Computer-Aided Studies for Novel Arylhydantoin 1,3,5-Triazine Derivatives as 5-HT6 Serotonin Receptor Ligands with Antidepressive-Like, Anxiolytic and Antiobesity Action In Vivo

 , , ,
, , , 
Abstract
1. Introduction
2. Results
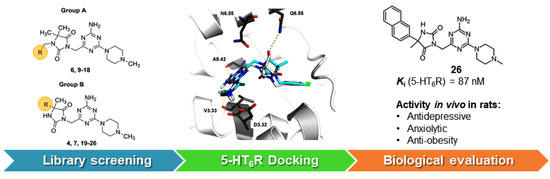
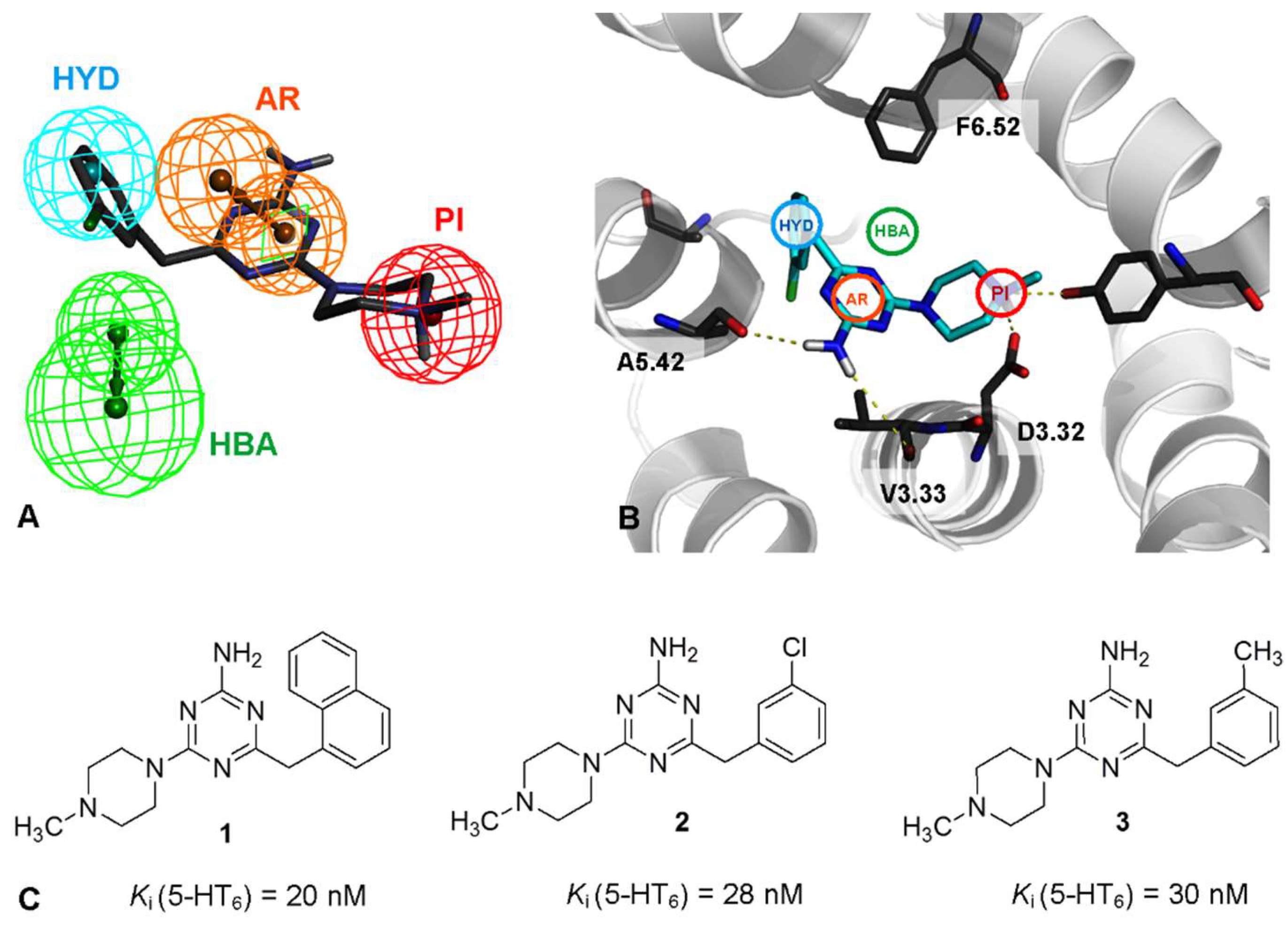
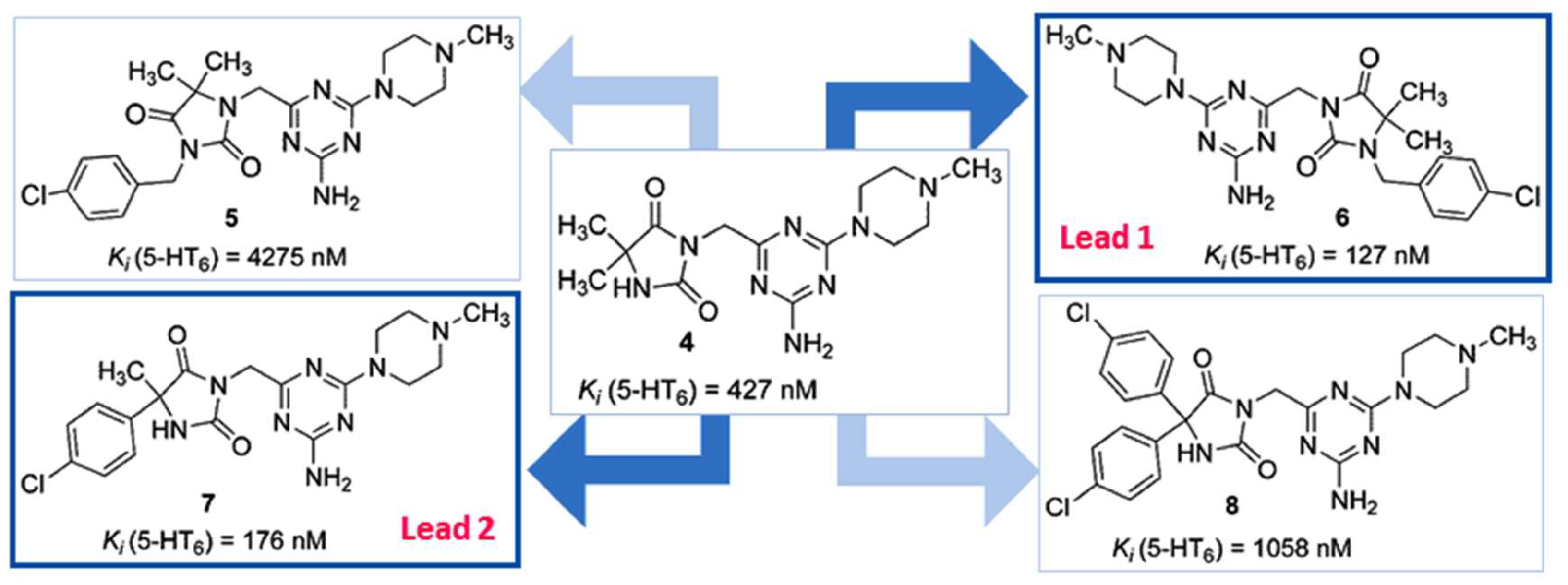
2.1. Identification of Lead Structures
2.2. Chemical Synthesis of Compounds 5–26
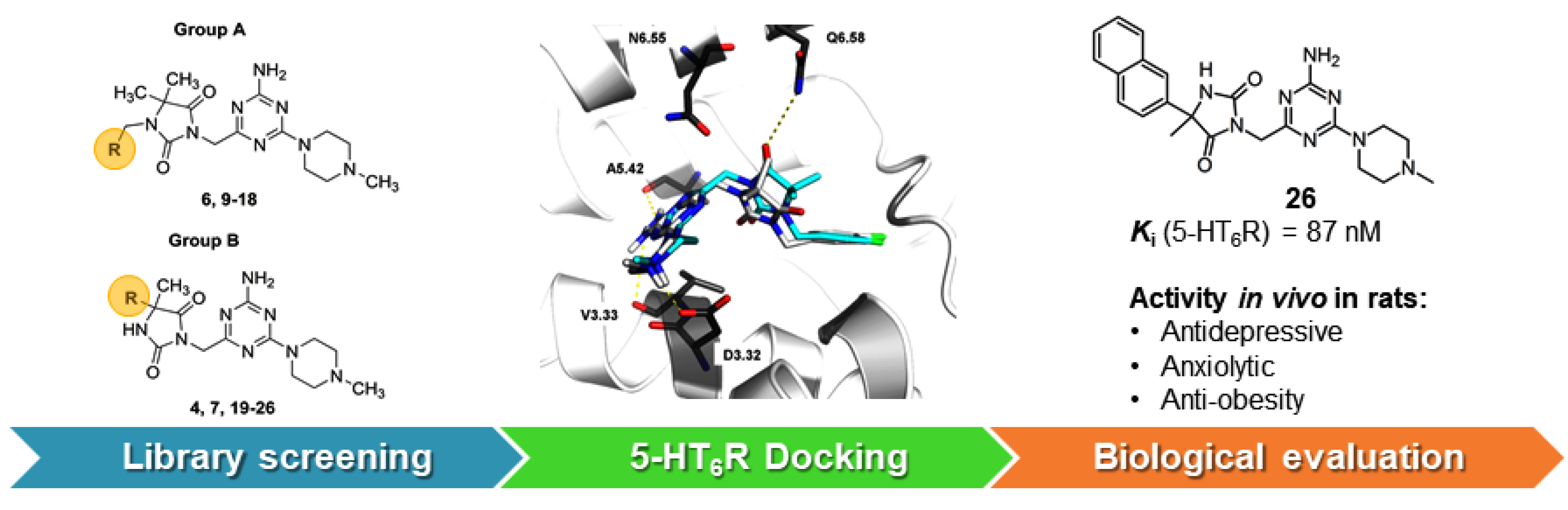
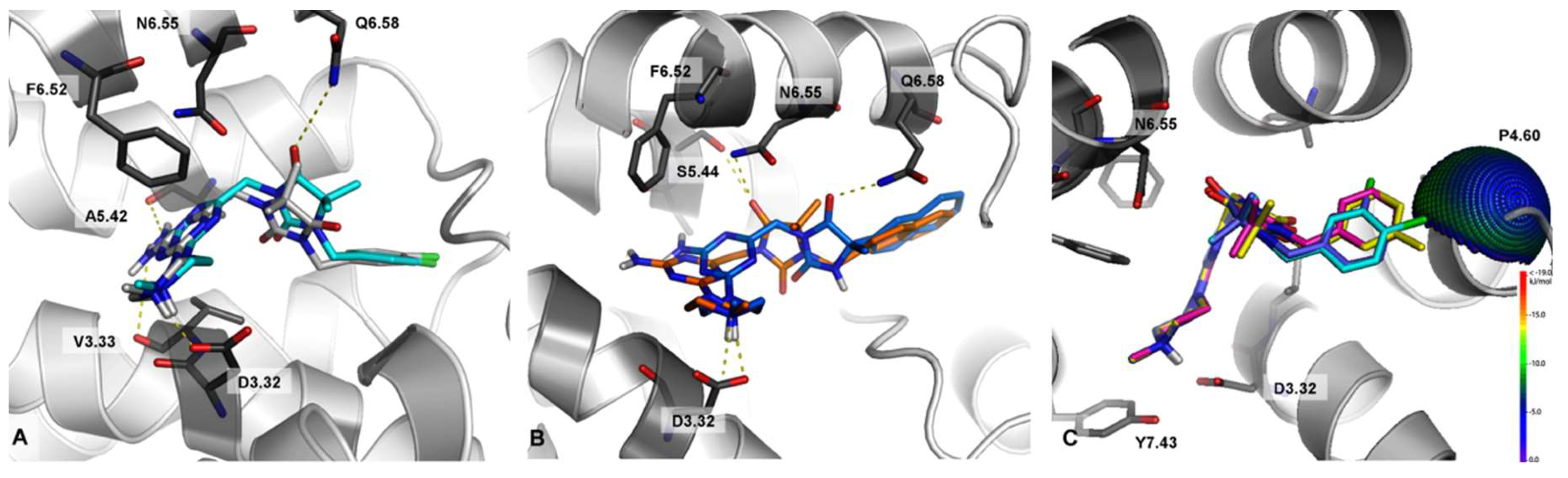
2.3. Molecular Modeling
2.4. Pharmacology
2.4.1. Radioligand Binding Assay
2.4.2. Behavioral Tests In Vivo
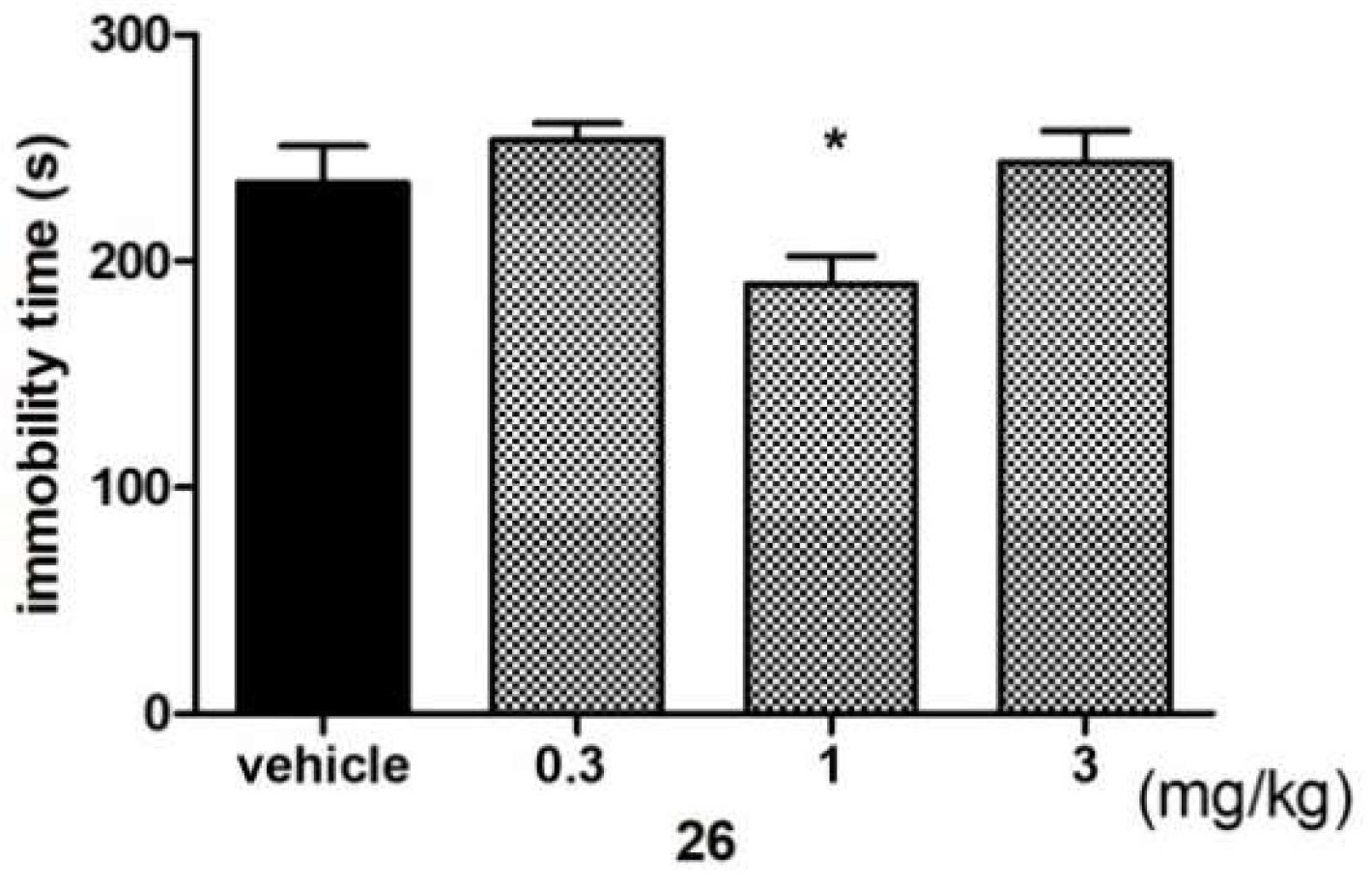
Antidepressant-Like Activity of Compound 26
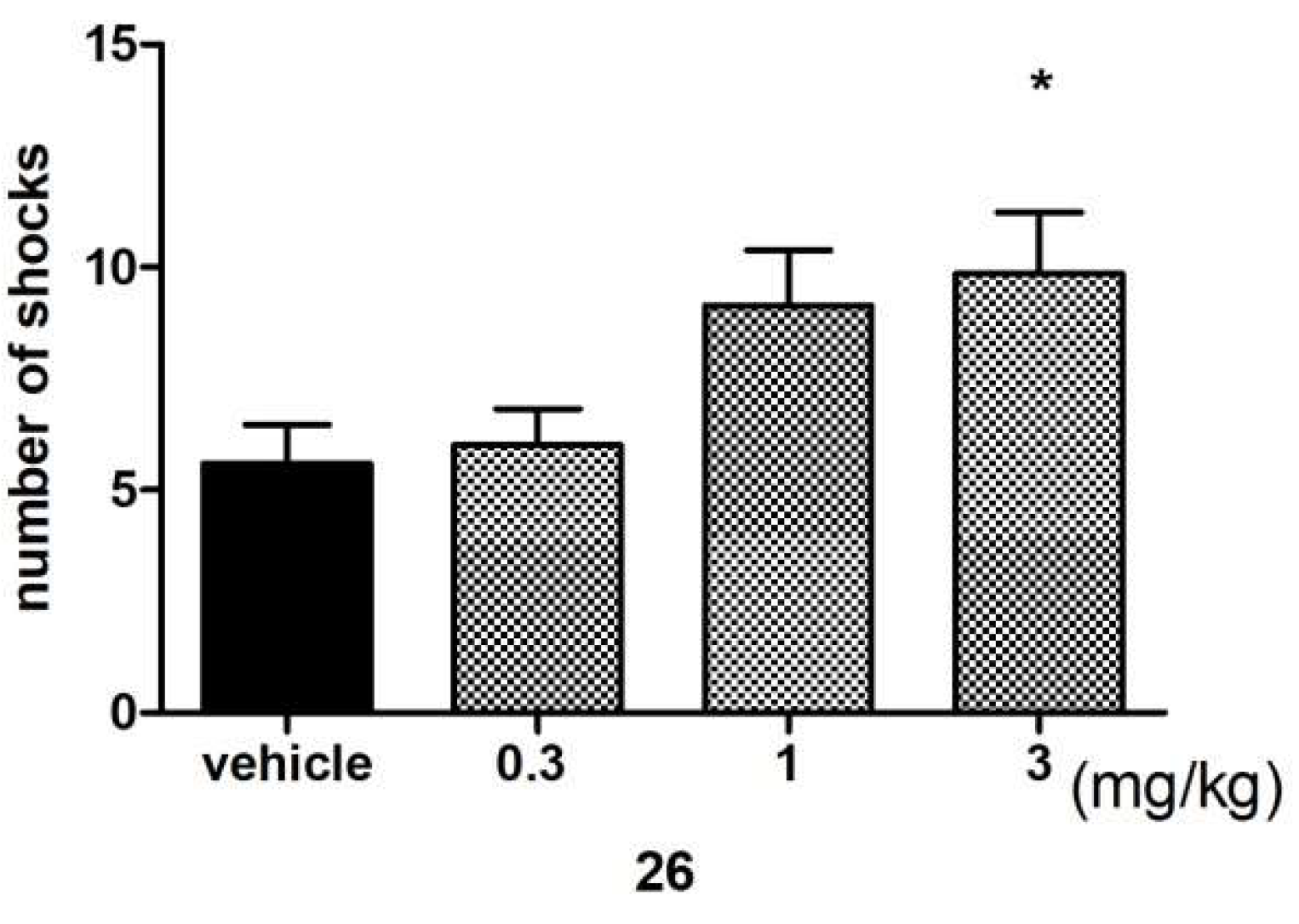
Anxiolytic-Like Activity of Compound 26
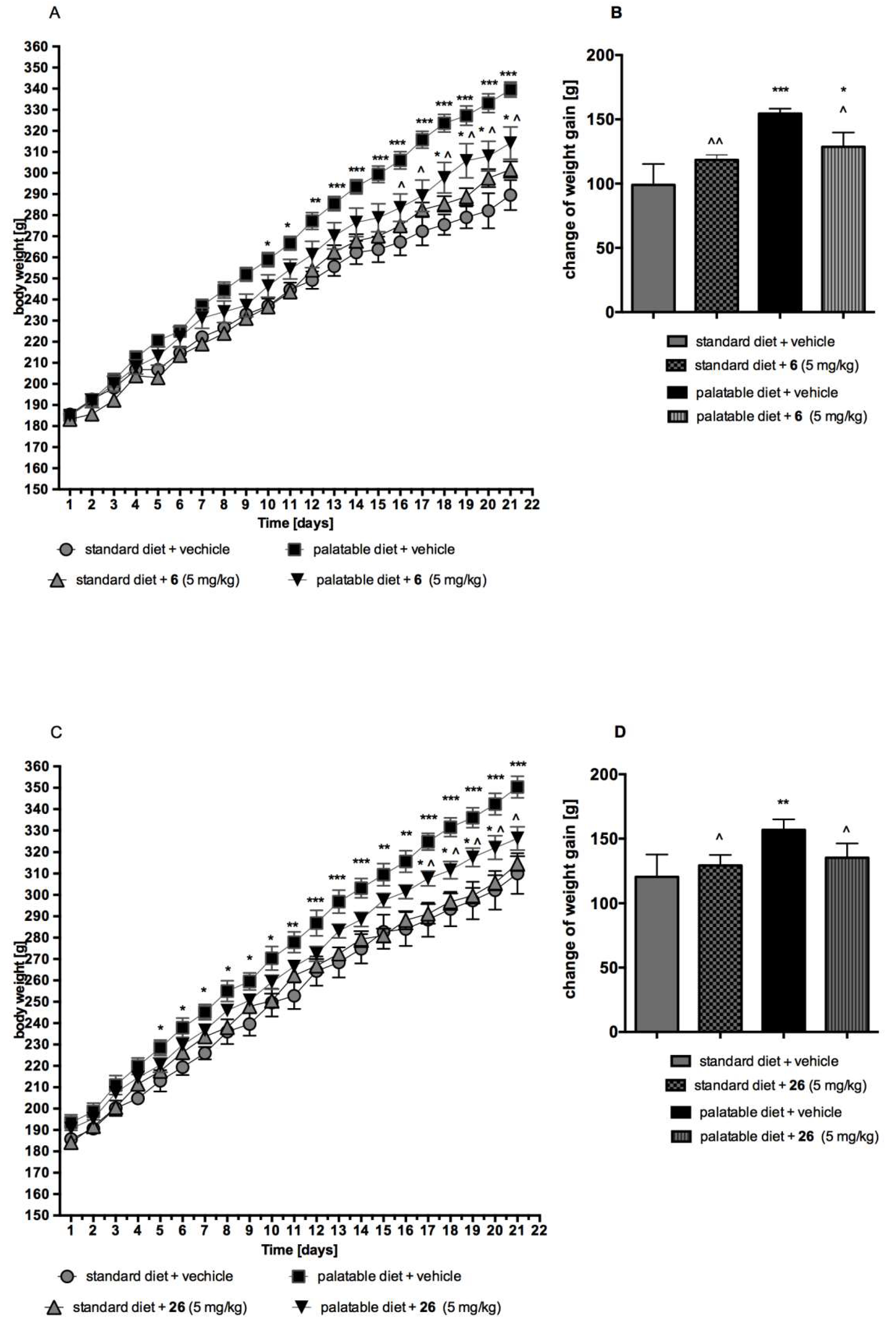
2.4.3. Metabolic Assays In Vivo
2.5. “Drug-Like” Properties
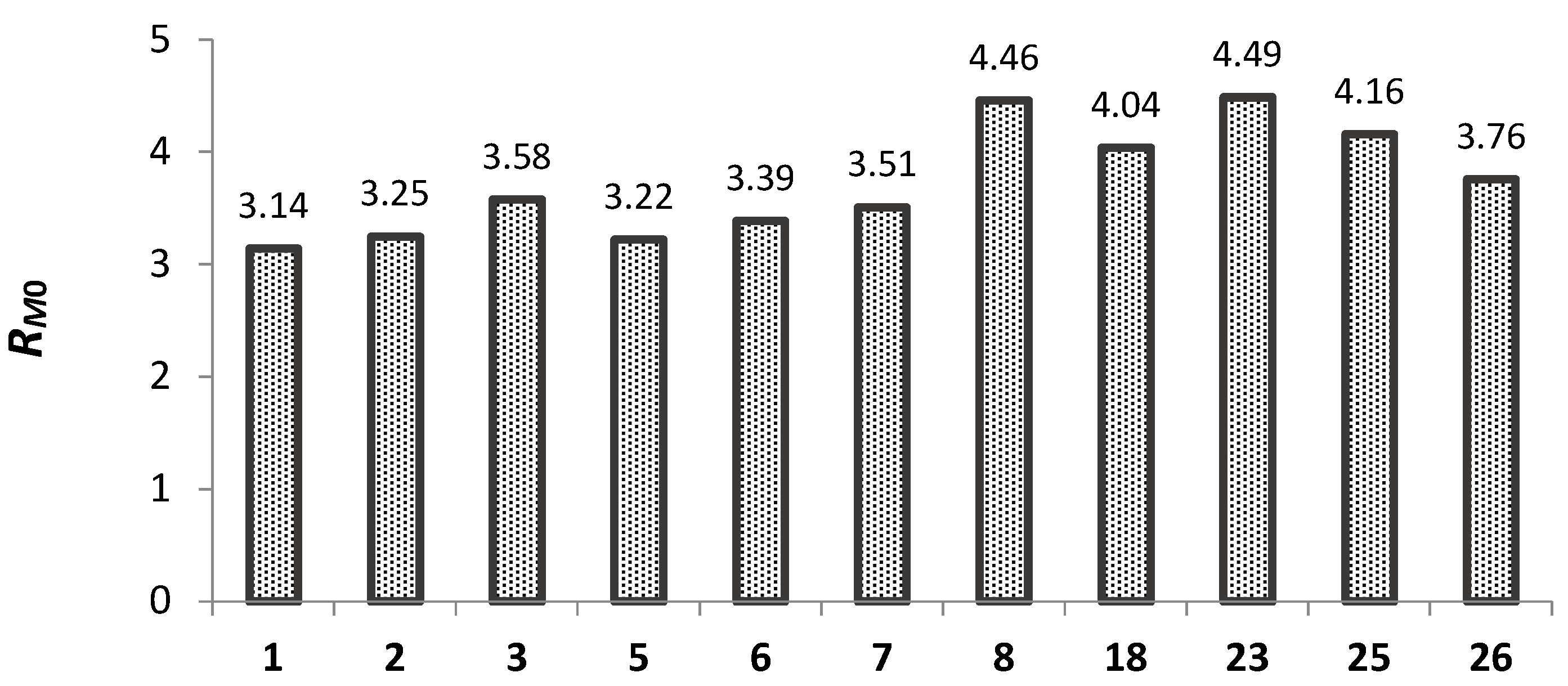
2.5.1. Lipophilicity
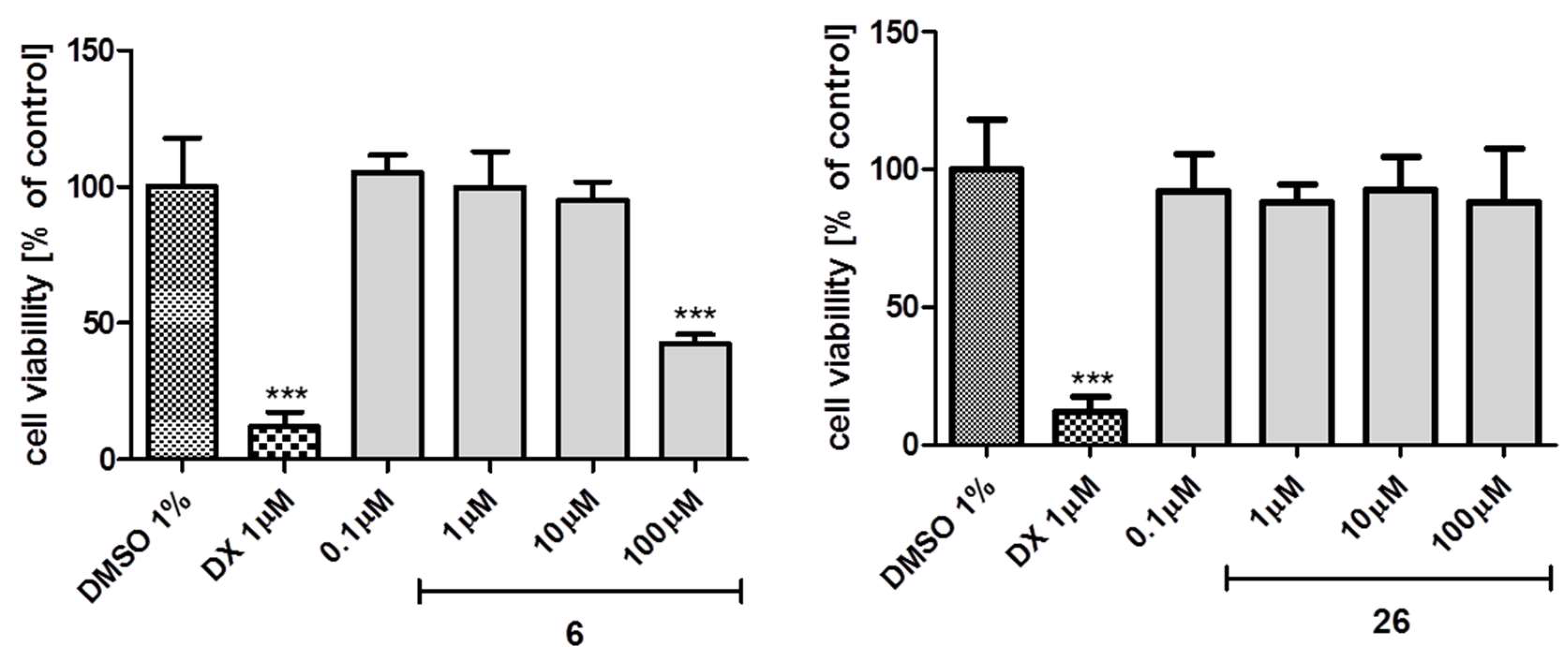
2.5.2. Toxicity In Vitro
2.5.3. Blood-Brain Barrier Permeability
3. Computer-Aided SAR Analysis
4. Experimental Section
4.1. Chemistry
4.1.1. Synthesis of Methyl 2-(3-(4-chlorobenzyl)-5,5-dimethyl-2,4-dioxoimidazolidin-1-yl)acetate (28)
4.1.2. General Procedure for the Synthesis of Alkyl 2-(1-arylmethyl-5,5-dimethyl-2,4-dioxoimidazolidin-3-yl)acetate (30–40)
4.1.3. General Procedures for the Synthesis of Methyl 2-(5-arylhydantoin-3-yl)acetates (60–67 and 71)
4.1.4. General Procedure for the Synthesis of 1,3,5-Triazine Final Products (5–26)
4.2. Molecular Modeling
4.2.1. Optimization of the 5-HT6R Binding Site Using Induced-Fit Docking Procedure
4.2.2. Molecular Docking
4.2.3. Plotting Interaction Spheres for Halogen Bonding
4.3. Lipophilicity Study
4.3.1. Thin-Layer Chromatography
4.3.2. Statistical Methods
4.4. Studies In Vitro
4.4.1. Radioligand Binding Assay
4.4.2. Toxicity In Vitro Test
4.5. Studies In Vivo
4.5.1. Animals
4.5.2. Drugs
4.5.3. Behavioral Procedures in Rats
Forced Swim Test (FST Test)
Vogel Conflict Drinking Test
4.5.4. Metabolic Assays In Vivo
The Effect of compound 6 or 26 on Body Weight by Non-Obese Rats Fed Palatable Diet (Model of Excessive Eating)
The Effect of compound 6 or 26 on Body Weight by Non-Obese Rats Fed Only with Standard Diet
4.6. Statistical Analysis
4.6.1. Lipophilicity
4.6.2. Studies In Vitro and In Vivo
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barnes, N.M.; Sharp, T. A review of central 5-HT receptors and their function. Neuropharmacology 1999, 38, 1083–1152. [Google Scholar] [CrossRef]
- Ruat, M.; Traiffort, E.; Arrang, J.M.; Tardivellacombe, J.; Diaz, J.; Leurs, R.; Schwartz, J.C. A Novel Rat Serotonin (5-HT6) Receptor: Molecular Cloning, Localization and Stimulation of cAMP Accumulation. Biochem. Biophys. Res. Commun. 1993, 193, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Kohen, R.; Metcalf, M.A.; Khan, N.; Druck, T.; Huebner, K.; Lachowicz, J.E.; Meltzer, H.Y.; Sibley, D.R.; Roth, B.L.; Hamblin, M.W. Cloning, Characterization, and Chromosomal Localization of a Human 5-HT6 Serotonin Receptor. J. Neurochem. 2002, 66, 47–56. [Google Scholar] [CrossRef]
- Liu, K.G.; Robichaud, A.J. 5-HT6 Medicinal Chemistry. Int. Rev. Neurobiol. 2010, 94, 1–34. [Google Scholar] [PubMed]
- Benhamú, B.; Martín-Fontecha, M.; Vázquez-Villa, H.; Pardo, L.; López-Rodríguez, M.L. Serotonin 5-HT6 Receptor Antagonists for the Treatment of Cognitive Deficiency in Alzheimer’s Disease. J. Med. Chem. 2014, 57, 7160–7181. [Google Scholar] [CrossRef] [PubMed]
- Wesołowska, A. Potential role of the 5-HT6 receptor in depression and anxiety: An overview of preclinical data. Pharmacol. Rep. 2010, 62, 564–577. [Google Scholar] [CrossRef]
- Heal, D.; Gosden, J.; Smith, S. The 5-HT6 Receptor as a Target for Developing Novel Antiobesity Drugs. Int. Rev. Neurobiol. 2011, 96, 73–109. [Google Scholar] [PubMed]
- Frassetto, A.; Zhang, J.; Lao, J.Z.; White, A.; Metzger, J.M.; Fong, T.M.; Chen, R.Z. Reduced sensitivity to diet-induced obesity in mice carrying a mutant 5-HT6 receptor. Brain Res. 2008, 1236, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Dudek, M.; Marcinkowska, M.; Bucki, A.; Olczyk, A.; Kołaczkowski, M. Idalopirdine—A small molecule antagonist of 5-HT6 with therapeutic potential against obesity. Metab. Brain Dis. 2015, 30, 1487–1494. [Google Scholar] [CrossRef] [PubMed]
- Krogsgaard-Larsen, N.; Jensen, A.A.; Schrøder, T.J.; Christoffersen, C.T.; Kehler, J. Novel Aza-analogous Ergoline Derived Scaffolds as Potent Serotonin 5-HT6 and Dopamine D2 Receptor Ligands. J. Med. Chem. 2014, 57, 5823–5828. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, T.; Martín-Fontecha, M.; Sallander, J.; Benhamú, B.; Campillo, M.; Medina, R.A.; Pellissier, L.P.; Claeysen, S.; Dumuis, A.; Pardo, L.; et al. Benzimidazole Derivatives as New Serotonin 5-HT6 Receptor Antagonists. Molecular Mechanisms of Receptor Inactivation. J. Med. Chem. 2010, 53, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Bojarski, A. Pharmacophore Models for Metabotropic 5-HT Receptor Ligands. Curr. Top. Med. Chem. 2006, 6, 2005–2026. [Google Scholar] [CrossRef] [PubMed]
- Łażewska, D.; Kurczab, R.; Więcek, M.; Kamińska, K.; Satała, G.; Jastrzębska-Więsek, M.; Partyka, A.; Bojarski, A.J.; Wesołowska, A.; Kieć-Kononowicz, K.; et al. The computer-aided discovery of novel family of the 5-HT6 serotonin receptor ligands among derivatives of 4-benzyl-1,3,5-triazine. Eur. J. Med. Chem. 2017, 135, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Latacz, G.; Kechagioglou, P.; Papi, R.; Łażewska, D.; Więcek, M.; Kamińska, K.; Wencel, P.; Karcz, T.; Schwed, J.S.; Stark, H.; et al. The Synthesis of 1,3,5-triazine Derivatives and JNJ7777120 Analogues with Histamine H 4 Receptor Affinity and Their Interaction with PTEN Promoter. Chem. Biol. Drug Des. 2016, 88, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Handzlik, J.; Bojarski, A.J.; Satała, G.; Kubacka, M.; Sadek, B.; Ashoor, A.; Siwek, A.; Więcek, M.; Kucwaj, K.; Filipek, B.; et al. SAR-studies on the importance of aromatic ring topologies in search for selective 5-HT7 receptor ligands among phenylpiperazine hydantoin derivatives. Eur. J. Med. Chem. 2014, 78, 324–339. [Google Scholar] [CrossRef] [PubMed]
- Łażewska, D.; Więcek, M.; Ner, J.; Kamińska, K.; Kottke, T.; Schwed, J.S.; Zygmunt, M.; Karcz, T.; Olejarz, A.; Kuder, K.; et al. Aryl-1,3,5-triazine derivatives as histamine H4 receptor ligands. Eur. J. Med. Chem. 2014, 83, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Grychowska, K.; Kurczab, R.; Śliwa, P.; Satała, G.; Dubiel, K.; Matłoka, M.; Moszczyński-Pętkowski, R.; Pieczykolan, J.; Bojarski, A.J.; Zajdel, P. Pyrroloquinoline scaffold-based 5-HT6R ligands: Synthesis, quantum chemical and molecular dynamic studies, and influence of nitrogen atom position in the scaffold on affinity. Bioorg. Med. Chem. 2018, 26, 3588–3595. [Google Scholar] [CrossRef] [PubMed]
- González-Vera, J.A.; Medina, R.A.; Martín-Fontecha, M.; Gonzalez, A.; de la Fuente, T.; Vázquez-Villa, H.; García-Cárceles, J.; Botta, J.; McCormick, P.J.; Benhamú, B.; et al. A new serotonin 5-HT6 receptor antagonist with procognitive activity—Importance of a halogen bond interaction to stabilize the binding. Sci. Rep. 2017, 7, 41293. [Google Scholar] [CrossRef] [PubMed]
- Borsini, F.; Meli, A. Is the forced swimming test a suitable model for revealing antidepressant activity? Psychopharmacology (Berlin) 1988, 94, 147–160. [Google Scholar] [CrossRef]
- Borsini, F. Role of the serotonergic system in the forced swimming test. Neurosci. Biobehav. Rev. 1995, 19, 377–395. [Google Scholar] [CrossRef]
- Wesołowska, A.; Nikiforuk, A.; Stachowicz, K. Anxiolytic-like and antidepressant-like effects produced by the selective 5-HT6 receptor antagonist SB-258585 after intrahippocampal administration to rats. Behav. Pharmacol. 2007, 18, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Soczewiński, E.; Wachtmeister, C.A. The relation between the composition of certain ternary two-phase solvent systems and RM values. J. Chromatogr. A 1962, 7, 311–320. [Google Scholar] [CrossRef]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Moving beyond rules: The development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010, 1, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2017-3: QikProp; Schrödinger LLC: New York, NY, USA, 2017.
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Zahn, S.; Boeckler, F.M. Using halogen bonds to address the protein backbone: A systematic evaluation. J. Comput. Aided Mol. Des. 2012, 26, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef] [PubMed]
- Matys, A.; Podlewska, S.; Witek, K.; Witek, J.; Bojarski, A.J.; Schabikowski, J.; Otrębska-Machaj, E.; Latacz, G.; Szymańska, E.; Kieć-Kononowicz, K.; et al. Imidazolidine-4-one derivatives in the search for novel chemosensitizers of Staphylococcus aureus MRSA: Synthesis, biological evaluation and molecular modeling studies. Eur. J. Med. Chem. 2015, 101, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Safari, J.; Naeimi, H.; Ghanbari, M.M.; Sabzi Fini, O. Preparation of phenytoin derivatives under solvent-free conditions using microwave irradiation. Russ. J. Org. Chem. 2009, 45, 477–479. [Google Scholar] [CrossRef]
- Werbel, L.M.; Elslager, E.F.; Islip, P.J.; Closier, M.D. Antischistosomal effects of 5-(2,4,5-trichlorophenyl)hydantoin and related compounds. J. Med. Chem. 1977, 20, 1569–1572. [Google Scholar] [CrossRef] [PubMed]
- Linol, J.; Coquerel, G. Influence of high energy milling on the kinetics of the polymorphic transition from the monoclinic form to the orthorhombic form of (±)5-methyl-5-(4′-methylphenyl)hydantoin. J. Therm. Anal. Calorim. 2007, 90, 367–370. [Google Scholar] [CrossRef]
- Keshtov, M.L.; Rusanov, A.L.; Belomoina, N.M.; Mikitaev, A.K. Improved synthesis of bis[p-(phenylethynyl)phenyl]hetarylenes. Russ. Chem. Bull. 1997, 46, 1794–1796. [Google Scholar] [CrossRef]
- Safari, J.; Javadian, L. Montmorillonite k-10 as a catalyst in the synthesis of 5,5-disubstituted hydantoins under ultrasound irradiation. J. Chem. Sci. 2013, 125, 981–987. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, A.J.; Kuhne, S.; De Esch, I.J.P.; Leurs, R.; De Graaf, C. A structural chemogenomics analysis of aminergic GPCRs: Lessons for histamine receptor ligand design. Br. J. Pharmacol. 2013, 170, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2017-3: LigPrep; Schrödinger LLC: New York, NY, USA, 2017.
- Schrödinger Release 2017-3: Epik; Schrödinger LLC: New York, NY, USA, 2017.
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2017-3: Glide; Schrödinger LLC: New York, NY, USA, 2017.
- Cheng, Y.-C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [PubMed]
- Porsolt, R.D.; Bertin, A.; Jalfre, M. “Behavioural despair” in rats and mice: Strain differences and the effects of imipramine. Eur. J. Pharmacol. 1978, 51, 291–294. [Google Scholar] [CrossRef]
- Vogel, J.R.; Beer, B.; Clody, D.E. A simple and reliable conflict procedure for testing anti-anxiety agents. Psychopharmacologia 1971, 21, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kotańska, M.; Lustyk, K.; Bucki, A.; Marcinkowska, M.; Śniecikowska, J.; Kołaczkowski, M. Idalopirdine, a selective 5-HT6 receptor antagonist, reduces food intake and body weight in a model of excessive eating. Metab. Brain Dis. 2018, 33, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Kotańska, M.; Śniecikowska, J.; Jastrzębska-Więsek, M.; Kołaczkowski, M.; Pytka, K. Metabolic and Cardiovascular Benefits and Risks of EMD386088—A 5-HT6 Receptor Partial Agonist and Dopamine Transporter Inhibitor. Front. Neurosci. 2017, 11, 50. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
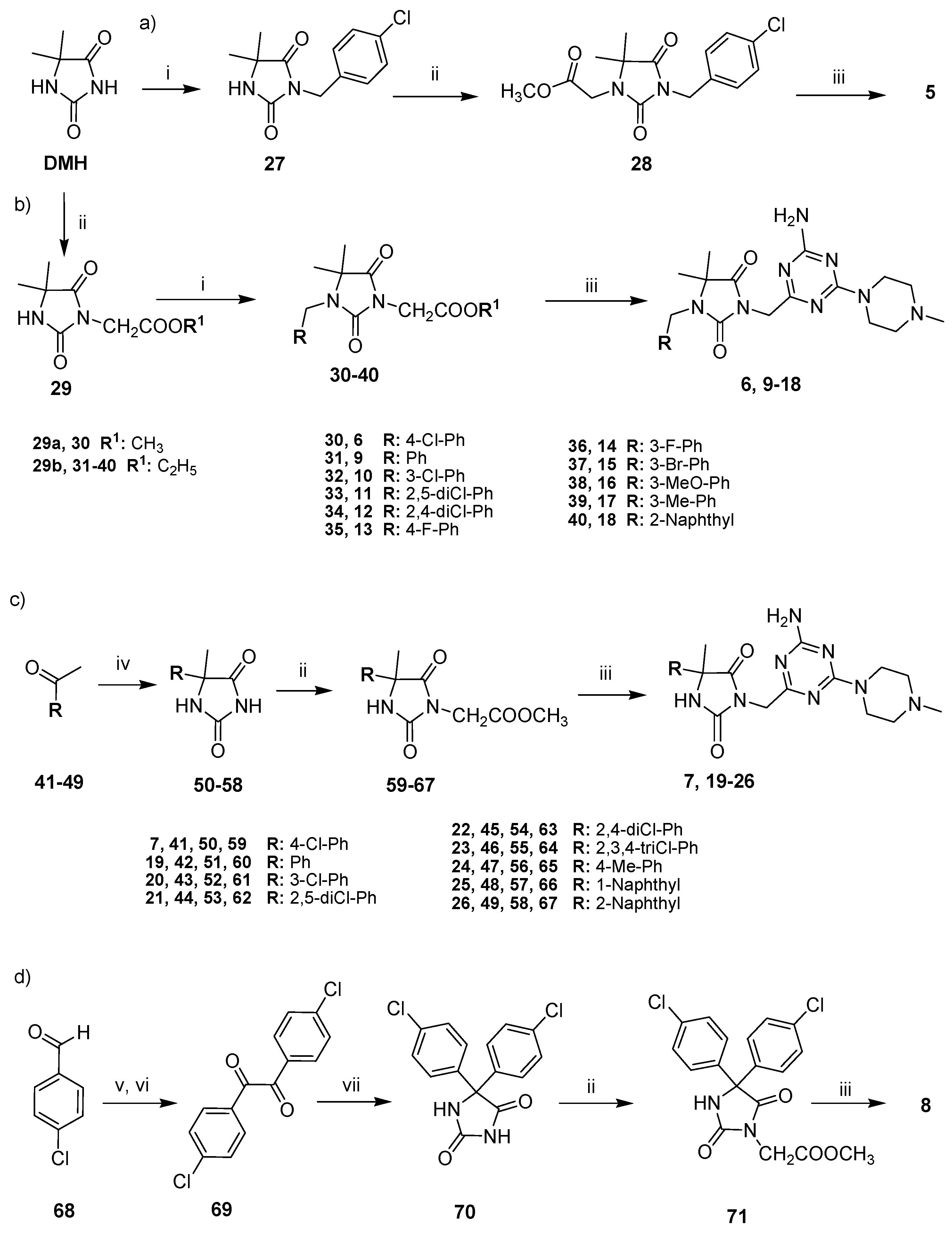{kind=link}

| Cpd | Gr | R | Ki (nM) a | ||||
|---|---|---|---|---|---|---|---|
| 5-HT6 [3H]-LSD | D2 [3H]-Raclopride | 5-HT1A [3H]-8-OH-DPAT | 5-HT2A [3H]-Ketanserin | 5-HT7 [3H]-5-CT | |||
| 4 | B | Methyl | 427 | 22,330 | 9.99 × 106 | nt | 35,790 |
| 5 | - | - | 4275 | 5007 | 15,430 | nt | 1620 |
| 6 | A | 4-Chlorophenyl | 127 | 4098 | 23,300 | nt | 3711 |
| 7 | B | 4-Chlorophenyl | 176 | 31,240 | 28,070 | nt | 938 |
| 8 | - | - | 1058 | 6855 | 3052 | nt | 594 |
| 9 | A | Phenyl | 592 | 8783 | 42,630 | 1596 | 350 |
| 10 | A | 3-Chlorophenyl | 271 | 4631 | 13,570 | 328 | 159 |
| 11 | A | 2,5-dichlorophenyl | 597 | 2867 | 4155 | 809 | 279 |
| 12 | A | 2,4-dichlorophenyl | 205 | 681 | 7693 | 3589 | 1116 |
| 13 | A | 4-Fluorophenyl | 663 | 6557 | 48,360 | 3548 | 953 |
| 14 | A | 3-Fluorophenyl | 447 | 6577 | 37,090 | 239 | 88 |
| 15 | A | 3-Bromophenyl | 570 | 6677 | 12,930 | 1327 | 571 |
| 16 | A | 3-Methoxyphenyl | 692 | 15,490 | 23,910 | 2036 | 3587 |
| 17 | A | 3-Methylphenyl | 895 | 5207 | 16,990 | 817 | 420 |
| 18 | A | 2-Naphthyl | 182 | 888 | 5718 | 1907 | 1525 |
| 19 | B | Phenyl | 726 | 41,040 | 31,620 | 12,150 | 1096 |
| 20 | B | 3-Chlorophenyl | 418 | 16,780 | 4101 | 7396 | 791 |
| 21 | B | 2,5-dichlorophenyl | 403 | 11,550 | 26,190 | 11,050 | 1565 |
| 22 | B | 2,4-dichlorophenyl | 457 | 15,090 | 13,980 | 10,770 | 10,130 |
| 23 | B | 2,3,4-trichlorophenyl | 195 | 4738 | 6028 | 3433 | 8905 |
| 24 | B | 4-Methylphenyl | 667 | 23,300 | 22,320 | 24,410 | 564 |
| 25 | B | 1-Naphthyl | 121 | 7811 | 7146 | 8080 | 3367 |
| 26 | B | 2-Naphthyl | 87 | 4247 | 14,160 | 17,170 | 514 |
| Ref. | 7 b | 9 b | 20 c | - | 18 d | ||
| Cpd | QPlogBB | Cpd | QPlogBB | Cpd | QPlogBB |
|---|---|---|---|---|---|
| 4 | −0.97 | 12 | −0.33 | 20 | −0.63 |
| 5 | −0.60 | 13 | −0.73 | 21 | −0.46 |
| 6 | −0.60 | 14 | −0.46 | 22 | −0.47 |
| 7 | −0.89 | 15 | −0.65 | 23 | −0.38 |
| 8 | −0.62 | 16 | −0.92 | 24 | −0.84 |
| 9 | −0.84 | 17 | −0.54 | 25 | −0.98 |
| 10 | −0.40 | 18 | −0.63 | 26 | −0.92 |
| 11 | −0.37 | 19 | −0.75 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurczab, R.; Ali, W.; Łażewska, D.; Kotańska, M.; Jastrzębska-Więsek, M.; Satała, G.; Więcek, M.; Lubelska, A.; Latacz, G.; Partyka, A.; et al. Computer-Aided Studies for Novel Arylhydantoin 1,3,5-Triazine Derivatives as 5-HT6 Serotonin Receptor Ligands with Antidepressive-Like, Anxiolytic and Antiobesity Action In Vivo. Molecules 2018, 23, 2529. https://doi.org/10.3390/molecules23102529
Kurczab R, Ali W, Łażewska D, Kotańska M, Jastrzębska-Więsek M, Satała G, Więcek M, Lubelska A, Latacz G, Partyka A, et al. Computer-Aided Studies for Novel Arylhydantoin 1,3,5-Triazine Derivatives as 5-HT6 Serotonin Receptor Ligands with Antidepressive-Like, Anxiolytic and Antiobesity Action In Vivo. Molecules. 2018; 23(10):2529. https://doi.org/10.3390/molecules23102529
Chicago/Turabian StyleKurczab, Rafał, Wesam Ali, Dorota Łażewska, Magdalena Kotańska, Magdalena Jastrzębska-Więsek, Grzegorz Satała, Małgorzata Więcek, Annamaria Lubelska, Gniewomir Latacz, Anna Partyka, and et al. 2018. "Computer-Aided Studies for Novel Arylhydantoin 1,3,5-Triazine Derivatives as 5-HT6 Serotonin Receptor Ligands with Antidepressive-Like, Anxiolytic and Antiobesity Action In Vivo" Molecules 23, no. 10: 2529. https://doi.org/10.3390/molecules23102529
APA StyleKurczab, R., Ali, W., Łażewska, D., Kotańska, M., Jastrzębska-Więsek, M., Satała, G., Więcek, M., Lubelska, A., Latacz, G., Partyka, A., Starek, M., Dąbrowska, M., Wesołowska, A., Jacob, C., Kieć-Kononowicz, K., & Handzlik, J. (2018). Computer-Aided Studies for Novel Arylhydantoin 1,3,5-Triazine Derivatives as 5-HT6 Serotonin Receptor Ligands with Antidepressive-Like, Anxiolytic and Antiobesity Action In Vivo. Molecules, 23(10), 2529. https://doi.org/10.3390/molecules23102529