Dystrophin is expressed in mdx skeletal muscle fibers after normal myoblast implantation. Am. J. Pathol. 1989, 135, 27–32. [Google Scholar]
- Gussoni, E.; Pavlath, G.K.; Lanctot, A.M.; Sharma, K.R.; Miller, R.G.; Steinman, L.; Blau, H.M. Normal dystrophin transcripts detected in Duchenne muscular dystrophy patients after myoblast transplantation. Nature 1992, 356, 435–438. [Google Scholar] [CrossRef]
- Huard, J.; Bouchard, J.P.; Roy, R.; Malouin, F.; Dansereau, G.; Labrecque, C.; Albert, N.; Richards, C.L.; Lemieux, B.; Tremblay, J.P. Human myoblast transplantation: Preliminary results of 4 cases. Muscle Nerve. 1992, 15, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Karpati, G.; Ajdukovic, D.; Arnold, D.; Gledhill, R.B.; Guttmann, R.; Holland, P.; Koch, P.A.; Shoubridge, E.; Spence, D.; Vanasse, M.; et al. Myoblast transfer in Duchenne muscular dystrophy. Ann. Neurol. 1993, 34, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Kissel, J.T.; Amato, A.A.; King, W.; Signore, L.; Prior, T.W.; Sahenk, Z.; Benson, S.; McAndrew, P.E.; Rice, R.; et al. Myoblast transfer in the treatment of Duchenne’s muscular dystrophy. N. Engl. J. Med. 1995, 333, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, I.; Vilquin, J.T.; Guerette, B.; Asselin, I.; Roy, R.; Tremblay, J.P. Very efficient myoblast allotransplantation in mice under FK506 immunosuppression. Muscle Nerve. 1994, 17, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, I.; Roy, R.; Dugre, F.J.; Gravel, C.; Roy, B.; Goulet, M.; Asselin, I.; Tremblay, J.P. Myoblast transplantation in monkeys: Control of immune response by FK506. J. Neuropathol. Exp. Neurol. 1996, 55, 687–697. [Google Scholar] [CrossRef]
- Skuk, D.; Roy, B.; Goulet, M.; Chapdelaine, P.; Bouchard, J.P.; Roy, R.; Dugre, F.J.; Lachance, J.G.; Deschenes, L.; Helene, S.; et al. Dystrophin expression in myofibers of Duchenne muscular dystrophy patients following intramuscular injections of normal myogenic cells. Mol. Ther. 2004, 9, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Skuk, D.; Goulet, M.; Roy, B.; Chapdelaine, P.; Bouchard, J.P.; Roy, R.; Dugre, F.J.; Sylvain, M.; Lachance, J.G.; Deschenes, L.; et al. Dystrophin expression in muscles of duchenne muscular dystrophy patients after high-density injections of normal myogenic cells. J. Neuropathol. Exp. Neurol. 2006, 65, 371–386. [Google Scholar] [CrossRef]
- Tremblay, J.P.; Skuk, D.; Palmieri, B.; Rothstein, D.M. A case for immunosuppression for myoblast transplantation in duchenne muscular dystrophy. Mol. Ther. 2009, 17, 1122–1124. [Google Scholar] [CrossRef]
- Quenneville, S.P.; Chapdelaine, P.; Rousseau, J.; Beaulieu, J.; Caron, N.J.; Skuk, D.; Mills, P.; Olivares, E.C.; Calos, M.P.; Tremblay, J.P. Nucleofection of muscle-derived stem cells and myoblasts with phiC31 integrase: Stable expression of a full-length-dystrophin fusion gene by human myoblasts. Mol. Ther. 2004, 10, 679–687. [Google Scholar] [CrossRef]
- Li, S.; Kimura, E.; Fall, B.M.; Reyes, M.; Angello, J.C.; Welikson, R.; Hauschka, S.D.; Chamberlain, J.S. Stable transduction of myogenic cells with lentiviral vectors expressing a minidystrophin. Gene. Ther. 2005, 12, 1099–1108. [Google Scholar] [CrossRef]
- Quenneville, S.P.; Chapdelaine, P.; Skuk, D.; Paradis, M.; Goulet, M.; Rousseau, J.; Xiao, X.; Garcia, L.; Tremblay, J.P. Autologous transplantation of muscle precursor cells modified with a lentivirus for muscular dystrophy: Human cells and primate models. Mol. Ther. 2007, 15, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.; Blau, H.M. Accelerated age-related decline in replicative life-span of Duchenne muscular dystrophy myoblasts: Implications for cell and gene therapy. Somat. Cell Mol. Genet. 1990, 16, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Ikemoto, M.; Fukada, S.; Uezumi, A.; Masuda, S.; Miyoshi, H.; Yamamoto, H.; Wada, M.R.; Masubuchi, N.; Miyagoe-Suzuki, Y.; Takeda, S. Autologous transplantation of SM/C-2.6(+) satellite cells transduced with micro-dystrophin CS1 cDNA by lentiviral vector into mdx mice. Mol. Ther. 2007, 15, 2178–2185. [Google Scholar] [CrossRef] [PubMed]
- Barthelemy, F.; Wein, N. Personalized gene and cell therapy for Duchenne Muscular Dystrophy. Neuromuscul Disord 2018, 28, 803–824. [Google Scholar] [CrossRef] [PubMed]
- Wilschut, K.J.; Ling, V.B.; Bernstein, H.S. Concise review: Stem cell therapy for muscular dystrophies. Stem Cells Transl. Med. 2012, 1, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Perie, S.; Trollet, C.; Mouly, V.; Vanneaux, V.; Mamchaoui, K.; Bouazza, B.; Marolleau, J.P.; Laforet, P.; Chapon, F.; Eymard, B.; et al. Autologous myoblast transplantation for oculopharyngeal muscular dystrophy: A phase I/IIa clinical study. Mol. Ther. 2014, 22, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Cossu, G.; Previtali, S.C.; Napolitano, S.; Cicalese, M.P.; Tedesco, F.S.; Nicastro, F.; Noviello, M.; Roostalu, U.; Natali Sora, M.G.; Scarlato, M.; et al. Intra-arterial transplantation of HLA-matched donor mesoangioblasts in Duchenne muscular dystrophy. Embo Mol. Med. 2015, 7, 1513–1528. [Google Scholar] [CrossRef]
- Gussoni, E.; Soneoka, Y.; Strickland, C.D.; Buzney, E.A.; Khan, M.K.; Flint, A.F.; Kunkel, L.M.; Mulligan, R.C. Dystrophin expression in the mdx mouse restored by stem cell transplantation. Nature 1999, 401, 390–394. [Google Scholar] [CrossRef]
- Jackson, K.A.; Mi, T.; Goodell, M.A. Hematopoietic potential of stem cells isolated from murine skeletal muscle. Proc. Natl. Acad. Sci. USA 1999, 96, 14482–14486. [Google Scholar] [CrossRef]
- Qu-Petersen, Z.; Deasy, B.; Jankowski, R.; Ikezawa, M.; Cummins, J.; Pruchnic, R.; Mytinger, J.; Cao, B.; Gates, C.; Wernig, A.; et al. Identification of a novel population of muscle stem cells in mice: Potential for muscle regeneration. J. Cell Biol. 2002, 157, 851–864. [Google Scholar] [CrossRef]
- Jiang, Y.; Vaessen, B.; Lenvik, T.; Blackstad, M.; Reyes, M.; Verfaillie, C.M. Multipotent progenitor cells can be isolated from postnatal murine bone marrow, muscle, and brain. Exp. Hematol. 2002, 30, 896–904. [Google Scholar] [CrossRef]
- Tamaki, T.; Akatsuka, A.; Ando, K.; Nakamura, Y.; Matsuzawa, H.; Hotta, T.; Roy, R.R.; Edgerton, V.R. Identification of myogenic-endothelial progenitor cells in the interstitial spaces of skeletal muscle. J. Cell Biol. 2002, 157, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Torrente, Y.; Belicchi, M.; Sampaolesi, M.; Pisati, F.; Meregalli, M.; D’Antona, G.; Tonlorenzi, R.; Porretti, L.; Gavina, M.; Mamchaoui, K.; et al. Human circulating AC133(+) stem cells restore dystrophin expression and ameliorate function in dystrophic skeletal muscle. J. Clin. Investig. 2004, 114, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Torrente, Y.; Belicchi, M.; Marchesi, C.; D’Antona, G.; Cogiamanian, F.; Pisati, F.; Gavina, M.; Giordano, R.; Tonlorenzi, R.; Fagiolari, G.; et al. Autologous transplantation of muscle-derived CD133+ stem cells in Duchenne muscle patients. Cell Transplant. 2007, 16, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Benchaouir, R.; Meregalli, M.; Farini, A.; D’Antona, G.; Belicchi, M.; Goyenvalle, A.; Battistelli, M.; Bresolin, N.; Bottinelli, R.; Garcia, L.; et al. Restoration of human dystrophin following transplantation of exon-skipping-engineered DMD patient stem cells into dystrophic mice. Cell Stem Cell 2007, 1, 646–657. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Chun, S.; Asfahani, R.; Lochmuller, H.; Muntoni, F.; Morgan, J. Human skeletal muscle-derived CD133(+) cells form functional satellite cells after intramuscular transplantation in immunodeficient host mice. Mol. Ther. 2014, 22, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Minasi, M.G.; Riminucci, M.; De Angelis, L.; Borello, U.; Berarducci, B.; Innocenzi, A.; Caprioli, A.; Sirabella, D.; Baiocchi, M.; De Maria, R.; et al. The meso-angioblast: A multipotent, self-renewing cell that originates from the dorsal aorta and differentiates into most mesodermal tissues. Development 2002, 129, 2773–2783. [Google Scholar] [PubMed]
- Sampaolesi, M.; Torrente, Y.; Innocenzi, A.; Tonlorenzi, R.; D’Antona, G.; Pellegrino, M.A.; Barresi, R.; Bresolin, N.; De Angelis, M.G.; Campbell, K.P.; et al. Cell therapy of alpha-sarcoglycan null dystrophic mice through intra-arterial delivery of mesoangioblasts. Science 2003, 301, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Sampaolesi, M.; Blot, S.; D’Antona, G.; Granger, N.; Tonlorenzi, R.; Innocenzi, A.; Mognol, P.; Thibaud, J.L.; Galvez, B.G.; Barthelemy, I.; et al. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature 2006, 444, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Dellavalle, A.; Sampaolesi, M.; Tonlorenzi, R.; Tagliafico, E.; Sacchetti, B.; Perani, L.; Innocenzi, A.; Galvez, B.G.; Messina, G.; Morosetti, R.; et al. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat. Cell Biol. 2007, 9, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Muntoni, F.; Morgan, J. CD133+ cells derived from skeletal muscles of Duchenne muscular dystrophy patients have a compromised myogenic and muscle regenerative capability. Stem Cell Res. 2018, 30, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Counsell, J.R.; Reza, M.; Laval, S.H.; Danos, O.; Thrasher, A.; Lochmuller, H.; Muntoni, F.; Morgan, J.E. Autologous skeletal muscle derived cells expressing a novel functional dystrophin provide a potential therapy for Duchenne Muscular Dystrophy. Sci. Rep. 2016, 6, 19750. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, K.M.; Campbell, K.; Viollet, L.; Wang, W.; Gomez, A.M.; Walker, C.M.; Mendell, J.R. Anti-dystrophin T cell responses in Duchenne muscular dystrophy: Prevalence and a glucocorticoid treatment effect. Hum. Gene. Ther. 2013, 24, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Mantegazza, R.; Hughes, S.M.; Mitchell, D.; Travis, M.; Blau, H.M.; Steinman, L. Modulation of MHC class II antigen expression in human myoblasts after treatment with IFN-gamma. Neurology 1991, 41, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Englund, P.; Lindroos, E.; Nennesmo, I.; Klareskog, L.; Lundberg, I.E. Skeletal muscle fibers express major histocompatibility complex class II antigens independently of inflammatory infiltrates in inflammatory myopathies. Am. J. Pathol. 2001, 159, 1263–1273. [Google Scholar] [CrossRef]
- Rosenberg, A.S.; Puig, M.; Nagaraju, K.; Hoffman, E.P.; Villalta, S.A.; Rao, V.A.; Wakefield, L.M.; Woodcock, J. Immune-mediated pathology in Duchenne muscular dystrophy. Sci Transl Med. 2015, 7, 299rv4. [Google Scholar] [CrossRef] [PubMed]
- English, K.; Tonlorenzi, R.; Cossu, G.; Wood, K.J. Mesoangioblasts suppress T cell proliferation through IDO and PGE-2-dependent pathways. Stem Cells Dev. 2013, 22, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Li, O.; English, K.; Tonlorenzi, R.; Cossu, G.; Saverio Tedesco, F.; Wood, K.J. Human iPSC-derived mesoangioblasts, like their tissue-derived counterparts, suppress T cell proliferation through IDO- and PGE-2-dependent pathways. F1000Research 2013, 2, 24. [Google Scholar] [CrossRef]
- Galvez, B.G.; Sampaolesi, M.; Brunelli, S.; Covarello, D.; Gavina, M.; Rossi, B.; Constantin, G.; Torrente, Y.; Cossu, G. Complete repair of dystrophic skeletal muscle by mesoangioblasts with enhanced migration ability. J. Cell Biol. 2006, 174, 231–243. [Google Scholar] [CrossRef]
- Furlani, D.; Ugurlucan, M.; Ong, L.; Bieback, K.; Pittermann, E.; Westien, I.; Wang, W.; Yerebakan, C.; Li, W.; Gaebel, R.; et al. Is the intravascular administration of mesenchymal stem cells safe? Mesenchymal stem cells and intravital microscopy. Microvasc. Res. 2009, 77, 370–376. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, F.S.; Gerli, M.F.; Perani, L.; Benedetti, S.; Ungaro, F.; Cassano, M.; Antonini, S.; Tagliafico, E.; Artusi, V.; Longa, E.; et al. Transplantation of genetically corrected human iPSC-derived progenitors in mice with limb-girdle muscular dystrophy. Sci. Transl. Med. 2012, 4, 140ra89. [Google Scholar] [CrossRef] [PubMed]
- Filareto, A.; Parker, S.; Darabi, R.; Borges, L.; Iacovino, M.; Schaaf, T.; Mayerhofer, T.; Chamberlain, J.S.; Ervasti, J.M.; McIvor, R.S.; et al. An ex vivo gene therapy approach to treat muscular dystrophy using inducible pluripotent stem cells. Nat. Commun. 2013, 4, 1549. [Google Scholar] [CrossRef] [PubMed]
- Young, C.S.; Hicks, M.R.; Ermolova, N.V.; Nakano, H.; Jan, M.; Younesi, S.; Karumbayaram, S.; Kumagai-Cresse, C.; Wang, D.; Zack, J.A.; et al. A Single CRISPR-Cas9 Deletion Strategy that Targets the Majority of DMD Patients Restores Dystrophin Function in hiPSC-Derived Muscle Cells. Cell Stem Cell 2016, 18, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Magli, A.; Chan, S.S.K.; Oliveira, V.K.P.; Wu, J.; Darabi, R.; Kyba, M.; Perlingeiro, R.C.R. Expansion and Purification Are Critical for the Therapeutic Application of Pluripotent Stem Cell-Derived Myogenic Progenitors. Stem Cell Rep. 2017, 9, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Hicks, M.R.; Hiserodt, J.; Paras, K.; Fujiwara, W.; Eskin, A.; Jan, M.; Xi, H.; Young, C.S.; Evseenko, D.; Nelson, S.F.; et al. ERBB3 and NGFR mark a distinct skeletal muscle progenitor cell in human development and hPSCs. Nat. Cell Biol. 2018, 20, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Matthias, N.; Lo, J.; Ortiz-Vitali, J.L.; Shieh, A.W.; Wang, S.H.; Darabi, R. A Myogenic Double-Reporter Human Pluripotent Stem Cell Line Allows Prospective Isolation of Skeletal Muscle Progenitors. Cell Rep. 2018, 25, 1966–1981.e4. [Google Scholar] [CrossRef]
- Magli, A.; Incitti, T.; Kiley, J.; Swanson, S.A.; Darabi, R.; Rinaldi, F.; Selvaraj, S.; Yamamoto, A.; Tolar, J.; Yuan, C.; et al. PAX7 Targets, CD54, Integrin alpha9beta1, and SDC2, Allow Isolation of Human ESC/iPSC-Derived Myogenic Progenitors. Cell Rep. 2017, 19, 2867–2877. [Google Scholar] [CrossRef]
- Montarras, D.; Morgan, J.; Collins, C.; Relaix, F.; Zaffran, S.; Cumano, A.; Partridge, T.; Buckingham, M. Direct isolation of satellite cells for skeletal muscle regeneration. Science 2005, 309, 2064–2067. [Google Scholar] [CrossRef] [PubMed]
- Sacco, A.; Doyonnas, R.; Kraft, P.; Vitorovic, S.; Blau, H.M. Self-renewal and expansion of single transplanted muscle stem cells. Nature 2008, 456, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Day, K.; Shefer, G.; Shearer, A.; Yablonka-Reuveni, Z. The depletion of skeletal muscle satellite cells with age is concomitant with reduced capacity of single progenitors to produce reserve progeny. Dev. Biol. 2010, 340, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Sacco, A.; Mourkioti, F.; Tran, R.; Choi, J.; Llewellyn, M.; Kraft, P.; Shkreli, M.; Delp, S.; Pomerantz, J.H.; Artandi, S.E.; et al. Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell 2010, 143, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Conboy, I.M.; Conboy, M.J.; Wagers, A.J.; Girma, E.R.; Weissman, I.L.; Rando, T.A. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 2005, 433, 760–764. [Google Scholar] [CrossRef]
- Ikemoto-Uezumi, M.; Uezumi, A.; Tsuchida, K.; Fukada, S.; Yamamoto, H.; Yamamoto, N.; Shiomi, K.; Hashimoto, N. Pro-Insulin-Like Growth Factor-II Ameliorates Age-Related Inefficient Regenerative Response by Orchestrating Self-Reinforcement Mechanism of Muscle Regeneration. Stem Cells 2015, 33, 2456–2468. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, D.; Gutierrez, J.; Cabello-Verrugio, C.; Morales, M.G.; Mezzano, S.; Fadic, R.; Casar, J.C.; Hancke, J.L.; Brandan, E. Andrographolide attenuates skeletal muscle dystrophy in mdx mice and increases efficiency of cell therapy by reducing fibrosis. Skelet Muscle 2014, 4, 6. [Google Scholar] [CrossRef]
- Brunelli, S.; Sciorati, C.; D’Antona, G.; Innocenzi, A.; Covarello, D.; Galvez, B.G.; Perrotta, C.; Monopoli, A.; Sanvito, F.; Bottinelli, R.; et al. Nitric oxide release combined with nonsteroidal antiinflammatory activity prevents muscular dystrophy pathology and enhances stem cell therapy. Proc. Natl. Acad. Sci. USA 2007, 104, 264–269. [Google Scholar] [CrossRef]
- Ohlstein, B.; Kai, T.; Decotto, E.; Spradling, A. The stem cell niche: Theme and variations. Curr. Opin. Cell Biol. 2004, 16, 693–699. [Google Scholar] [CrossRef]
- Scadden, D.T. The stem-cell niche as an entity of action. Nature 2006, 441, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.C. Notch signaling: Control of cell communication and cell fate. Development 2004, 131, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Mourikis, P.; Sambasivan, R.; Castel, D.; Rocheteau, P.; Bizzarro, V.; Tajbakhsh, S. A critical requirement for notch signaling in maintenance of the quiescent skeletal muscle stem cell state. Stem Cells 2012, 30, 243–252. [Google Scholar] [CrossRef]
- Bjornson, C.R.; Cheung, T.H.; Liu, L.; Tripathi, P.V.; Steeper, K.M.; Rando, T.A. Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells 2012, 30, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Conboy, I.M.; Rando, T.A. The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev. Cell 2002, 3, 397–409. [Google Scholar] [CrossRef]
- Parker, M.H.; Loretz, C.; Tyler, A.E.; Duddy, W.J.; Hall, J.K.; Olwin, B.B.; Bernstein, I.D.; Storb, R.; Tapscott, S.J. Activation of Notch signaling during ex vivo expansion maintains donor muscle cell engraftment. Stem Cells 2012, 30, 2212–2220. [Google Scholar] [CrossRef] [PubMed]
- Vieira, N.M.; Elvers, I.; Alexander, M.S.; Moreira, Y.B.; Eran, A.; Gomes, J.P.; Marshall, J.L.; Karlsson, E.K.; Verjovski-Almeida, S.; Lindblad-Toh, K.; et al. Jagged 1 Rescues the Duchenne Muscular Dystrophy Phenotype. Cell 2015, 163, 1204–1213. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Fukuda, S.; Nakamura, M.; Uezumi, A.; Noguchi, Y.T.; Sato, T.; Morita, M.; Yamada, H.; Tsuchida, K.; Tajbakhsh, S.; et al. Notch ligands regulate the muscle stem-like state ex vivo but are not sufficient for retaining regenerative capacity. PLoS ONE 2017, 12, 0177516. [Google Scholar] [CrossRef] [PubMed]
- Christov, C.; Chretien, F.; Abou-Khalil, R.; Bassez, G.; Vallet, G.; Authier, F.J.; Bassaglia, Y.; Shinin, V.; Tajbakhsh, S.; Chazaud, B.; et al. Muscle satellite cells and endothelial cells: Close neighbors and privileged partners. Mol. Biol. Cell 2007, 18, 1397–1409. [Google Scholar] [CrossRef]
- Abou-Khalil, R.; Le Grand, F.; Pallafacchina, G.; Valable, S.; Authier, F.J.; Rudnicki, M.A.; Gherardi, R.K.; Germain, S.; Chretien, F.; Sotiropoulos, A.; et al. Autocrine and paracrine angiopoietin 1/Tie-2 signaling promotes muscle satellite cell self-renewal. Cell Stem Cell 2009, 5, 298–309. [Google Scholar] [CrossRef]
- Verma, M.; Asakura, Y.; Murakonda, B.S.R.; Pengo, T.; Latroche, C.; Chazaud, B.; McLoon, L.K.; Asakura, A. Muscle Satellite Cell Cross-Talk with a Vascular Niche Maintains Quiescence via VEGF and Notch Signaling. Cell Stem Cell 2018, 23, 530–543. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Asakura, Y.; Hirai, H.; Watanabe, S.; Tastad, C.; Fong, G.H.; Ema, M.; Call, J.A.; Lowe, D.A.; Asakura, A. Flt-1 haploinsufficiency ameliorates muscular dystrophy phenotype by developmentally increased vasculature in mdx mice. Hum. Mol. Genet. 2010, 19, 4145–4159. [Google Scholar] [CrossRef] [PubMed]
- Miyatake, M.; Miike, T.; Zhao, J.; Yoshioka, K.; Uchino, M.; Usuku, G. Possible systemic smooth muscle layer dysfunction due to a deficiency of dystrophin in Duchenne muscular dystrophy. J. Neurol. Sci. 1989, 93, 11–17. [Google Scholar] [CrossRef]
- Ito, K.; Kimura, S.; Ozasa, S.; Matsukura, M.; Ikezawa, M.; Yoshioka, K.; Ueno, H.; Suzuki, M.; Araki, K.; Yamamura, K.; et al. Smooth muscle-specific dystrophin expression improves aberrant vasoregulation in mdx mice. Hum. Mol. Genet. 2006, 15, 2266–2275. [Google Scholar] [CrossRef] [PubMed]
- Loufrani, L.; Dubroca, C.; You, D.; Li, Z.; Levy, B.; Paulin, D.; Henrion, D. Absence of dystrophin in mice reduces NO-dependent vascular function and vascular density: Total recovery after a treatment with the aminoglycoside gentamicin. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Matsakas, A.; Yadav, V.; Lorca, S.; Narkar, V. Muscle ERRgamma mitigates Duchenne muscular dystrophy via metabolic and angiogenic reprogramming. FASEB J. 2013, 27, 4004–4016. [Google Scholar] [CrossRef]
- Kherif, S.; Lafuma, C.; Dehaupas, M.; Lachkar, S.; Fournier, J.G.; Verdiere-Sahuque, M.; Fardeau, M.; Alameddine, H.S. Expression of matrix metalloproteinases 2 and 9 in regenerating skeletal muscle: A study in experimentally injured and mdx muscles. Dev. Biol. 1999, 205, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, N.; Uezumi, A.; Yada, E.; Fukada, S.; Fukushima, K.; Imaizumi, K.; Miyagoe-Suzuki, Y.; Takeda, S. Muscle CD31(−) CD45(−) side population cells promote muscle regeneration by stimulating proliferation and migration of myoblasts. Am. J. Pathol. 2008, 173, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Takahashi, R.; Adachi, E.; Kondo, S.; Kuratomi, S.; Noma, A.; Alexander, D.B.; Motoda, H.; Okada, A.; Seiki, M.; et al. Mutations in two matrix metalloproteinase genes, MMP-2 and MT1-MMP, are synthetic lethal in mice. Oncogene 2004, 23, 5041–5048. [Google Scholar] [CrossRef]
- El Fahime, E.; Torrente, Y.; Caron, N.J.; Bresolin, M.D.; Tremblay, J.P. In vivo migration of transplanted myoblasts requires matrix metalloproteinase activity. Exp. Cell Res. 2000, 258, 279–287. [Google Scholar] [CrossRef]
- Gargioli, C.; Coletta, M.; De Grandis, F.; Cannata, S.M.; Cossu, G. PlGF-MMP-9-expressing cells restore microcirculation and efficacy of cell therapy in aged dystrophic muscle. Nat. Med. 2008, 14, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Fry, C.S.; Lee, J.D.; Mula, J.; Kirby, T.J.; Jackson, J.R.; Liu, F.; Yang, L.; Mendias, C.L.; Dupont-Versteegden, E.E.; McCarthy, J.J.; et al. Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nat. Med. 2015, 21, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Desguerre, I.; Mayer, M.; Leturcq, F.; Barbet, J.P.; Gherardi, R.K.; Christov, C. Endomysial fibrosis in Duchenne muscular dystrophy: A marker of poor outcome associated with macrophage alternative activation. J. Neuropathol. Exp. Neurol. 2009, 68, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Huebner, K.D.; Jassal, D.S.; Halevy, O.; Pines, M.; Anderson, J.E. Functional resolution of fibrosis in mdx mouse dystrophic heart and skeletal muscle by halofuginone. Am. J. Physiology. Heart Circ. Physiol. 2008, 294, H1550–H1561. [Google Scholar] [CrossRef] [PubMed]
- Shimizu-Motohashi, Y.; Miyatake, S.; Komaki, H.; Takeda, S.; Aoki, Y. Recent advances in innovative therapeutic approaches for Duchenne muscular dystrophy: From discovery to clinical trials. Am. J. Transl. Res. 2016, 8, 2471–2489. [Google Scholar] [PubMed]
- Fakhfakh, R.; Lamarre, Y.; Skuk, D.; Tremblay, J.P. Losartan enhances the success of myoblast transplantation. Cell Transplant. 2012, 21, 139–152. [Google Scholar] [CrossRef]
- Lee, E.M.; Kim, A.Y.; Lee, E.J.; Park, J.K.; Lee, M.M.; Hwang, M.; Kim, C.Y.; Kim, S.Y.; Jeong, K.S. Therapeutic effects of mouse adipose-derived stem cells and losartan in the skeletal muscle of injured mdx mice. Cell Transplant. 2015, 24, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Morales, M.G.; Gutierrez, J.; Cabello-Verrugio, C.; Cabrera, D.; Lipson, K.E.; Goldschmeding, R.; Brandan, E. Reducing CTGF/CCN2 slows down mdx muscle dystrophy and improves cell therapy. Hum. Mol. Genet. 2013, 22, 4938–4951. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.J.; Hansen, J.M.; Merrell, A.J.; Murphy, M.M.; Lawson, J.A.; Hutcheson, D.A.; Hansen, M.S.; Angus-Hill, M.; Kardon, G. Connective tissue fibroblasts and Tcf4 regulate myogenesis. Development 2011, 138, 371–384. [Google Scholar] [CrossRef]
- Murphy, M.M.; Lawson, J.A.; Mathew, S.J.; Hutcheson, D.A.; Kardon, G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 2011, 138, 3625–3637. [Google Scholar] [CrossRef]
- Urciuolo, A.; Quarta, M.; Morbidoni, V.; Gattazzo, F.; Molon, S.; Grumati, P.; Montemurro, F.; Tedesco, F.S.; Blaauw, B.; Cossu, G.; et al. Collagen VI regulates satellite cell self-renewal and muscle regeneration. Nat. Commun. 2013, 4, 1964. [Google Scholar] [CrossRef]
- Baghdadi, M.B.; Castel, D.; Machado, L.; Fukada, S.I.; Birk, D.E.; Relaix, F.; Tajbakhsh, S.; Mourikis, P. Reciprocal signalling by Notch-Collagen V-CALCR retains muscle stem cells in their niche. Nature 2018, 557, 714–718. [Google Scholar] [CrossRef]
- Miner, J.H.; Yurchenco, P.D. Laminin functions in tissue morphogenesis. Annu. Rev. Cell Dev. Biol. 2004, 20, 255–284. [Google Scholar] [CrossRef] [PubMed]
- Rooney, J.E.; Gurpur, P.B.; Yablonka-Reuveni, Z.; Burkin, D.J. Laminin-111 restores regenerative capacity in a mouse model for alpha7 integrin congenital myopathy. Am. J. Pathol. 2009, 174, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Rayagiri, S.S.; Ranaldi, D.; Raven, A.; Mohamad Azhar, N.I.F.; Lefebvre, O.; Zammit, P.S.; Borycki, A.G. Basal lamina remodeling at the skeletal muscle stem cell niche mediates stem cell self-renewal. Nat. Commun. 2018, 9, 1075. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.; Sakurai, H.; Suzuki, N.; Mabuchi, Y.; Sekiya, I.; Sekiguchi, K.; Akazawa, C. Recapitulation of Extracellular LAMININ Environment Maintains Stemness of Satellite Cells In Vitro. Stem Cell Rep. 2018, 10, 568–582. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; Wang, Y.X.; von Maltzahn, J.; Soleimani, V.D.; Yin, H.; Rudnicki, M.A. Fibronectin regulates Wnt7a signaling and satellite cell expansion. Cell Stem Cell 2013, 12, 75–87. [Google Scholar] [CrossRef]
- Gilbert, P.M.; Havenstrite, K.L.; Magnusson, K.E.; Sacco, A.; Leonardi, N.A.; Kraft, P.; Nguyen, N.K.; Thrun, S.; Lutolf, M.P.; Blau, H.M. Substrate elasticity regulates skeletal muscle stem cell self-renewal in culture. Science 2010, 329, 1078–1081. [Google Scholar] [CrossRef]
- Quarta, M.; Cromie, M.; Chacon, R.; Blonigan, J.; Garcia, V.; Akimenko, I.; Hamer, M.; Paine, P.; Stok, M.; Shrager, J.B.; et al. Bioengineered constructs combined with exercise enhance stem cell-mediated treatment of volumetric muscle loss. Nat. Commun. 2017, 8, 15613. [Google Scholar] [CrossRef]
- Bernet, J.D.; Doles, J.D.; Hall, J.K.; Kelly Tanaka, K.; Carter, T.A.; Olwin, B.B. p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat. Med. 2014, 20, 265–271. [Google Scholar] [CrossRef]
- Cosgrove, B.D.; Gilbert, P.M.; Porpiglia, E.; Mourkioti, F.; Lee, S.P.; Corbel, S.Y.; Llewellyn, M.E.; Delp, S.L.; Blau, H.M. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat. Med. 2014, 20, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Price, F.D.; von Maltzahn, J.; Bentzinger, C.F.; Dumont, N.A.; Yin, H.; Chang, N.C.; Wilson, D.H.; Frenette, J.; Rudnicki, M.A. Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat. Med. 2014, 20, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Judson, R.N.; Quarta, M.; Oudhoff, M.J.; Soliman, H.; Yi, L.; Chang, C.K.; Loi, G.; Vander Werff, R.; Cait, A.; Hamer, M.; et al. Inhibition of Methyltransferase Setd7 Allows the In Vitro Expansion of Myogenic Stem Cells with Improved Therapeutic Potential. Cell Stem Cell 2018, 22, 177–190.e7. [Google Scholar] [CrossRef] [PubMed]
- Lev, R.; Seliktar, D. Hydrogel biomaterials and their therapeutic potential for muscle injuries and muscular dystrophies. J. R. Soc. Interface 2018, 15. [Google Scholar] [CrossRef] [PubMed]
- Rossi, C.A.; Flaibani, M.; Blaauw, B.; Pozzobon, M.; Tallo, E.; Reggiani, C.; Vitiello, L.; Elvassore, N.; De Coppi, P. In vivo tissue engineering of functional skeletal muscle by freshly isolated satellite cells embedded in a photopolymerizable hydrogel. FASEB J. 2011, 25, 2296–2304. [Google Scholar] [CrossRef]
- Fuoco, C.; Salvatori, M.L.; Biondo, A.; Shapira-Schweitzer, K.; Santoleri, S.; Antonini, S.; Bernardini, S.; Tedesco, F.S.; Cannata, S.; Seliktar, D.; et al. Injectable polyethylene glycol-fibrinogen hydrogel adjuvant improves survival and differentiation of transplanted mesoangioblasts in acute and chronic skeletal-muscle degeneration. Skelet Muscle 2012, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- DeQuach, J.A.; Lin, J.E.; Cam, C.; Hu, D.; Salvatore, M.A.; Sheikh, F.; Christman, K.L. Injectable skeletal muscle matrix hydrogel promotes neovascularization and muscle cell infiltration in a hindlimb ischemia model. Eur. Cells Mater. 2012, 23, 400–412. [Google Scholar] [CrossRef]
- Wang, L.; Cao, L.; Shansky, J.; Wang, Z.; Mooney, D.; Vandenburgh, H. Minimally invasive approach to the repair of injured skeletal muscle with a shape-memory scaffold. Mol. Ther. 2014, 22, 1441–1449. [Google Scholar] [CrossRef]
- Fuoco, C.; Rizzi, R.; Biondo, A.; Longa, E.; Mascaro, A.; Shapira-Schweitzer, K.; Kossovar, O.; Benedetti, S.; Salvatori, M.L.; Santoleri, S.; et al. In vivo generation of a mature and functional artificial skeletal muscle. Embo Mol. Med. 2015, 7, 411–422. [Google Scholar] [CrossRef]
- Maffioletti, S.M.; Sarcar, S.; Henderson, A.B.H.; Mannhardt, I.; Pinton, L.; Moyle, L.A.; Steele-Stallard, H.; Cappellari, O.; Wells, K.E.; Ferrari, G.; et al. Three-Dimensional Human iPSC-Derived Artificial Skeletal Muscles Model Muscular Dystrophies and Enable Multilineage Tissue Engineering. Cell Rep. 2018, 23, 899–908. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Fokkema, I.; Verschuuren, J.; Ginjaar, I.; van Deutekom, J.; van Ommen, G.J.; den Dunnen, J.T. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 2009, 30, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Nagata, T.; Yokota, T.; Nakamura, A.; Wood, M.J.; Partridge, T.; Takeda, S. Highly efficient in vivo delivery of PMO into regenerating myotubes and rescue in laminin-alpha2 chain-null congenital muscular dystrophy mice. Hum. Mol. Genet. 2013, 22, 4914–4928. [Google Scholar] [CrossRef] [PubMed]
- Lukjanenko, L.; Karaz, S.; Stuelsatz, P.; Gurriaran-Rodriguez, U.; Michaud, J.; Dammone, G.; Sizzano, F.; Mashinchian, O.; Ancel, S.; Migliavacca, E.; et al. Aging Disrupts Muscle Stem Cell Function by Impairing Matricellular WISP1 Secretion from Fibro-Adipogenic Progenitors. Cell Stem Cell 2019, 24, 433–446 e7. [Google Scholar] [CrossRef] [PubMed]
- Hogarth, M.W.; Defour, A.; Lazarski, C.; Gallardo, E.; Manera, J.D.; Partridge, T.A.; Nagaraju, K.; Jaiswal, J.K. Fibroadipogenic progenitors are responsible for muscle loss in limb girdle muscular dystrophy 2B. Nat. Commun. 2019, 10, 2430. [Google Scholar] [CrossRef] [PubMed]
- Uezumi, A.; Fukada, S.; Yamamoto, N.; Takeda, S.; Tsuchida, K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol. 2010, 12, 143–152. [Google Scholar] [CrossRef]
- Joe, A.W.; Yi, L.; Natarajan, A.; Le Grand, F.; So, L.; Wang, J.; Rudnicki, M.A.; Rossi, F.M. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 2010, 12, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.R.; Ruckdeschel Smith, R.; Marban, E. Human cardiospheres are a source of stem cells with cardiomyogenic potential. Stem Cells 2010, 28, 903–904. [Google Scholar] [CrossRef]
- Makkar, R.R.; Smith, R.R.; Cheng, K.; Malliaras, K.; Thomson, L.E.; Berman, D.; Czer, L.S.; Marban, L.; Mendizabal, A.; Johnston, P.V.; et al. Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): A prospective, randomised phase 1 trial. Lancet 2012, 379, 895–904. [Google Scholar] [CrossRef]
- Malliaras, K.; Li, T.S.; Luthringer, D.; Terrovitis, J.; Cheng, K.; Chakravarty, T.; Galang, G.; Zhang, Y.; Schoenhoff, F.; Van Eyk, J.; et al. Safety and efficacy of allogeneic cell therapy in infarcted rats transplanted with mismatched cardiosphere-derived cells. Circulation 2012, 125, 100–112. [Google Scholar] [CrossRef]
- Ibrahim, A.G.; Cheng, K.; Marban, E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Rep. 2014, 2, 606–619. [Google Scholar] [CrossRef]
- Aminzadeh, M.A.; Rogers, R.G.; Fournier, M.; Tobin, R.E.; Guan, X.; Childers, M.K.; Andres, A.M.; Taylor, D.J.; Ibrahim, A.; Ding, X.; et al. Exosome-Mediated Benefits of Cell Therapy in Mouse and Human Models of Duchenne Muscular Dystrophy. Stem Cell Rep. 2018, 10, 942–955. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motohashi, N.; Shimizu-Motohashi, Y.; Roberts, T.C.; Aoki, Y. Potential Therapies Using Myogenic Stem Cells Combined with Bio-Engineering Approaches for Treatment of Muscular Dystrophies. Cells 2019, 8, 1066. https://doi.org/10.3390/cells8091066
Motohashi N, Shimizu-Motohashi Y, Roberts TC, Aoki Y. Potential Therapies Using Myogenic Stem Cells Combined with Bio-Engineering Approaches for Treatment of Muscular Dystrophies. Cells. 2019; 8(9):1066. https://doi.org/10.3390/cells8091066
Chicago/Turabian StyleMotohashi, Norio, Yuko Shimizu-Motohashi, Thomas C. Roberts, and Yoshitsugu Aoki. 2019. "Potential Therapies Using Myogenic Stem Cells Combined with Bio-Engineering Approaches for Treatment of Muscular Dystrophies" Cells 8, no. 9: 1066. https://doi.org/10.3390/cells8091066
APA StyleMotohashi, N., Shimizu-Motohashi, Y., Roberts, T. C., & Aoki, Y. (2019). Potential Therapies Using Myogenic Stem Cells Combined with Bio-Engineering Approaches for Treatment of Muscular Dystrophies. Cells, 8(9), 1066. https://doi.org/10.3390/cells8091066