
The Underexplored Landscape of Hypoxia-Inducible Factor 2 Alpha and Potential Roles in Tumor Macrophages: A Review



Abstract
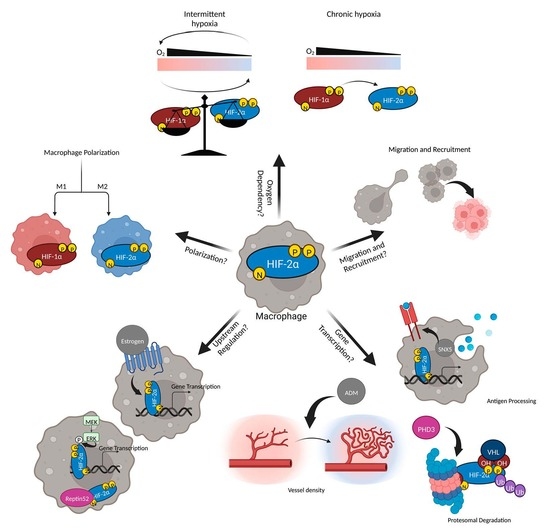
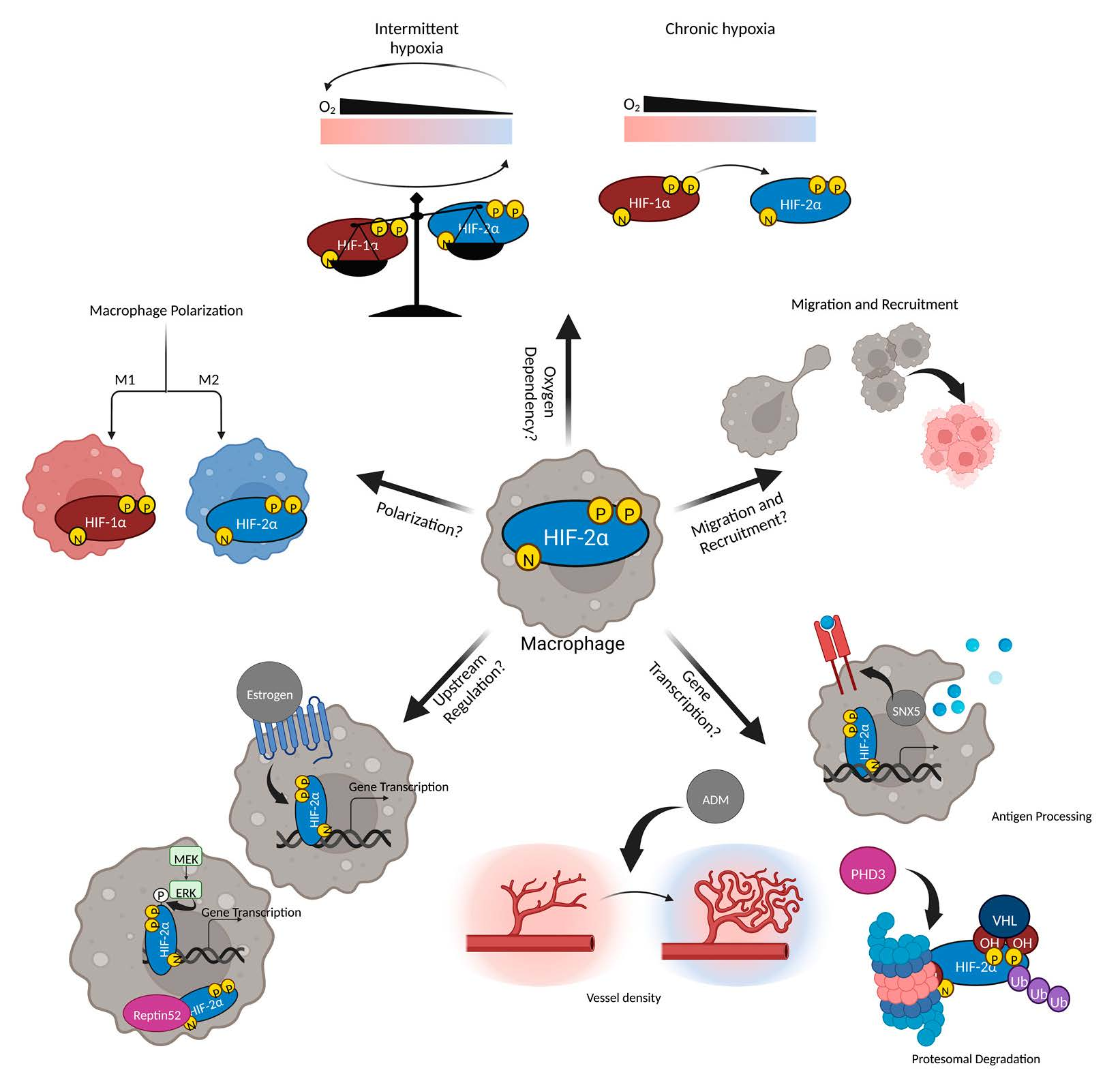
1. Introduction
2. HIF-2α in Different Cell Types
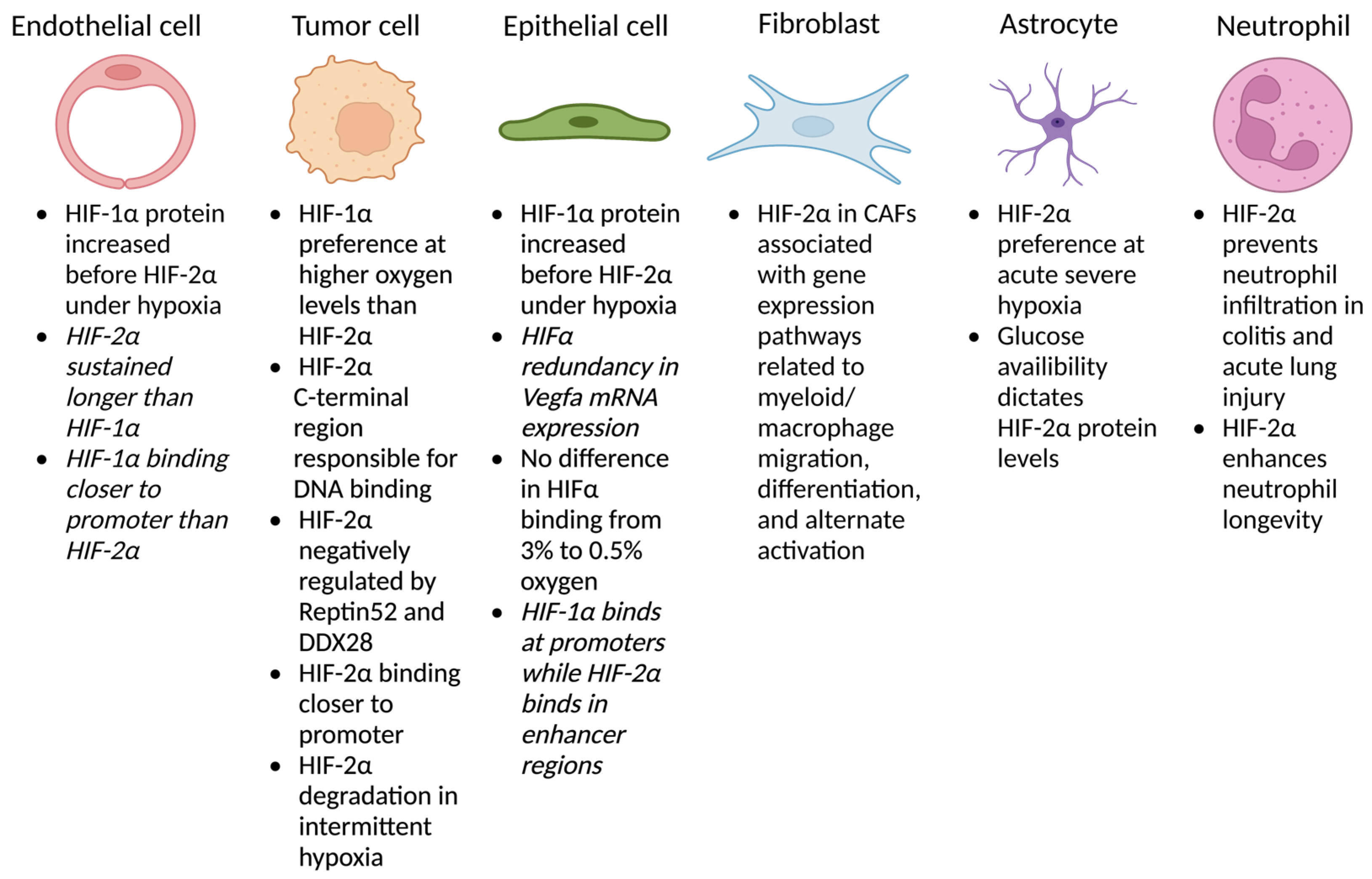
2.1. Endothelium
2.2. Tumor Cells
2.3. Epithelium
2.4. Fibroblasts
2.5. Astrocytes
2.6. Myeloid Cells
2.7. Other
3. Oxygen Dependent Macrophage HIF-2α Functions
3.1. In Vitro
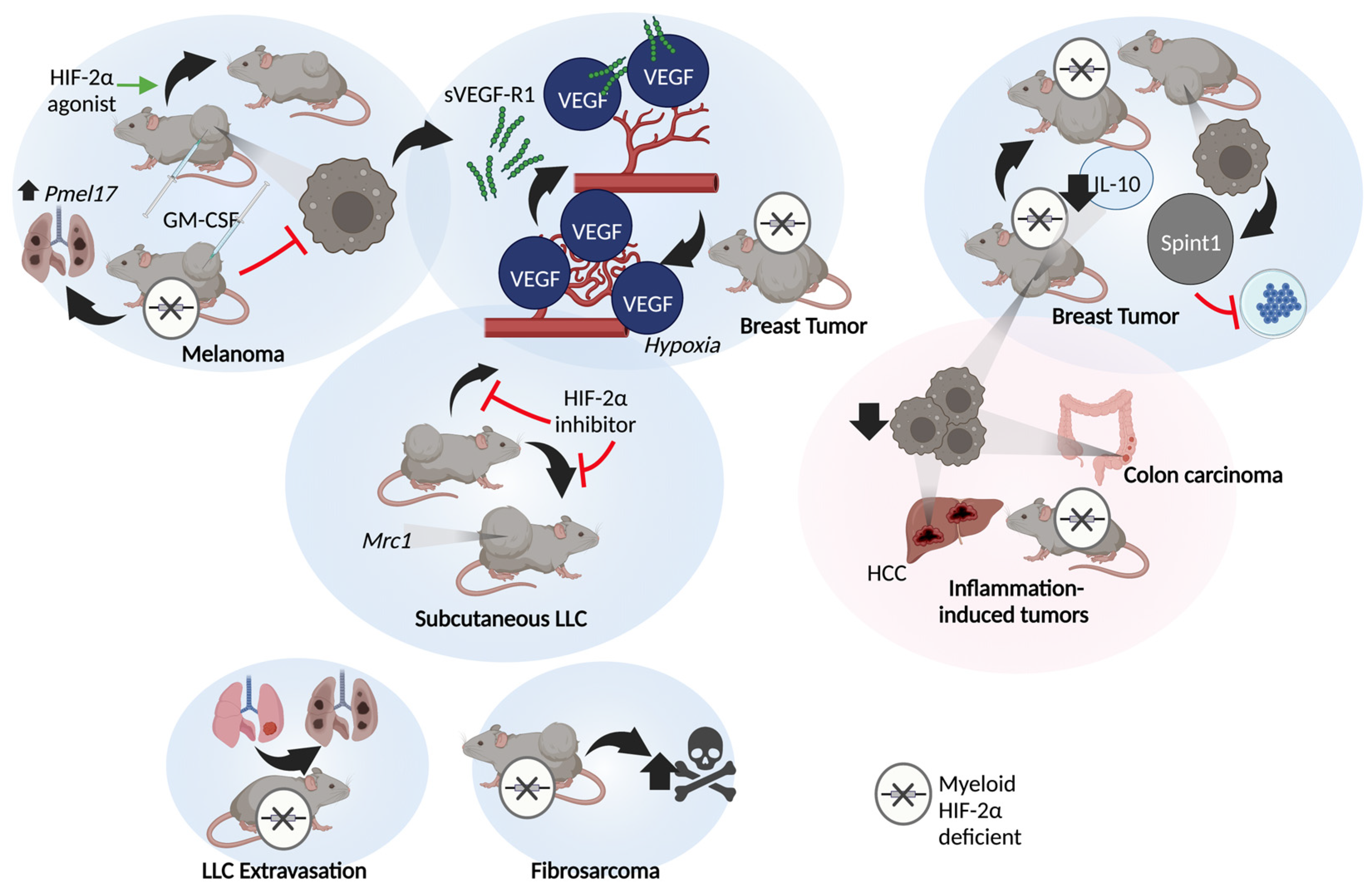
3.2. In Vivo
4. Oxygen Independent Macrophage HIF-2α Functions
4.1. Non-Human Studies
4.2. Human Studies
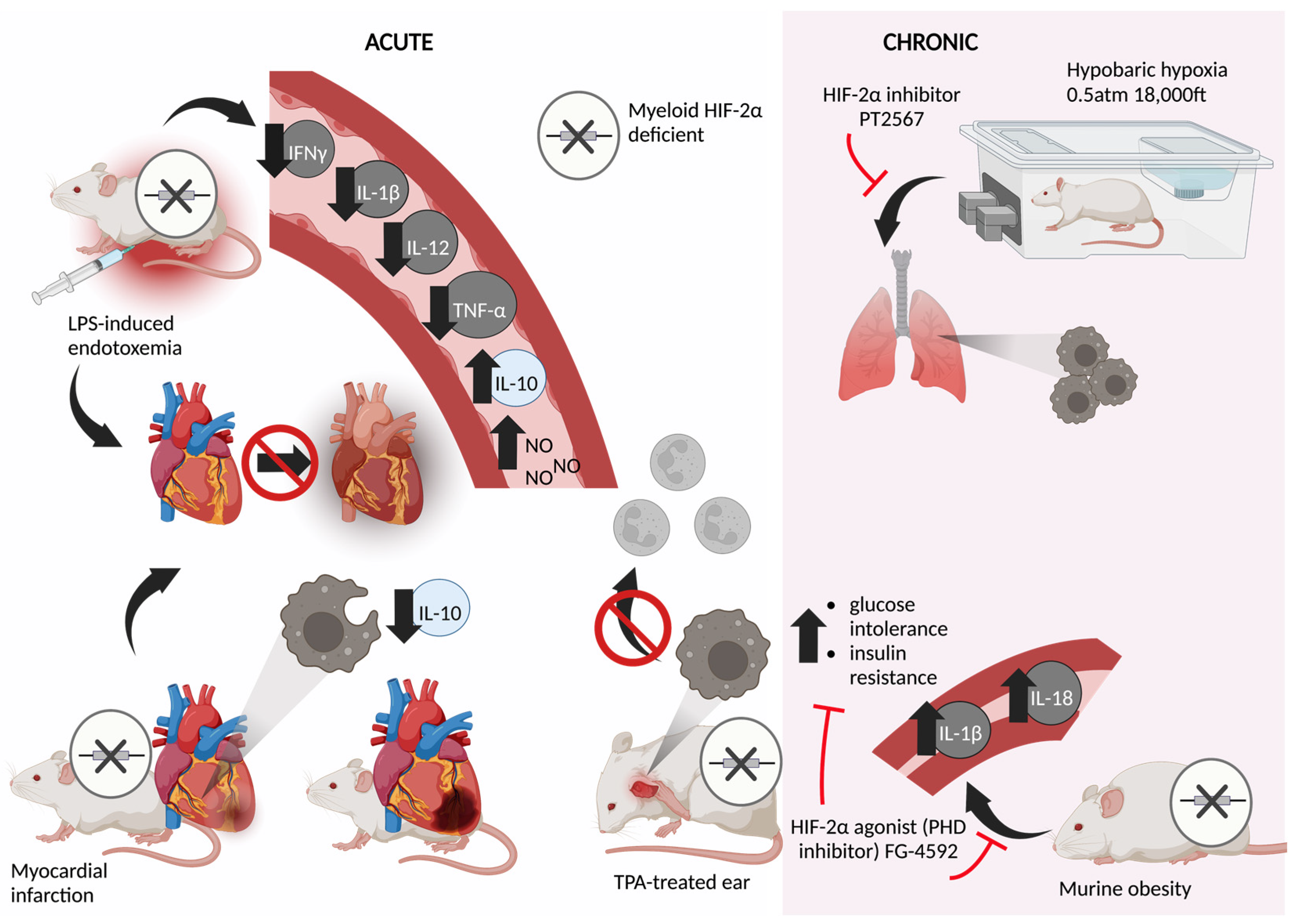
5. Acute Models
6. Chronic Models
7. Targeting HIF-2α Clinically
8. Conclusions
9. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McKeown, S.R. Defining Normoxia, Physoxia and Hypoxia in Tumours—Implications for Treatment Response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef] [PubMed]
- Siemann, D.W. The Unique Characteristics of Tumor Vasculature and Preclinical Evidence for Its Selective Disruption by Tumor-Vascular Disrupting Agents. Cancer Treat. Rev. 2011, 37, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.S.; Lee, J.A.; Underwood, J.C.E.; Harris, A.L.; Lewis, C.E. Macrophage Responses to Hypoxia: Relevance to Disease Mechanisms; Macrophage Responses to Hypoxia: Relevance to Disease Mechanisms. J. Leukoc. Biol. 1999, 66, 889–900. [Google Scholar] [CrossRef]
- Walmsley, S.R.; Print, C.; Farahi, N.; Peyssonnaux, C.; Johnson, R.S.; Cramer, T.; Sobolewski, A.; Condliffe, A.M.; Cowburn, A.S.; Johnson, N.; et al. Hypoxia-Induced Neutrophil Survival Is Mediated by HIF-1α–Dependent NF-ΚB Activity. J. Exp. Med. 2005, 201, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF Transcription Factors, Inflammation, and Immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Haschemi, A.; Kosma, P.; Gille, L.; Evans, C.R.; Burant, C.F.; Starkl, P.; Knapp, B.; Haas, R.; Schmid, J.A.; Jandl, C.; et al. The Sedoheptulose Kinase CARKL Directs Macrophage Polarization through Control of Glucose Metabolism. Cell Metab. 2012, 15, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Firth, J.D.; Ebert, B.L.; Pugh, C.W.; Ratcliffe, P.J. Oxygen-Regulated Control Elements in the Phosphoglycerate Kinase 1 and Lactate Dehydrogenase A Genes: Similarities with the Erythropoietin 3′ Enhancer. Proc. Natl. Acad. Sci. USA 1994, 9, 6496–6500. [Google Scholar] [CrossRef]
- Ahn, G.-O.; Seita, J.; Hong, B.-J.; Kim, Y.-E.; Bok, S.; Lee, C.-J.; Kim, K.S.; Lee, J.C.; Leeper, N.J.; Cooke, J.P.; et al. Transcriptional Activation of Hypoxia-Inducible Factor-1 (HIF-1) in Myeloid Cells Promotes Angiogenesis through VEGF and S100A8. Proc. Nati. Acad. Sci. USA 2014, 111, 2698–2703. [Google Scholar] [CrossRef]
- Steinberger, K.J.; Forget, M.A.; Bobko, A.A.; Mihalik, N.E.; Gencheva, M.; Roda, J.M.; Cole, S.L.; Mo, X.; Hoblitzell, E.H.; Evans, R.; et al. Hypoxia-Inducible Factor α Subunits Regulate Tie2-Expressing Macrophages That Influence Tumor Oxygen and Perfusion in Murine Breast Cancer. J. Immunol. 2020, 205, 2301–2311. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen Sensing, Homeostasis, and Disease. New Engl. J. Med. 2011, 365, 537–547. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen Sensing, Hypoxia-Inducible Factors, and Disease Pathophysiology. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G.; Ratcliffe, P.J. Oxygen Sensing by Metazoans: The Central Role of the HIF Hydroxylase Pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Strowitzki, M.J.; Cummins, E.P.; Taylor, C.T. Protein Hydroxylation by Hypoxia-Inducible Factor (HIF) Hydroxylases: Unique or Ubiquitous? Cells 2019, 8, 384. [Google Scholar] [CrossRef] [PubMed]
- Salceda, S.; Caro, J. Hypoxia-Inducible Factor 1α (HIF-1α) Protein Is Rapidly Degraded by the Ubiquitin-Proteasome System under Normoxic Conditions. Its Stabilization by Hypoxia Depends on Redox-Induced Changes. J. Biol. Chem. 1997, 272, 22642–22647. [Google Scholar] [CrossRef]
- Appelhoffl, R.J.; Tian, Y.M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J.M. Differential Function of the Prolyl Hydroxylases PHD1, PHD2, and PHD3 in the Regulation of Hypoxia-Inducible Factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef]
- Milano, G.; Fujii, Y.; Shibasaki, F.; Hashimoto, T. Hypoxia-Inducible Factor as an Angiogenic Master Switch. Front. Pediatr. 2015, 3, 33. [Google Scholar] [CrossRef]
- Kajimura, S.; Aida, K.; Duan, C. Understanding Hypoxia-Induced Gene Expression in Early Development: In Vitro and In Vivo Analysis of Hypoxia-Inducible Factor 1-Regulated Zebra Fish Insulin-Like Growth Factor Binding Protein 1 Gene Expression. Mol. Cell. Biol. 2006, 26, 1142–1155. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cox, S.R.; Morita, T.; Kourembanas, S. Hypoxia Regulates Vascular Endothelial Growth Factor Gene Expression in Endothelial Cells: Identification of a 5′ Enhancer. Circ. Res. 1995, 77, 638–643. [Google Scholar] [CrossRef]
- Semenza, G.L. Defining the Role of Hypoxia-Inducible Factor 1 in Cancer Biology and Therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef]
- Hara, S.; Hamada, J.; Kobayashi, C.; Kondo, Y.; Imura, N. Expression and Characterization of Hypoxia-Inducible Factor (HIF)-3α in Human Kidney: Suppression of HIF-Mediated Gene Expression by HIF-3α. Biochem. Biophys. Res. Commun. 2001, 287, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Maynard, M.A.; Qi, H.; Chung, J.; Lee, E.H.L.; Kondo, Y.; Hara, S.; Conaway, R.C.; Conaway, J.W.; Ohh, M. Multiple Splice Variants of the Human HIF-3α Locus Are Targets of the von Hippel-Lindau E3 Ubiquitin Ligase Complex. J. Biol. Chem. 2003, 278, 11032–11040. [Google Scholar] [CrossRef]
- Blake, D.T.; Strata, F.; Churchland, A.K.; Merzenich, M.M. Neural Correlates of Instrumental Learning in Primary Auditory Cortex. Proc. Natl. Acad. Sci. USA 2002, 99, 10114–10119. [Google Scholar] [CrossRef] [PubMed]
- Tudela, J.; Martínez, M.; Valdivia, R.; Romo, J.; Portillo, M.; Rangel, R. Dominant-Negative HIF-3 Alpha 4 Suppresses VHL-Null Renal Cell Carcinoma Progression. Nature 2010, 388, 539–547. [Google Scholar]
- Zhang, P.; Lu, L.; Yao, Q.; Li, Y.; Zhou, J.; Liu, Y.; Duan, C. Molecular, Functional, and Gene Expression Analysis of Zebrafish Hypoxia-Inducible Factor-3α. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R1165–R1174. [Google Scholar] [CrossRef] [PubMed]
- Makino, Y.; Cao, R.; Svensson, K.; Bertilsson, G.; Asman, M.; Tanaka, H.; Cao, Y.; Berkenstam, A.; Poellinger, L. Inhibitory PAS Domain Protein Is a Negative Regulator of Hypoxia-Inducible Gene Expression. Nature 2001, 414, 550–554. [Google Scholar] [CrossRef]
- Yamashita, T.; Ohneda, O.; Nagano, M.; Iemitsu, M.; Makino, Y.; Tanaka, H.; Miyauchi, T.; Goto, K.; Ohneda, K.; Fujii-Kuriyama, Y.; et al. Abnormal Heart Development and Lung Remodeling in Mice Lacking the Hypoxia-Inducible Factor-Related Basic Helix-Loop-Helix PAS Protein NEPAS. Mol. Cell Biol. 2008, 28, 1285–1297. [Google Scholar] [CrossRef]
- Milovanova, T.N.; Bhopale, V.M.; Sorokina, E.M.; Moore, J.S.; Hunt, T.K.; Hauer-Jensen, M.; Velazquez, O.C.; Thom, S.R. Hyperbaric Oxygen Stimulates Vasculogenic Stem Cell Growth and Differentiation in Vivo. J. Appl. Physiol. 2009, 106, 711–728. [Google Scholar] [CrossRef]
- Zhang, P.; Yao, Q.; Lu, L.; Li, Y.; Chen, P.J.; Duan, C. Hypoxia-Inducible Factor 3 Is an Oxygen-Dependent Transcription Activator and Regulates a Distinct Transcriptional Response to Hypoxia. Cell Rep. 2014, 6, 1110–1121. [Google Scholar] [CrossRef]
- Janaszak-Jasiecka, A.; Bartoszewska, S.; Kochan, K.; Piotrowski, A.; Kalinowski, L.; Kamysz, W.; Ochocka, R.J.; Bartoszewski, R.; Collawn, J.F. MiR-429 Regulates the Transition between Hypoxia-Inducible Factor (HIF)1A and HIF3A Expression in Human Endothelial Cells. Sci. Rep. 2016, 6, 22775. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-Inducible Factor 1: Control of Oxygen Homeostasis in Health and Disease. Pediatr. Res. 2001, 49, 614–617. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of Hypoxia-Inducible Factor 1alpha in Common Human Cancers and Their Metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar] [PubMed]
- Giatromanolaki, A.; Koukourakis, M.I.; Simopoulos, C.; Polychronidis, A.; Gatter, K.C.; Harris, A.L.; Sivridis, E. C-ErbB-2 Related Aggressiveness in Breast Cancer Is Hypoxia Inducible Factor-1 Dependent. Cell Biol. 2001, 21, 3995–4005. [Google Scholar]
- Dales, J.-P.; Garcia, S.; Meunier-Carpentier, S.; Andrac-Meyer, L.; Haddad, O.; Lavaut, M.-N.; Allasia, C.; Bonnier, P.; Charpin, C. Overexpression of hypoxia-inducible factor HIF-1α predicts early relapse in breast cancer: Retrospective study in a series of 745 patients. Int. J. Cancer 2005, 116, 734–739. [Google Scholar] [CrossRef]
- Vleugel, M.M.; Greijer, A.E.; Shvarts, A.; Van Der Groep, P.; Van Berkel, M.; Aarbodem, Y.; Van Tinteren, H.; Harris, A.L.; Van Diest, P.J.; Van, E. Differential Prognostic Impact of Hypoxia Induced and Diffuse HIF-1a Expression in Invasive Breast Cancer. J Clin Pathol. 2005, 58, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Generali, D.; Berruti, A.; Brizzi, M.P.; Campo, L.; Bonardi, S.; Wigfield, S.; Bersiga, A.; Allevi, G.; Milani, M.; Aguggini, S.; et al. Hypoxia-Inducible Factor-1α Expression Predicts a Poor Response to Primary Chemoendocrine Therapy and Disease-Free Survival in Primary Human Breast Cancer. Clin. Cancer Res. 2006, 12, 4562–4568. [Google Scholar] [CrossRef]
- Kronblad, Å.; Jirström, K.; Rydén, L.; Nordenskjöld, B.; Landberg, G. Hypoxia Inducible Factor-1α Is a Prognostic Marker in Premenopausal Patients with Intermediate to Highly Differentiated Breast Cancer but Not a Predictive Marker for Tamoxifen Response. Int. J. Cancer 2006, 118, 2609–2616. [Google Scholar] [CrossRef] [PubMed]
- Trastour, C.; Benizri, E.; Ettore, F.; Ramaioli, A.; Chamorey, E.; Pouyss Egur, J.; Berra, E. HIF-1a and CA IX Staining in Invasive Breast Carcinomas: Prognosis and Treatment Outcome. Int. J. Cancer 2007, 120, 1451–1458. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Mutsuko, A.E.; Ae, I.; Okumura, Y.; Teru, A.E.; Ae, K.; Kai, K.; Kenichi, A.E.; Ae, I.; Iwase, H. Hypoxia-Inducible Factor 1a Is Closely Linked to an Aggressive Phenotype in Breast Cancer. Breast Cancer Res. Treat 2008, 110, 465–475. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Moszyńska, A.; Serocki, M.; Cabaj, A.; Polten, A.; Ochocka, R.; Dell’Italis, L.; Bartoszewska, S.; Króliczewski, J.; Dąbrowski, M.J.; et al. Primary Endothelial Cell-Specific Regulation of Hypoxia-Inducible Factor (HIF)-1 and HIF-2 and Their Target Gene Expression Profiles during Hypoxia. FASEB J. 2019, 33, 7929–7941. [Google Scholar] [CrossRef]
- Moszyńska, A.; Jaśkiewicz, M.; Serocki, M.; Cabaj, A.; Crossman, D.K.; Bartoszewska, S.; Gebert, M.; Dąbrowski, M.; Collawn, J.F.; Bartoszewski, R. The Hypoxia-Induced Changes in MiRNA-MRNA in RNA-Induced Silencing Complexes and HIF-2 Induced MiRNAs in Human Endothelial Cells. FASEB J. 2022, 36, e22412. [Google Scholar] [CrossRef]
- Kimura, H.; Weisz, A.; Ogura, T.; Hitomi, Y.; Kurashima, Y.; Hashimoto, K.; D’Acquisto, F.; Makuuchi, M.; Esumi, H. Identification of Hypoxia-Inducible Factor 1 Ancillary Sequence and Its Function in Vascular Endothelial Growth Factor Gene Induction by Hypoxia and Nitric Oxide. J. Biol. Chem. 2001, 276, 2292–2298. [Google Scholar] [CrossRef] [PubMed]
- Smythies, J.A.; Sun, M.; Masson, N.; Salama, R.; Simpson, P.D.; Murray, E.; Neumann, V.; Cockman, M.E.; Choudhry, H.; Ratcliffe, P.J.; et al. Inherent DNA -binding Specificities of the HIF -1α and HIF -2α Transcription Factors in Chromatin. EMBO Rep. 2019, 20, e46401. [Google Scholar] [CrossRef] [PubMed]
- Elbarghatia, L.; Murdoch, C.; Lewis, C.E. Effects of Hypoxia on Transcription Factor Expression in Human Monocytes and Macrophages. Immunobiology 2008, 213, 899–908. [Google Scholar] [CrossRef]
- Mimura, I.; Nangaku, M.; Kanki, Y.; Tsutsumi, S.; Inoue, T.; Kohro, T.; Yamamoto, S.; Fujita, T.; Shimamura, T.; Suehiro, J.; et al. Dynamic Change of Chromatin Conformation in Response to Hypoxia Enhances the Expression of GLUT3 (SLC2A3) by Cooperative Interaction of Hypoxia-Inducible Factor 1 and KDM3A. Mol. Cell Biol. 2012, 32, 3018–3032. [Google Scholar] [CrossRef]
- Tiana, M.; Acosta-Iborra, B.; Puente-Santamaría, L.; Hernansanz-Agustin, P.; Worsley-Hunt, R.; Masson, N.; García-Rio, F.; Mole, D.; Ratcliffe, P.; Wasserman, W.W.; et al. The SIN3A Histone Deacetylase Complex Is Required for a Complete Transcriptional Response to Hypoxia. Nucleic Acids Res. 2018, 46, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Tausendschön, M.; Rehli, M.; Dehne, N.; Schmidl, C.; Döring, C.; Hansmann, M.L.; Brüne, B. Genome-Wide Identification of Hypoxia-Inducible Factor-1 and -2 Binding Sites in Hypoxic Human Macrophages Alternatively Activated by IL-10. Biochim. Biophys. Acta Gene Regul. Mech. 2015, 1849, 10–22. [Google Scholar] [CrossRef]
- Cabaj, A.; Moszyńska, A.; Charzyńska, A.; Bartoszewski, R.; Dąbrowski, M. Functional and HRE Motifs Count Analysis of Induction of Selected Hypoxia-Responsive Genes by HIF-1 and HIF-2 in Human Umbilical Endothelial Cells. Cell Signal. 2022, 90, 110209. [Google Scholar] [CrossRef]
- Larsen, H.; Muz, B.; Khong, T.L.; Feldmann, M.; Paleolog, E.M. Differential Effects of Th1 versus Th2 Cytokines in Combination with Hypoxia on HIFs and Angiogenesis in RA. Arthritis Res. Ther. 2012, 14, R180. [Google Scholar] [CrossRef]
- Gonzalez-Flores, A.; Aguilar-Quesada, R.; Siles, E.; Pozo, S.; Rodríguez-Lara, M.I.; López-Jiménez, L.; López-Rodríguez, M.; Peralta-Leal, A.; Villar, D.; Martín-Oliva, D.; et al. Interaction between PARP-1 and HIF-2α in the Hypoxic Response _ Enhanced Reader.Pdf. Oncogene 2014, 33, 891–898. [Google Scholar]
- Wang, J.; Ikeda, R.; Che, X.F.; Ooyama, A.; Yamamoto, M.; Furukawa, T.; Hasui, K.; Zheng, C.L.; Tajitsu, Y.; Oka, T.; et al. VEGF Expression Is Augmented by Hypoxia-Induced PGIS in Human Fibroblasts. Int. J. Oncol. 2013, 43, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.J.; Poth, J.M.; Zhang, H.; Flockton, A.; Laux, A.; Kumar, S.; McKeon, B.; Mouradian, G.; Li, M.; Riddle, S.; et al. Suppression of HIF2 Signalling Attenuates the Initiation of Hypoxia-Induced Pulmonary Hypertension. Eur. Respir. J. 2019, 54, 1900378. [Google Scholar] [CrossRef]
- Wenger, R.H.; Fuady, J.; Bordoli, M.; Abreu Rodriguez, I.; Kristiansen, G.; Hoogewijs, D.; Stiehl, D. Hypoxia-Inducible Factor-Mediated Induction of WISP-2 Contributes to Attenuated Progression of Breast Cancer. Hypoxia 2014, 23, 1900378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Qin, Y.; Martinez, M.; Flores-Bellver, M.; Rodrigues, M.; Dinabandhu, A.; Cao, X.; Deshpande, M.; Qin, Y.; Aparicio-Domingo, S.; et al. HIF-1α and HIF-2α Redundantly Promote Retinal Neovascularization in Patients with Ischemic Retinal Disease. J. Clin. Investig. 2021, 131, e139202. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; O’Dea, E.L.; Doedens, A.; Kim, J.W.; Weidemann, A.; Stockmann, C.; Asagiri, M.; Simon, M.C.; Hoffmann, A.; Johnson, R.S. Differential Activation and Antagonistic Function of HIF-α Isoforms in Macrophages Are Essential for NO Homeostasis. Genes Dev. 2010, 24, 491–501. [Google Scholar] [CrossRef]
- Choe, S.S.; Shin, K.C.; Ka, S.; Lee, Y.K.; Chun, J.S.; Kim, J.B. Macrophage HIF-2α Ameliorates Adipose Tissue Inflammation and Insulin Resistance in Obesity. Diabetes 2014, 63, 3359–3371. [Google Scholar] [CrossRef]
- Niu, Y.; Bao, L.; Chen, Y.; Wang, C.; Luo, M.; Zhang, B.; Zhou, M.; Wang, J.E.; Fang, Y.V.; Kumar, A.; et al. HIF2-Induced Long Noncoding RNA RAB11B-AS1 Promotes Hypoxia-Mediated Angiogenesis and Breast Cancer Metastasis. Cancer Res. 2020, 80, 964–975. [Google Scholar] [CrossRef] [PubMed]
- García García, C.J.; Acevedo Diaz, A.C.; Kumari, N.; Govindaraju, S.; de la Cruz Bonilla, M.; San Lucas, F.A.; Nguyen, N.D.; Jiménez Sacarello, I.; Piwnica-Worms, H.; Maitra, A.; et al. HIF2 Regulates Intestinal Wnt5a Expression. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Susen, R.M.; Bauer, R.; Olesch, C.; Fuhrmann, D.C.; Fink, A.F.; Dehne, N.; Jain, A.; Ebersberger, I.; Schmid, T.; Brüne, B. Macrophage HIF-2α Regulates Tumor-Suppressive Spint1 in the Tumor Microenvironment. Mol. Carcinog. 2019, 58, 2127–2138. [Google Scholar] [CrossRef]
- Fuhrmann, D.C.; Tausendschön, M.; Wittig, I.; Steger, M.; Ding, M.G.; Schmid, T.; Dehne, N.; Brüne, B. Inactivation of Tristetraprolin in Chronic Hypoxia Provokes the Expression of Cathepsin B. Mol. Cell Biol. 2015, 35, 619–630. [Google Scholar] [CrossRef]
- Fang, H.Y.; Hughes, R.; Murdoch, C.; Coffelt, S.B.; Biswas, S.K.; Harris, A.L.; Johnson, R.S.; Imityaz, H.Z.; Simon, M.C.; Fredlund, E.; et al. Hypoxia-Inducible Factors 1 and 2 Are Important Transcriptional Effectors in Primary Macrophages Experiencing Hypoxia. Blood 2009, 114, 844–859. [Google Scholar] [CrossRef] [PubMed]
- Imtiyaz, H.Z.; Williams, E.P.; Hickey, M.M.; Patel, S.A.; Durham, A.C.; Yuan, L.J.; Hammond, R.; Gimotty, P.A.; Keith, B.; Simon, M.C. Hypoxia-Inducible Factor 2α Regulates Macrophage Function in Mouse Models of Acute and Tumor Inflammation. J. Clin. Investig. 2010, 120, 2699–2714. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, M.; Lee, J.W.; Seo, S.W.; Brodsky, K.S.; Kreth, S.; Yang, I.V.; Buttrick, P.M.; Eckle, T.; Eltzschig, H.K. Hypoxia-Inducible Factor 2-Alpha-Dependent Induction of Amphiregulin Dampens Myocardial Ischemia-Reperfusion Injury. Nat. Commun. 2018, 9, 816. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Koeppen, M.; Seo, S.W.; Bowser, J.L.; Yuan, X.; Li, J.; Sibilia, M.; Ambardekar, A.V.; Zhang, X.; Eckle, T.; et al. Transcription-Independent Induction of ERBB1 through Hypoxia-Inducible Factor 2A Provides Cardioprotection during Ischemia and Reperfusion. Anesthesiology 2020, 132, 763–780. [Google Scholar] [CrossRef] [PubMed]
- Cowburn, A.S.; Crosby, A.; Macias, D.; Branco, C.; Colaço, R.D.D.R.; Southwood, M.; Toshner, M.; Alexander, L.E.C.; Morrell, N.W.; Chilvers, E.R.; et al. HIF2α-Arginase Axis Is Essential for the Development of Pulmonary Hypertension. Proc. Natl. Acad. Sci. USA 2016, 113, 8801–8806. [Google Scholar] [CrossRef]
- Bouthelier, A.; Meléndez-Rodríguez, F.; Urrutia, A.A.; Aragonés, J. Differential Contribution of N-and c-Terminal Regions of Hif1α and Hif2α to Their Target Gene Selectivity. Int. J. Mol. Sci. 2020, 21, 9401. [Google Scholar] [CrossRef] [PubMed]
- Wohlrab, C.; Kuiper, C.; Vissers, M.C.; Phillips, E.; Robinson, B.A.; Dachs, G.U. Ascorbate Modulates the Hypoxic Pathway by Increasing Intracellular Activity of the HIF Hydroxylases in Renal Cell Carcinoma Cells. Hypoxia 2019, 7, 17–31. [Google Scholar] [CrossRef]
- He, M.; Yang, H.; Shi, H.; Hu, Y.; Chang, C.; Liu, S.; Yeh, S. Sunitinib Increases the Cancer Stem Cells and Vasculogenic Mimicry Formation via Modulating the LncRNA-ECVSR/ERβ/Hif2-α Signaling. Cancer Lett. 2022, 524, 15–28. [Google Scholar] [CrossRef]
- Hu, C.J.; Sataur, A.; Wang, L.; Chen, H.; Simon, M.C. The N-Terminal Transactivation Domain Confers Target Gene Specificity of Hypoxia-Inducible Factors HIF-1α and HIF-2α. Mol. Biol. Cell 2007, 18, 4528–4542. [Google Scholar] [CrossRef]
- O’Rourke, J.F.; Tian, Y.M.; Ratcliffe, P.J.; Pugh, C.W. Oxygen-Regulated and Transactivating Domains in Endothelial PAS Protein 1: Comparison with Hypoxia-Inducible Factor-1α. J. Biol. Chem. 1999, 274, 2060–2071. [Google Scholar] [CrossRef]
- Roda, J.M.; Wang, Y.; Sumner, L.A.; Phillips, G.S.; Marsh, C.B.; Eubank, T.D. Stabilization of HIF-2α Induces SVEGFR-1 Production from Tumor-Associated Macrophages and Decreases Tumor Growth in a Murine Melanoma Model. J. Immunol. 2012, 189, 3168–3177. [Google Scholar] [CrossRef] [PubMed]
- Singhal, R.; Mitta, S.R.; Das, N.K.; Kerk, S.A.; Sajjakulnukit, P.; Solanki, S.; Andren, A.; Kumar, R.; Olive, K.P.; Banerjee, R.; et al. HIF-2α Activation Potentiates Oxidative Cell Death in Colorectal Cancers by Increasing Cellular Iron. J. Clin. Investig. 2021, 131, e143691. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Palte, M.J.; Deik, A.A.; Li, H.; Eaton, J.K.; Wang, W.; Tseng, Y.Y.; Deasy, R.; Kost-Alimova, M.; Dančík, V.; et al. A GPX4-Dependent Cancer Cell State Underlies the Clear-Cell Morphology and Confers Sensitivity to Ferroptosis. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gkotinakou, I.M.; Befani, C.; Samiotaki, M.; Panayotou, G.; Liakos, P. Novel HIF-2α Interaction with Reptin52 Impairs HIF-2 Transcriptional Activity and EPO Secretion. Biochem. Biophys. Res. Commun. 2021, 557, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Evagelou, S.L.; Bebenek, O.; Specker, E.J.; Uniacke, J. DEAD Box Protein Family Member DDX28 Is a Negative Regulator of Hypoxia-Inducible Factor 2α- and Eukaryotic Initiation Factor 4E2-Directed Hypoxic Translation. Mol. Cell. Biol. 2020, 40, e00610-19. [Google Scholar] [CrossRef]
- Jarman, E.J.; Ward, C.; Turnbull, A.K.; Martinez-Perez, C.; Meehan, J.; Xintaropoulou, C.; Sims, A.H.; Langdon, S.P. HER2 Regulates HIF-2α and Drives an Increased Hypoxic Response in Breast Cancer. Breast Cancer Res. 2019, 21, 1–18. [Google Scholar] [CrossRef]
- Chaturvedi, P.; Gilkes, D.M.; Takano, N.; Semenza, G.L. Hypoxia-Inducible Factor-Dependent Signaling between Triple-Negative Breast Cancer Cells and Mesenchymal Stem Cells Promotes Macrophage Recruitment. Proc. Natl. Acad. Sci. USA 2014, 111, E2120–E2129. [Google Scholar] [CrossRef]
- Roig, E.M.; Groot, A.J.; Yaromina, A.; Hendrickx, T.C.; Barbeau, L.M.O.; Giuranno, L.; Dams, G.; Ient, J.; Pimentel, V.O.; Van Gisbergen, M.W.; et al. HIF-1α and HIF-2alpha Differently Regulate the Radiation Sensitivity of NSCLC Cells. Cells 2019, 8, 45. [Google Scholar] [CrossRef]
- Gkotinakou, I.M.; Befani, C.; Simos, G.; Liakos, P. ERK1/2 Phosphorylates HIF-2α and Regulates Its Activity by Controlling Its CRM1-Dependent Nuclear Shuttling. J. Cell Sci. 2019, 132, jcs225698. [Google Scholar] [CrossRef] [PubMed]
- Nanduri, J.; Wang, N.; Yuan, G.; Khan, S.A.; Souvannakitti, D.; Peng, Y.J.; Kumar, G.K.; Garcia, J.A.; Prabhakar, N.R. Intermittent Hypoxia Degrades HIF-2α via Calpains Resulting in Oxidative Stress: Implications for Recurrent Apnea-Induced Morbidities. Proc. Natl. Acad. Sci. USA 2009, 106, 1199–1204. [Google Scholar] [CrossRef]
- Xia, P.; Yang, Y.; Liu, R.; Feng, Z.; Lin, Y.; Tang, H.; Du, J.; Cheng, Y.; Cai, J.; Hu, H.; et al. FG-4592 Alleviates Radiation-Induced Intestinal Injury by Facilitating Recovery of Intestinal Stem Cell and Reducing Damage of Intestinal Epithelial. Toxicol. Lett. 2022, 357, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Triner, D.; Xue, X.; Schwartz, A.J.; Jung, I.; Colacino, J.A.; Shah, Y.M. Epithelial Hypoxia-Inducible Factor 2α Facilitates the Progression of Colon Tumors through Recruiting Neutrophils. Mol. Cell Biol. 2017, 37, e00481-16. [Google Scholar] [CrossRef] [PubMed]
- Garcia Garcia, C.J.; Huang, Y.; Fuentes, N.R.; Turner, M.C.; Monberg, M.E.; Lin, D.; Nguyen, N.D.; Fujimoto, T.N.; Zhao, J.; Lee, J.J.; et al. Stromal HIF2 Regulates Immune Suppression in the Pancreatic Cancer Microenvironment. Gastroenterology 2022, 162, 2018–2031. [Google Scholar] [CrossRef]
- Ousman, S.S.; Kubes, P. Immune Surveillance in the Central Nervous System. Nat. Neurosci. 2012, 15, 1096–1101. [Google Scholar] [CrossRef]
- Guo, M.; Ma, X.; Feng, Y.; Han, S.; Dong, Q.; Cui, M.; Zhao, Y. In Chronic Hypoxia, Glucose Availability and Hypoxic Severity Dictate the Balance between HIF-1 and HIF-2 in Astrocytes. FASEB J. 2019, 33, 11123–11136. [Google Scholar] [CrossRef]
- Kerber, E.L.; Padberg, C.; Koll, N.; Schuetzhold, V.; Fandrey, J.; Winning, S. The Importance of Hypoxia-Inducible Factors (Hif-1 and Hif-2) for the Pathophysiology of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2020, 21, 8551. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.A.R.; Elks, P.M.; Marriott, H.M.; Eamsamarng, S.; Higgins, K.R.; Lewis, A.; Williams, L.; Parmar, S.; Shaw, G.; McGrath, E.E.; et al. Hypoxia-Inducible Factor 2α Regulates Key Neutrophil Functions in Humans, Mice, and Zebrafish. Blood 2014, 123, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Huntula, S.; Saegusa, H.; Wang, X.; Zong, S.; Tanabe, T. Involvement of N-Type Ca2+ Channel in Microglial Activation and Its Implications to Aging-Induced Exaggerated Cytokine Response. Cell Calcium 2019, 82, 102059. [Google Scholar] [CrossRef] [PubMed]
- Matak, P.; Heinis, M.; Mathieu, J.R.R.; Corriden, R.; Cuvellier, S.; Delga, S.; Mounier, R.; Rouquette, A.; Raymond, J.; Lamarque, D.; et al. Myeloid HIF-1 Is Protective in Helicobacter Pylori-Mediated Gastritis. J. Immunol. 2015, 194, 3259–3266. [Google Scholar] [CrossRef]
- Meng, C.; Liu, G.; Mu, H.; Zhou, M.; Zhang, S.; Xu, Y. Amphiregulin May Be a New Biomarker of Classically Activated Macrophages. Biochem. Biophys. Res. Commun. 2015, 466, 393–399. [Google Scholar] [CrossRef]
- Liu, S.; Geng, R.; Lin, E.; Zhao, P.; Chen, Y. ERBB1/2/3 Expression, Prognosis, and Immune Infiltration in Cutaneous Melanoma. Front. Genet. 2021, 12, 602160. [Google Scholar] [CrossRef] [PubMed]
- Eubank, T.D.; Roberts, R.; Galloway, M.; Wang, Y.; Cohn, D.E.; Marsh, C.B. GM-CSF Induces Expression of Soluble VEGF Receptor-1 from Human Monocytes and Inhibits Angiogenesis in Mice. Immunity 2004, 21, 831–842. [Google Scholar] [CrossRef]
- Eubank, T.D.; Roda, J.M.; Liu, H.; O’Neil, T.; Marsh, C.B. Opposing Roles for HIF-1α and HIF-2α in the Regulation of Angiogenesis by Mononuclear Phagocytes. Blood 2011, 117, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Roda, J.M.; Sumner, L.A.; Evans, R.; Phillips, G.S.; Marsh, C.B.; Eubank, T.D. Hypoxia-Inducible Factor-2α Regulates GM-CSF–Derived Soluble Vascular Endothelial Growth Factor Receptor 1 Production from Macrophages and Inhibits Tumor Growth and Angiogenesis. J. Immunol. 2011, 187, 1970–1976. [Google Scholar] [CrossRef] [PubMed]
- Henke, N.; Ferreirós, N.; Geisslinger, G.; Ding, M.G.; Essler, S.; Fuhrmann, D.C.; Geis, T.; Namgaladze, D.; Dehne, N.; Brüne, B. Loss of HIF-1α in Macrophages Attenuates AhR/ARNT-Mediated Tumorigenesis in a PAH-Driven Tumor Model. Oncotarget 2016, 7, 25915–25929. [Google Scholar] [CrossRef]
- Leek, R.; Talks, K.; Pezzella, F.; Turley, H.; Campo, L.; Brown, N.; Bicknell, R.; Taylor, M.; Gatter, K.C.; Harris, A. Relation of Hypoxia-Inducible Factor-2 Alpha (HIF-2 Alpha) Expression in Tumor-Infiltrative Macrophages to Tumor Angiogenesis and the Oxidative Thymidine Phosphorylase Pathway in Human Breast Cancer. Cancer Res. 2002, 62, 1326–1329. [Google Scholar]
- Liu, N.; Luo, J.; Kuang, D.; Xu, S.; Duan, Y.; Xia, Y.; Wei, Z.; Xie, X.; Yin, B.; Chen, F.; et al. Lactate Inhibits ATP6V0d2 Expression in Tumor-Associated Macrophages to Promote HIF-2α–Mediated Tumor Progression. J. Clin. Investig. 2019, 129, 631–646. [Google Scholar] [CrossRef]
- Dennis, K.L.; Blatner, N.R.; Gounari, F.; Khazaie, K. Current Status of Interleukin-10 and Regulatory T-Cells in Cancer. Curr. Opin. Oncol. 2013, 25, 637–645. [Google Scholar] [CrossRef]
- Guo, Y.; Xie, Y.Q.; Gao, M.; Zhao, Y.; Franco, F.; Wenes, M.; Siddiqui, I.; Bevilacqua, A.; Wang, H.; Yang, H.; et al. Metabolic Reprogramming of Terminally Exhausted CD8+ T Cells by IL-10 Enhances Anti-Tumor Immunity. Nat. Immunol. 2021, 22, 746–756. [Google Scholar] [CrossRef]
- Hanna, B.S.; Llaó-Cid, L.; Iskar, M.; Roessner, P.M.; Klett, L.C.; Wong, J.K.L.; Paul, Y.; Ioannou, N.; Öztürk, S.; Mack, N.; et al. Interleukin-10 Receptor Signaling Promotes the Maintenance of a PD-1int TCF-1+ CD8+ T Cell Population That Sustains Anti-Tumor Immunity. Immunity 2021, 54, 2825–2841. [Google Scholar] [CrossRef]
- Lu, X.; Prodger, A.; Sim, J.; Evans, C.E. Pulmonary Thrombosis Promotes Tumorigenesis via Myeloid Hypoxia-Inducible Factors. Biomolecules 2022, 12, 1354. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Mulder, K.; Patel, A.A.; Kong, W.T.; Piot, C.; Halitzki, E.; Dunsmore, G.; Khalilnezhad, S.; Irac, S.E.; Dubuisson, A.; Chevrier, M.; et al. Cross-Tissue Single-Cell Landscape of Human Monocytes and Macrophages in Health and Disease. Immunity 2021, 54, 1883–1900.e5. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.Y.; Black, A.; Qian, B.Z. Macrophage Diversity in Cancer Revisited in the Era of Single-Cell Omics. Trends Immunol. 2022, 43, 546–563. [Google Scholar] [CrossRef]
- Xiu, F.; Diao, L.; Qi, P.; Catapano, M.; Jeschke, M.G. Palmitate Differentially Regulates the Polarization of Differentiating and Differentiated Macrophages. Immunology 2016, 147, 82–96. [Google Scholar] [CrossRef]
- Kobayashi, H.; Gilbert, V.; Liu, Q.; Kapitsinou, P.P.; Unger, T.L.; Rha, J.; Rivella, S.; Schlöndorff, D.; Haase, V.H. Myeloid Cell-Derived Hypoxia-Inducible Factor Attenuates Inflammation in Unilateral Ureteral Obstruction-Induced Kidney Injury. J. Immunol. 2012, 188, 5106–5115. [Google Scholar] [CrossRef]
- Wang, Q.; He, Z.; Huang, M.; Liu, T.; Wang, Y.; Xu, H.; Duan, H.; Ma, P.; Zhang, L.; Zamvil, S.S.; et al. Vascular Niche IL-6 Induces Alternative Macrophage Activation in Glioblastoma through HIF-2α. Nat. Commun. 2018, 9, 559. [Google Scholar] [CrossRef]
- Xu, Z.; Zhao, L.; Zhu, L.Y.; He, M.; Zheng, L.; Wu, Y. MicroRNA-17, 20a Regulates the Proangiogenic Function of Tumor-Associated Macrophages via Targeting Hypoxia-Inducible Factor 2α. PLoS One 2013, 8, e77890. [Google Scholar] [CrossRef]
- Burke, B.; Tang, N.; Corke, K.P.; Tazzyman, D.; Ameri, K.; Wells, M.; Lewis, C.E. Expression of HIF-Iα by Human Macrophages: Implications for the Use of Macrophages in Hypoxia-Regulated Cancer Gene Therapy. J. Pathol. 2002, 196, 204–212. [Google Scholar] [CrossRef]
- Talks, K.L.; Turley, H.; Gatter, K.C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J.; Harris, A.L. The Expression and Distribution of the Hypoxia-Inducible Factors HIF-1α and HIF-2α in Normal Human Tissues, Cancers, and Tumor-Associated Macrophages. Am. J. Pathol. 2000, 157, 411–421. [Google Scholar] [CrossRef]
- Pietras, A.; Gisselsson, D.; Øra, I.; Noguera, R.; Beckman, S.; Navarro, S.; Påhlman, S. High Levels of HIF-2α Highlight an Immature Neural Crest-like Neuroblastoma Cell Cohort Located in a Perivascular Niche. J. Pathol. 2008, 214, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Villacampa, P.; Liyanage, S.E.; Klaska, I.P.; Cristante, E.; Menger, K.E.; Sampson, R.D.; Barlow, M.; Abelleira-Hervas, L.; Duran, Y.; Smith, A.J.; et al. Stabilization of Myeloid-Derived HIFs Promotes Vascular Regeneration in Retinal Ischemia. Angiogenesis 2020, 23, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, X.; Xia, J.; Zhang, L.; Chen, B.; Lian, G.; Yun, C.; Yang, J.; Yan, Y.; Wang, P.; et al. Macrophage HIF-2α Suppresses NLRP3 Inflammasome Activation and Alleviates Insulin Resistance. Cell Rep. 2021, 36, 109607. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The Chemokine System in Diverse Forms of Macrophage Activation and Polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Aras, S.; Raza Zaidi, M. TAMeless Traitors: Macrophages in Cancer Progression and Metastasis. Br. J. Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(Lps+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Cramer, T.; Yamanishi, Y.; Clausen, B.E.; Förster, I.; Pawlinski, R.; Mackman, N.; Haase, V.H.; Jaenisch, R.; Corr, M.; Nizet, V.; et al. Erratum: HIF-1α Is Essential for Myeloid Cell-Mediated Inflammation (Cell 112:5). Cell 2003, 113, 419. [Google Scholar] [CrossRef]
- Cramer, T.; Wiedenmann, B.; Höcker, M. HIF–1alpha Regulates the Bactericidal Capacity of Phagocytes. Z. Gastroenterol. 2005, 43, P122. [Google Scholar] [CrossRef]
- DeBerge, M.; Lantz, C.; Dehn, S.; Sullivan, D.P.; van der Laan, A.M.; Niessen, H.W.M.; Flanagan, M.E.; Brat, D.J.; Feinstein, M.J.; Kaushal, S.; et al. Hypoxia-Inducible Factors Individually Facilitate Inflammatory Myeloid Metabolism and Inefficient Cardiac Repair. J. Exp. Med. 2021, 218, e20200667. [Google Scholar] [CrossRef]
- Li, X.; Cui, X.-X.; Chen, Y.-J.; Wu, T.-T.; Xu, H.; Yin, H.; Wu, Y.-C. Therapeutic Potential of a Prolyl Hydroxylase Inhibitor FG-4592 for Parkinson’s Diseases in Vitro and in Vivo: Regulation of Redox Biology and Mitochondrial Function. Front. Aging Neurosci. 2018, 10, 121. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhao, M.; Cheng, X.; Zhao, T.; Feng, Z.; Zhao, Y.; Fan, M.; Zhu, L. FG-4592 Improves Depressive-Like Behaviors through HIF-1-Mediated Neurogenesis and Synapse Plasticity in Rats. Neurotherapeutics 2020, 17, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Poblete, J.M.S.; Ballinger, M.N.; Bao, S.; Alghothani, M.; Nevado, J.B.; Eubank, T.D.; Christman, J.W.; Magalang, U.J. Macrophage HIF-1α Mediates Obesity-Related Adipose Tissue Dysfunction via Interleukin-1 Receptor-Associated Kinase M. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E689–E700. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gao, Y.; Li, Y.; Wang, C.; Chen, D.; Gao, Y.; Ran, X. Severe Intermittent Hypoxia Modulates the Macrophage Phenotype and Impairs Wound Healing Through Downregulation of HIF-2α. Nat. Sci. Sleep 2022, 14, 1511–1520. [Google Scholar] [CrossRef]
- Ahmed, S.; Ayscough, A.; Barker, G.R.; Canning, H.E.; Davenport, R.; Downham, R.; Harrison, D.; Jenkins, K.; Kinsella, N.; Livermore, D.G.; et al. 1,2,4-Triazolo-[1,5-a]Pyridine HIF Prolylhydroxylase Domain-1 (PHD-1) Inhibitors with a Novel Monodentate Binding Interaction. J. Med. Chem. 2017, 60, 5663–5672. [Google Scholar] [CrossRef]
- Yu, Y.; Yang, F.; Yu, Q.; Liu, S.; Wu, C.; Su, K.; Yang, L.; Bao, X.; Li, Z.; Li, X.; et al. Discovery of a Potent and Orally Bioavailable Hypoxia-Inducible Factor 2α (HIF-2α) Agonist and Its Synergistic Therapy with Prolyl Hydroxylase Inhibitors for the Treatment of Renal Anemia. J. Med. Chem. 2021, 64, 17384–17402. [Google Scholar] [CrossRef]
- Watts, E.R.; Walmsley, S.R. Inflammation and Hypoxia: HIF and PHD Isoform Selectivity. Trends Mol. Med. 2019, 25, 33–46. [Google Scholar] [CrossRef]
- Singh, A.K.; Szczech, L.; Tang, K.L.; Barnhart, H.; Sapp, S.; Wolfson, M.; Reddan, D. Correction of Anemia with Epoetin Alfa in Chronic Kidney Disease. N. Engl. J. Med. 2006, 355, 2085–2098. [Google Scholar] [CrossRef]
- Courtney, K.D.; Ma, Y.; de Leon, A.D.; Christie, A.; Xie, Z.; Woolford, L.; Singla, N.; Joyce, A.; Hill, H.; Madhuranthakam, A.J.; et al. HIF-2 Complex Dissociation, Target Inhibition, and Acquired Resistance with PT2385, a First-in-Class HIF-2 Inhibitor, in Patients with Clear Cell Renal Cell Carcinoma. Clin. Cancer Res. 2020, 26, 793–803. [Google Scholar] [CrossRef]
- Courtney, K.D.; Infante, J.R.; Lam, E.T.; Figlin, R.A.; Rini, B.I.; Brugarolas, J.; Zojwalla, N.J.; Lowe, A.M.; Wang, K.; Wallace, E.M.; et al. Phase I Dose-Escalation Trial of PT2385, a First-in-Class Hypoxia-Inducible Factor-2a Antagonist in Patients with Previously Treated Advanced Clear Cell Renal Cell Carcinoma. J. Clin. Oncol. 2018, 36, 867–874. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Bauer, T.M.; Papadopoulos, K.P.; Plimack, E.R.; Merchan, J.R.; McDermott, D.F.; Michaelson, M.D.; Appleman, L.J.; Thamake, S.; Perini, R.F.; et al. Inhibition of Hypoxia-Inducible Factor-2α in Renal Cell Carcinoma with Belzutifan: A Phase 1 Trial and Biomarker Analysis. Nat. Med. 2021, 27, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Koizume, S.; Miyagi, Y. Diverse Mechanisms of Sp1-Dependent Transcriptional Regulation Potentially Involved in the Adaptive Response of Cancer Cells to Oxygen-Deficient Conditions. Cancers 2015, 8, 2. [Google Scholar] [CrossRef]
- Schödel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-Resolution Genome-Wide Mapping of HIF-Binding Sites by ChIP-Seq. Blood 2011, 117, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.C.; Huang, H.J.; Chang, T.H.; Huang, H.C.; Hsieh, S.Y.; Chen, Y.S.; Chou, W.Y.; Chiang, C.H.; Lai, C.H.; Shiau, C.Y. Genome-Wide Analysis of HIF-2α Chromatin Binding Sites under Normoxia in Human Bronchial Epithelial Cells (BEAS-2B) Suggests Its Diverse Functions. Sci. Rep. 2016, 6, 29311. [Google Scholar] [CrossRef]
- Ohtori, S.; Yamashita, M.; Inoue, G.; Yamauchi, K.; Koshi, T.; Suzuki, M.; Takaso, M.; Orita, S.; Eguchi, Y.; Ochiai, N.; et al. Rotational Hypermobility of Disc Wedging Using Kinematic CT: Preliminary Study to Investigate the Instability of Discs in Degenerated Scoliosis in the Lumbar Spine. Eur. Spine J. 2010, 19, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Bader, S.B.; Dewhirst, M.W.; Hammond, E.M. Review Cyclic Hypoxia: An Update on Its Characteristics, Methods to Measure It and Biological Implications in Cancer. Cancers 2021, 13, 23. [Google Scholar] [CrossRef] [PubMed]
- Doedens, A.L.; Stockmann, C.; Rubinstein, M.P.; Liao, D.; Zhang, N.; Denardo, D.G.; Coussens, L.M.; Karin, M.; Goldrath, A.W.; Johnson, R.S. Macrophage Expression of HIF-1alpha Suppresses T Cell Function and Promotes Tumor Progression. Cancer Res. 2010, 70, 7465–7475. [Google Scholar] [CrossRef]
- Lin, C.; Yang, H.; Zhao, W.; Wang, W. CTSB+ Macrophage Repress Memory Immune Hub in the Liver Metastasis Site of Colorectal Cancer Patient Revealed by Multi-Omics Analysis. Biochem. Biophys. Res. Commun. 2022, 626, 8–14. [Google Scholar] [CrossRef]
- Lim, J.P.; Teasdale, R.D.; Gleeson, P.A. SNX5 Is Essential for Efficient Macropinocytosis and Antigen Processing in Primary Macrophages. Biol. Open 2012, 1, 904–914. [Google Scholar] [CrossRef]
- Kramer, P.R.; Winger, V.; Kramer, S.F. 17β-Estradiol Utilizes the Estrogen Receptor to Regulate CD16 Expression in Monocytes. Mol. Cell Endocrinol. 2007, 279, 16–25. [Google Scholar] [CrossRef]
- Zhang, R.; Cheung, C.Y.; Seo, S.U.; Liu, H.; Pardeshi, L.; Wong, K.H.; Chow, L.M.C.; Chau, M.P.; Wang, Y.; Lee, A.R.; et al. RUVBL1/2 Complex Regulates Pro-Inflammatory Responses in Macrophages via Regulating Histone H3K4 Trimethylation. Front. Immunol. 2021, 12, 679184. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Huang, Y.; Bong, R.; Ding, Y.; Song, N.; Wang, X.; Song, X.; Luo, Y. Tumor-Associated Macrophages Promote Angiogenesis and Melanoma Growth via Adrenomedullin in a Paracrine and Autocrine Manner. Clin. Cancer Res. 2011, 17, 7230–7239. [Google Scholar] [CrossRef] [PubMed]
- Duong, T.; Koopman, P.; Francois, M. Tumor Lymphangiogenesis as a Potential Therapeutic Target. J. Oncol. 2012, 2012, 204946. [Google Scholar] [CrossRef] [PubMed]
- Oehler, M.K.; Fischer, D.C.; Orlowska-Volk, M.; Herrle, F.; Kieback, D.G.; Rees, M.; Bicknell, R. Tissue and Plasma Expression of the Angiogenic Peptide Adrenomedullin in Breast Cancer. Br. J. Cancer 2003, 89, 1927–1933. [Google Scholar] [CrossRef]
- Ran, S.; Montgomery, K.E. Macrophage-Mediated Lymphangiogenesis: The Emerging Role of Macrophages as Lymphatic Endothelial Progenitors. Cancers 2012, 4, 618–657. [Google Scholar] [CrossRef] [PubMed]
- Zudaire, E.; Martínez, A.; Cuttitta, F. Adrenomedullin and Cancer. Regul. Pept. 2003, 112, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Bunton, D.C.; Petrie, M.C.; Hillier, C.; Johnston, F.; McMurray, J.J.V. The Clinical Relevance of Adrenomedullin: A Promising Profile? Pharmacol. Ther. 2004, 103, 179–201. [Google Scholar] [CrossRef]
- Kubot, A.; Minamino, N.; Isumi, Y.; Katafuchi, T.; Kangawa, K.; Dohi, K.; Matsuo, H. Production of Adrenomedullin in Macrophage Cell Line and Peritoneal Macrophage. J. Biol. Chem. 1998, 273, 16730–16738. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, Z.; Zhang, A.; Gupte, A.A.; Hamilton, D.J. The Role of Calcium Signaling in Melanoma. Int. J. Mol. Sci. 2022, 23, 1010. [Google Scholar] [CrossRef]
- Chen, Y.-F.; Chen, Y.-T.; Chiu, W.-T.; Shen, M.-R. Remodeling of Calcium Signaling in Tumorprogression. J. Biomed. Sci. 2013, 1, 23. [Google Scholar] [CrossRef]
- Roberts-Thomson, S.J.; Chalmers, S.B.; Monteith, G.R. The Calcium-Signaling Toolkit in Cancer: Remodeling and Targeting. Cold Spring Harb. Perspect. Biol. 2019, 11, a035204. [Google Scholar] [CrossRef] [PubMed]
- Consonni, S.V.; Maurice, M.M.; Bos, J.L. DEP Domains: Structurally Similar but Functionally Different. Nat. Rev. Mol. Cell Biol. 2014, 15, 357–362. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, E.L.; Ferravante, A.; Scudiero, I.; Zotti, T.; Reale, C.; Pizzulo, M.; De La Motte, L.R.; De Maio, C.; Mazzone, P.; Telesio, G.; et al. The Dishevelled, EGL-10 and Pleckstrin (DEP) Domain-Containing Protein DEPDC7 Binds to CARMA2 and CARMA3 Proteins, and Regulates NF-ΚB Activation. PLoS ONE 2014, 9, e116062. [Google Scholar] [CrossRef]
- Liao, Z.; Wang, X.; Wang, X.; Li, L.; Lin, D. DEPDC7 Inhibits Cell Proliferation, Migration and Invasion in Hepatoma Cells. Oncol. Lett. 2017, 14, 7332–7338. [Google Scholar] [CrossRef][Green Version]
- Poth, J.M.; Brodsky, K.; Ehrentraut, H.; Grenz, A.; Eltzschig, H.K. Transcriptional Control of Adenosine Signaling by Hypoxia-Inducible Transcription Factors during Ischemic or Inflammatory Disease. J. Mol. Med. 2013, 91, 183–193. [Google Scholar] [CrossRef]
- Kong, T.; Westerman, K.A.; Faigle, M.; Eltzschig, H.K.; Colgan, S.P. HIF-dependent Induction of Adenosine A2B Receptor in Hypoxia. FASEB J. 2006, 20, 2242–2250. [Google Scholar] [CrossRef]
- Yuan, X.; Mills, T.; Doursout, M.F.; Evans, S.E.; Vidal Melo, M.F.; Eltzschig, H.K. Alternative Adenosine Receptor Activation: The Netrin-Adora2b Link. Front. Pharmacol. 2022, 13, 944994. [Google Scholar] [CrossRef]
- Ahmad, A.; Ahmad, S.; Glover, L.; Miller, S.M.; Shannon, J.M.; Guo, X.; Franklin, W.A.; Bridges, J.P.; Schaack, J.B.; Colgan, S.P.; et al. Adenosine A2A Receptor Is a Unique Angiogenic Target of HIF-2α in Pulmonary Endothelial Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 10684–10689. [Google Scholar] [CrossRef]
- Hadi, T.; Boytard, L.; Silvestro, M.; Alebrahim, D.; Jacob, S.; Feinstein, J.; Barone, K.; Spiro, W.; Hutchison, S.; Simon, R.; et al. Macrophage-Derived Netrin-1 Promotes Abdominal Aortic Aneurysm Formation by Activating MMP3 in Vascular Smooth Muscle Cells. Nat. Commun. 2018, 9, 5022. [Google Scholar] [CrossRef]
- Ramkhelawon, B.; Yang, Y.; van Gils, J.M.; Hewing, B.; Rayner, K.J.; Parathath, S.; Guo, L.; Oldebeken, S.; Feig, J.L.; Fisher, E.A.; et al. Hypoxia Induces Netrin-1 and Unc5b in Atherosclerotic Plaques. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1180–1188. [Google Scholar] [CrossRef]
- Cekic, C.; Day, Y.J.; Sag, D.; Linden, J. Myeloid Expression of Adenosine A2A Receptor Suppresses T and NK Cell Responses in the Solid Tumor Microenvironment. Cancer Res. 2014, 74, 7250–7259. [Google Scholar] [CrossRef]
- Ohta, A.; Sitkovsky, M. Role of G-Protein-Coupled Adenosine Receptors in Downregulation of Inflammation and Protection from Tissue Damage. Nat. für Gastroenterol. 2001, 414, 916–920. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Ibla, J.C.; Furuta, G.T.; Leonard, M.O.; Jacobson, K.A.; Enjyoji, K.; Robson, S.C.; Colgan, S.R. Coordinated Adenine Nucleotide Phosphohydrolysis and Nucleoside Signaling in Posthypoxic Endothelium: Role of Ectonucleotidases and Adenosine A 2B Receptors. J. Exp. Med. 2003, 198, 783–796. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Thompson, L.F.; Karhausen, J.; Cotta, R.J.; Ibla, J.C.; Robson, S.C.; Colgan, S.P. Endogenous Adenosine Produced during Hypoxia Attenuates Neutrophil Accumulation: Coordination by Extracellular Nucleotide Metabolism. Blood 2004, 104, 3986–3992. [Google Scholar] [CrossRef]
- Eckle, T.; Kewley, E.M.; Brodsky, K.S.; Tak, E.; Bonney, S.; Gobel, M.; Anderson, D.; Glover, L.E.; Riegel, A.K.; Colgan, S.P.; et al. Identification of Hypoxia-Inducible Factor HIF-1A as Transcriptional Regulator of the A2B Adenosine Receptor during Acute Lung Injury. J. Immunol. 2014, 192, 1249–1256. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HIF-2α Regulated Genes | Prior Literature |
|---|---|
| ADM | Induced by hypoxia in HUVECs [45,46] and macrophages [47] |
| ANGPTL4 | Induced by hypoxia in HUVECs and human synoviocytes [45,46,49], regulated by HIF-2α in mouse embryonic fibroblasts [50] |
| C1orf21 | Induced by hypoxia in HUVECs [45,46] |
| MAGI1 | Induced by hypoxia in HUVECs [45,46] |
| PTGIS | Induced by hypoxia in HUVECs [32,33] and fibroblasts [51] |
| LUCAT1 | Induced by hypoxia in *HUVECs [48] |
| MIR210HG | Induced by hypoxia in *HUVECs [48] |
| BNIP3L | Induced by hypoxia in *HUVECs [48] |
| EGLN3 | Induced by hypoxia in *HUVECs [45,46,48] and mouse macrophages [47] |
| SDF1 | Induced by hypoxia in human pulmonary ECs [52] |
| CXCR4 | Induced by hypoxia in human pulmonary ECs [52] |
| ICAM1 | Induced by hypoxia in human pulmonary ECs [52] |
| TGFA | Induced by hypoxia in human pulmonary ECs [52] |
| WISP2 | Induced by hypoxia in breast cancer cell lines [53] |
| Vegfa | Induced by hypoxia in *retinal organoids [54] and * BMDMs [9] |
| Arg-1 | Reduced in * peritoneal macrophages [55] from HIF-2αflox/flox;Tekcre+/− or peritoneal macrophages [56] treated with HIF-2α siRNA |
| RAB11B-AS1 | Hypoxia-induced expression reduced in HIF-2α knockout human breast cancer cell lines [57] |
| WNT5 | Luciferase reporter assays in AD-293 cells showed HIF-2α directly activates the WNT5A promoter [58] |
| Spint1 | HIF-2α-deficient BMDMs produce less Spint1 than controls [59] |
| ADORA2A | mRNA expression downregulated by HIF-2α siRNA in human MDMs [60] |
| CTSB | Hypoxic induction prevented in primary human macrophages transfected with HIF-2α siRNA [60] |
| SNX5 | Hypoxic induction prevented in primary human macrophages transfected with HIF-2α siRNA [60] |
| IL-1 | Hypoxic induction prevented in human MDMs transfected with HIF-2α siRNA [61] |
| Cxcl2 | Reduced expression in hypoxia-treated murine BMDMs deficient in HIF-2α [62] |
| Areg | Reduced in murine ischemic cardiac tissue of Hif2aloxP/loxPMyosin-Cre+ [63] |
| ERBB1 | Reduced in human cardiac myocytes using HIF-2α shRNA [64] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steinberger, K.J.; Eubank, T.D. The Underexplored Landscape of Hypoxia-Inducible Factor 2 Alpha and Potential Roles in Tumor Macrophages: A Review. Oxygen 2023, 3, 45-76. https://doi.org/10.3390/oxygen3010005
Steinberger KJ, Eubank TD. The Underexplored Landscape of Hypoxia-Inducible Factor 2 Alpha and Potential Roles in Tumor Macrophages: A Review. Oxygen. 2023; 3(1):45-76. https://doi.org/10.3390/oxygen3010005
Chicago/Turabian StyleSteinberger, Kayla J., and Timothy D. Eubank. 2023. "The Underexplored Landscape of Hypoxia-Inducible Factor 2 Alpha and Potential Roles in Tumor Macrophages: A Review" Oxygen 3, no. 1: 45-76. https://doi.org/10.3390/oxygen3010005
APA StyleSteinberger, K. J., & Eubank, T. D. (2023). The Underexplored Landscape of Hypoxia-Inducible Factor 2 Alpha and Potential Roles in Tumor Macrophages: A Review. Oxygen, 3(1), 45-76. https://doi.org/10.3390/oxygen3010005