Abstract
Metabolic engineering presents the possibility of creating novel and practical whole-cell biocatalysts. The practice of metabolic engineering is achieved first by in vitro DNA assembly, followed by the introduction of the newly constructed DNA into industrial microorganisms to create a novel phenotype. Although this approach of in vitro DNA assembly has been studied extensively, generation of unwanted recombinant DNA products remains a possibility. In this study, a recombinant DNA, namely pGRN02, was constructed using the sequence- and ligation- independent cloning. However, this DNA assembly method had a low success rate (5%). Unexpectedly, we identified an un-wanted recombinant DNA product as a major recombinant product (70%). DNA sequencing of this product indicated that it should not have been formed during in vitro DNA assembly, but rather post in vitro assembly. This study aims to report and discuss profound results of the DNA assembly reaction. The standard SLIC design using 20 bp homology arms is theoretically sufficient for correct assembly under typical conditions. However, longer unexpected repeats, such as the 44 bp internal homology observed here, can outcompete the designed junctions and dominate the recombination outcome.
1. Introduction
DNA assembly plays a key role in constructing gene expression systems and even whole chromosomes [1]. The development of DNA assembly methods is important in synthetic biology and metabolic engineering, and since the early 1970s, significant efforts have been taken to develop these methods. Presently, DNA manipulation has become a manageable practice. Different methods with improved manipulation efficiency, fidelity, and modularity and simpler and faster protocols have been proposed [2]. While some of them—for example, the Golden Gate method—employ the traditional restriction digestion and ligation technique [3], others—such as the Gibson assembly [4] and DNA assembler [5]—are based on a different principle.
Sequence- and ligation-independent cloning (SLIC) is an in vitro DNA assembly method. Typically, short DNA sequences can be engineered to attach to the ends of DNA fragments by polymerase chain reaction (PCR) These short sequences must be carefully designed to form a homology relay for the assembly reaction. Subsequently, the 3′ ends of the double-stranded linearized fragments are digested by T4 DNA polymerase in the absence of dNTPs to generate 5′ overhangs. These 5′ overhangs can find unique intermolecular homologous sequences for annealing. Lastly, the circular DNA (with nicks) formed during the annealing process is introduced into competent cells. In this process, nicks are ligated in vivo [6,7,8,9]. Notably, the supplementation of RecA and ATP may further help the annealing process.
The principle of DNA assembly has been extensively studied. However, the crosstalk between the different DNA manipulation processes, such as DNA replication, repair, and recombination, potentially causes mutations or decrease the success rate of DNA assembly. This study reports an unexpected but highly specific DNA recombination process occurring during the course of a metabolic engineering technique, which broadens our understanding of the crosstalk between the different processes involved in DNA recombination and provides essential information for further improvements in the designing of DNA assembly methods.
2. Results
2.1. Construction of pGRN02
After introducing the SLIC reaction mixture into competent Escherichia coli DH5α cells, the total number of the colony forming units of E. coli DH5α on the SmR Luria agar plate was 208. Twenty colonies were randomly selected and transformants were verified using colony PCR. The accurate size of the colony PCR product for pGRN02 was 1620 bp, as shown in Lane 2 of Figure 1. This clearly differed from the 756 bp PCR product of the control reaction consisting of the vector pCDFDuet-FRT as the template (Lane C, Figure 1). Recombinant pGRN02 was isolated and validated by DNA sequencing.
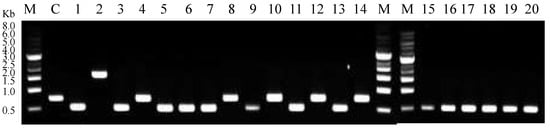
Figure 1.
Analysis of colony PCR products by DNA gel electrophoresis. M: 1 kb DNA ladder; C: control (pCDFDeut-FRT as template); 1–14: sample trials. Negative results: 756 bp (Lanes C, 4, 8, 10, 12, 14). pGRN02: 1620 bp (Lane 2). Unknown product: 500 bp (Lanes 1, 3, 5–7, 9, 11, 13, 15–20).
Our results indicated that 25% (5/20) of the colonies harbored only the vector pCDFDuet-FRT, which was 756 bp in size (Lanes 4, 8, 10, 12, 14, Figure 1). This could be attributed to a carry-over issue occurring during vector preparation. Overall, 30% of the colony PCR results were explainable. Unexpectedly, we also observed (Figure 1) unknown bands approximately the size of 500 bp that accounted for 70% of the total PCR products (14/20 colonies). This unexpected major PCR product indicated that this occurrence could be a result of an unknown but specific side reaction.
2.2. DNA Sequence of the Unknown Product
To obtain more insight into the side reaction, the 500 bp colony-PCR product was isolated and subjected to DNA sequencing. Upon aligning this 500 bp sequence with the vector pCDFDuet-FRT sequence (Figure S1), we observed that the sequence between the two PT7-lacO regions in the vector was deleted in the 500 bp sequence (the deletion was 44 bp long and thus was named HR44 in this study) (Figure 2), forming an unexpected circular recombinant plasmid that we named p500 (refer to Discussion for further details).
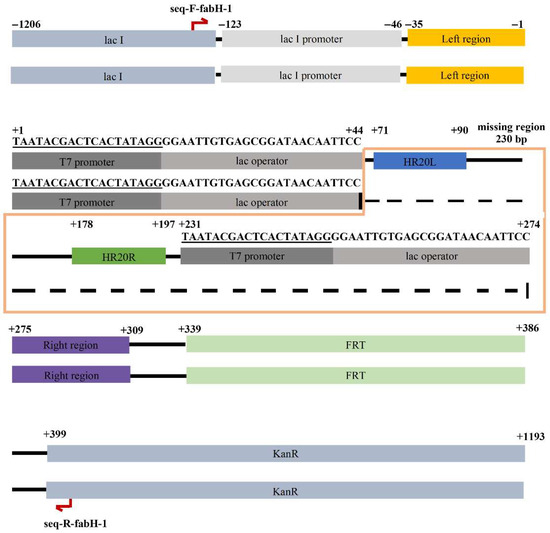
Figure 2.
Alignment of p500 with pCDFDuet-FRT, where the top is the vector and bottom strain is p500. A deletion mutation was found in p500. HR44 region covers the T7 promoter and lac operator.
3. Discussion
DNA assembly is a technique frequently performed in metabolic engineering. Although various feasible assembly methods have been proposed and demonstrated, performing this technique remains challenging. If no recombinant plasmid is obtained in an experiment, the only practical solution is to perform a new assembly trial, which is often frustrating.
As shown in Figure 3, when the insert and the vector fragments were assembled by SLIC, we expected to observe two products in our colony PCR analysis. We determined that the 756 bp and 1620 bp products were derived from the vector construct pCDFDeut-FRT (resulting from carry-over during vector preparation) and the target construct pGRN02, respectively. However, while these accounted for only 30% of the total products, an unexpected product of approximately 500 bp constituted the majority (Figure 3a). The sequence alignment of the 500 bp unknown product with the vector pCDFDuet-FRT indicated a deletion between two PT7-lacO regions in the former (Figure 2), resulting in the formation of the circular recombinant plasmid, p500 (Figure 3a). Thus, we inferred that pGRN02 could be constructed, albeit with a low success rate.
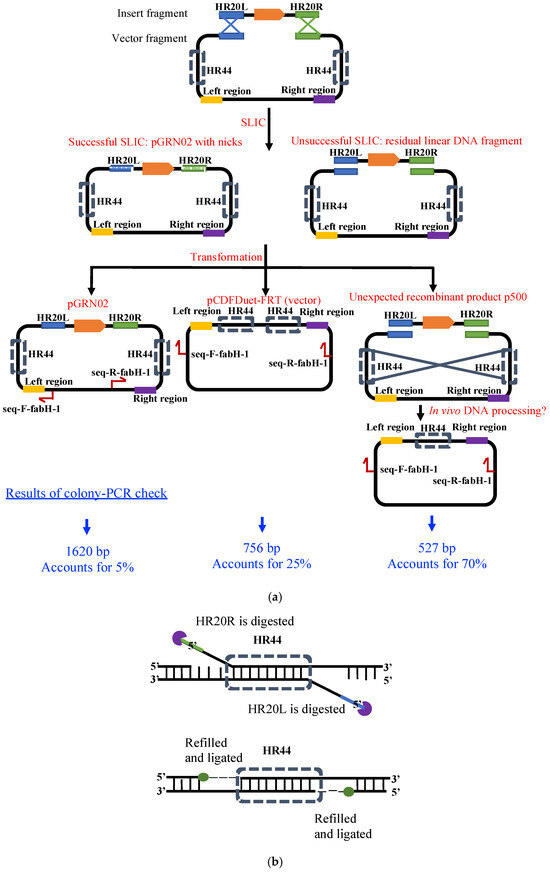
Figure 3.
Proposed mechanism for the formation of pGRN02 and the major by-product p500. (a) Expected SLIC assembly generates pGRN02 after exonuclease processing of HR20L and HR20R followed by annealing, gap filling, and ligation. However, annealing between the internal homologous HR44 sequences promotes formation of the truncated recombinant plasmid p500. (b) After HR44 pairing, in vivo exonucleases remove the remaining non-paired HR20L and HR20R overhangs, followed by gap filling and ligation to yield the circular plasmid p500.
We now turn our attention to the low success rate of pGRN02 in this study. The formation of p500, suggested that the formation was not via the in vitro SLIC method, as shown in Figure 3a. We believe that p500 formation is specific and causes a low success rate for pGRN02 formation. To construct pGRN02 in vitro by SLIC, or any other DNA assembly method, the generation of single-stranded DNA for the subsequent homologous annealing is critical and depends on the exonuclease activity of T4 DNA polymerase. The exonuclease activity of T4 DNA polymerase is 40 bp/min [10]. We assessed from our study results that for in vitro p500 formation by SLIC, a reaction time of at least 2.5 min for T4 DNA polymerase was needed to generate the single-stranded HR20R-HR44 region (total length is 97 bp, Figure 2). The use of 1 min for T4 DNA polymerase treatment in this study should not have generated p500 as the major recombinant product; therefore, an alternative other than in vitro reaction is suggested to explain p500 formation.
In vivo DNA processing and assembly has been investigated for a long time [11], although its practical application is not as popular as that of in vitro DNA assembly. The formation of p500 can be attributed to the possibility of in vivo DNA processing and assembly. Upon introducing the linearized DNA fragment with a 5′-overhang into the competent cell, the 5′-overhang was further extended by digestion of the complementary strand by in vivo exonucleases (Figure 3a). The total length of the homologous region in p500 is 44 bp (HR44 in Figure 3a) and serves as a strong target for annealing. When HR44 is annealed, the overhangs of HR20R and HR20L (shown in Figure 3b) are digested in vivo, as suggested in a previous study [11]. Therefore, p500 was found to be the major recombinant product in this study. Recombination between the HR44 sequences demonstrates the substantial effect of side reactions, which is suggested to occur in the cytosol. The findings of this study thus reveal the possible important role of in vivo DNA assembly in determining the success rates of in vitro DNA assembly. It is suggested that the peripheral sequences around the selected assembly sites for DNA assembly should be carefully examined for sequence similarity to avoid possible in vivo DNA assembly.
The predominance of p500 indicates that short internal homologies can outcompete designed SLIC junctions during assembly. In our system, a 44 bp internal homology dominated over the intended 20 bp homology arms. Based on the commonly cited T4 DNA polymerase exonuclease rate (40 bp/min), a 1 min treatment would be expected to incompletely expose the full 44 bp region; however, this value is only a reference and can vary with reaction conditions. Therefore, we cannot exclude that the 44 bp single-stranded region may also form during the in vitro step. Rather than strictly distinguishing in vitro from in vivo formation, this observation highlights a practical design consideration: exonuclease digestion time and enzyme concentration should be treated as tunable parameters to control the effective single-stranded exposure length. Together with avoiding internal sequence homology near assembly junctions, such control may reduce dominant unwanted recombination and improve SLIC fidelity.
In practice, any vector containing cryptic repeats or duplicated functional parts (e.g., terminators, RBS cassettes, or regulatory modules), or homology introduced during insert design, could enable an analogous rearrangement. Consequently, minimizing internal sequence homology should be considered a general design principle to improve assembly fidelity. It should be noted that this study does not define a universal cutoff; rather, it demonstrates a relative competition effect in which a 44 bp internal homology dominated over the intended 20 bp SLIC homology arms.
4. Materials and Methods
4.1. Strains and Plasmids
All strains and plasmids used are listed in Table 1. Escherichia coli DH5α was used as a host for the construction of pGRN02.

Table 1.
Strains and plasmids used in this study.
4.2. Construction of pGRN02 by Sequence- and Ligation-Independent Cloning (SLIC)
The recombinant plasmid pGRN02 was constructed by combining the insert fragment [fabH of E. coli BL21 (DE3)] with the vector pCDFDeutFRT-1 [our unpublished laboratory construct where FRT-Kam-FRT fragment (1311 bp) of pKD13 was inserted into pCDFDuet-1 MilliporeSigma (Burlington, MA, USA). through NdeI and XhoI sites]. The vector for pGRN02 was prepared by PCR (Q5 polymerase, NEB, Ipswich, MA, USA) using SLIC-F-pCDFduetFRT-1 and SLIC-R-pCDFduetFRT-1 primers (Table 2) and pCDFDeutFRT-1 as the template. The insert fragment containing the fabH gene was prepared by PCR (Q5 polymerase, NEB, Ipswich, MA, USA) using SLIC-F-FabH-1 and SLIC-R-FabH-1 primers (Table 2) and E. coli BL21(DE3) chromosome as the template. Two DNA fragments, insert fragments, and vector fragments were subjected to DNA clean-up (Protech, Taiwan). The PCR programs for the vector and insert fragments are displayed in Table 3 and Table 4, respectively.
Table 2.
List of primers used for pGRN02 construction.
Table 3.
PCR program for vector preparation for pGRN02 assembly.
Table 4.
PCR program for insert preparation for pGRN02 assembly.
To conduct in vitro SLIC [12], the vector and insert fragments were mixed at molar ratios of 1:0.5 to 1:2 with 100 ng of the vector DNA. NEB 2.1 commercial buffer (NEB, Ipswich, MA, USA) (1 µL) and bovine serum albumin (1 µL, 10×) were added to the DNA mixture solution. Subsequently, the DNA solution was diluted to 9.5 µL, and 0.5 µL of T4 polymerase was added to it. The SLIC reaction was performed at 37 °C for 1 min. The reaction was quenched by immediately placing the reaction mixture in an ice bath for 10 min. The single-stranded homologous region between the vector fragment and the insert fragment was annealed. Finally, the reaction solution was introduced into E. coli DH5α competent cells using a chemical transformation method [13].
4.3. Verification of pGRN02 Transformants by Colony PCR
pGRN02 transformants were verified by colony PCR using the primers seq-F-fabH-1 and seq-R-fabH-1 (Table 2). Taq polymerase (Protech, Taiwan) was used. The program for the colony PCR is shown in Table 5.
Table 5.
PCR program for colony PCR.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/synbio4010006/s1, Figure S1: Sequence of unexpected colony PCR product, p500. A deletion mutation is found in p500. HR 44 region covers T7 promoter and lac operator.
Author Contributions
Conceptualization, methodology, original draft preparation: S.-C.C. and S.-Y.L.; formal analysis, S.-C.C., Y.-J.L., B.S. and S.-Y.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the “Innovation and Development Center of Sustainable Agriculture” from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan. This work was also financially supported by the Ministry of Science and Technology (MOST 108-2628-E-005-002-MY3 and 109-2622-E-005-010-CC2) in Taiwan.
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Gibson, D.G.; Glass, J.I.; Lartigue, C.; Noskov, V.N.; Chuang, R.-Y.; Algire, M.A.; Benders, G.A.; Montague, M.G.; Ma, L.; Moodie, M.M.; et al. Creation of a Bacterial Cell Controlled by a Chemically Synthesized Genome. Science 2010, 329, 52. [Google Scholar] [CrossRef] [PubMed]
- Chao, R.; Yuan, Y.; Zhao, H. Recent advances in DNA assembly technologies. FEMS Yeast Res. 2015, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Engler, C.; Kandzia, R.; Marillonnet, S. A one pot, one step, precision cloning method with high throughput capability. PLoS ONE 2008, 3, e3647. [Google Scholar] [CrossRef] [PubMed]
- Weber, E.; Engler, C.; Gruetzner, R.; Werner, S.; Marillonnet, S. A Modular Cloning System for Standardized Assembly of Multigene Constructs. PLoS ONE 2011, 6, e16765. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Zhao, H.; Zhao, H. DNA assembler, an in vivo genetic method for rapid construction of biochemical pathways. Nucleic Acids Res. 2009, 37, e16. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Kim, H.U.; Chae, T.U.; Cho, J.S.; Kim, J.W.; Shin, J.H.; Kim, D.I.; Ko, Y.-S.; Jang, W.D.; Jang, Y.-S. A comprehensive metabolic map for production of bio-based chemicals. Nat. Catal. 2019, 2, 18–33, Correction in Nat. Catal. 2019, 2, 942–944. https://doi.org/10.1038/s41929-019-0358-8. [Google Scholar] [CrossRef]
- Chen, F.Y.; Jung, H.W.; Tsuei, C.Y.; Liao, J.C. Converting Escherichia coli to a Synthetic Methylotroph Growing Solely on Methanol. Cell 2020, 182, 933–946.e914. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wei, Y.; Lin, L.; Teng, L.; Yin, J.; Lu, Q.; Chen, J.; Zheng, Y.; Li, Y.; Xu, R.; et al. Two radical-dependent mechanisms for anaerobic degradation of the globally abundant organosulfur compound dihydroxypropanesulfonate. Proc. Natl. Acad. Sci. USA 2020, 117, 15599. [Google Scholar] [CrossRef] [PubMed]
- Tseng, I.T.; Chen, Y.-L.; Chen, C.-H.; Shen, Z.-X.; Yang, C.-H.; Li, S.-Y. Exceeding the theoretical fermentation yield in mixotrophic Rubisco-based engineered Escherichia coli. Metab. Eng. 2018, 47, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Rittié, L.; Perbal, B. Enzymes used in molecular biology: A useful guide. J. Cell Commun. Signal 2008, 2, 25–45. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.F.; García-Nafría, J. In vivo DNA assembly using common laboratory bacteria: A re-emerging tool to simplify molecular cloning. J. Biol. Chem. 2019, 294, 15271–15281. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.-Y.; Yim, H.-S.; Ryu, J.-Y.; Lee, H.S.; Lee, J.-H.; Seen, D.-S.; Kang, S.G. One-Step Sequence- and Ligation-Independent Cloning as a Rapid and Versatile Cloning Method for Functional Genomics Studies. Appl. Environ. Microbiol. 2012, 78, 5440–5443. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.