
Comparison of Oxygen Electrode Chronoamperometry and Spectrophotometry for Determination of Catalase Activity



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Spectrophotometry Measurements of Catalase Activity
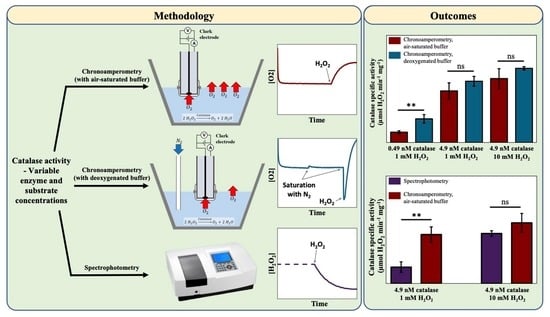
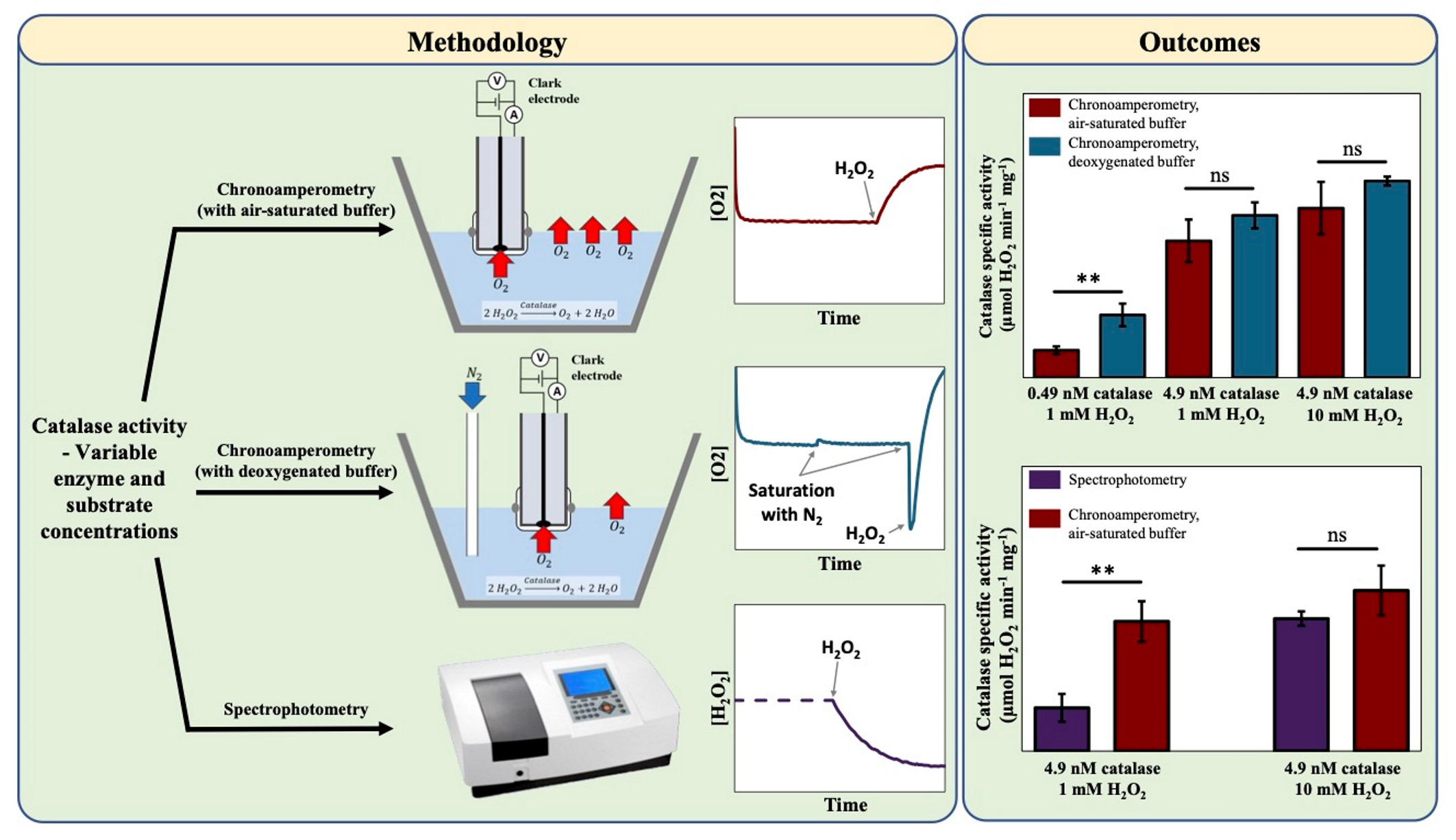
2.3. Preparation of the Oxygen Electrode
2.4. Chronoamperometry Meaurements of Catalase Activity
2.5. Calculation of Specific Catalase Activity
2.6. Statistics
3. Results
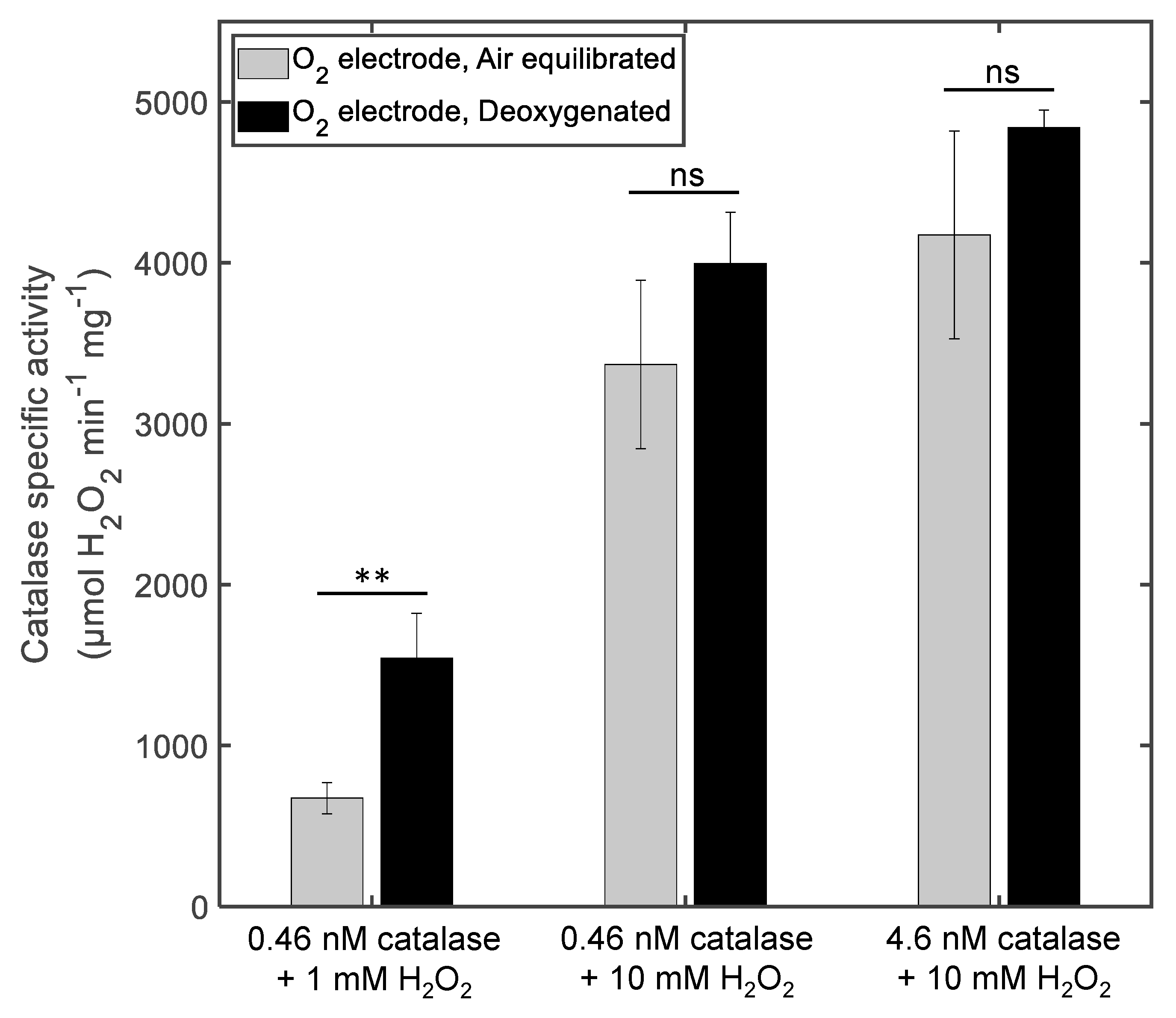
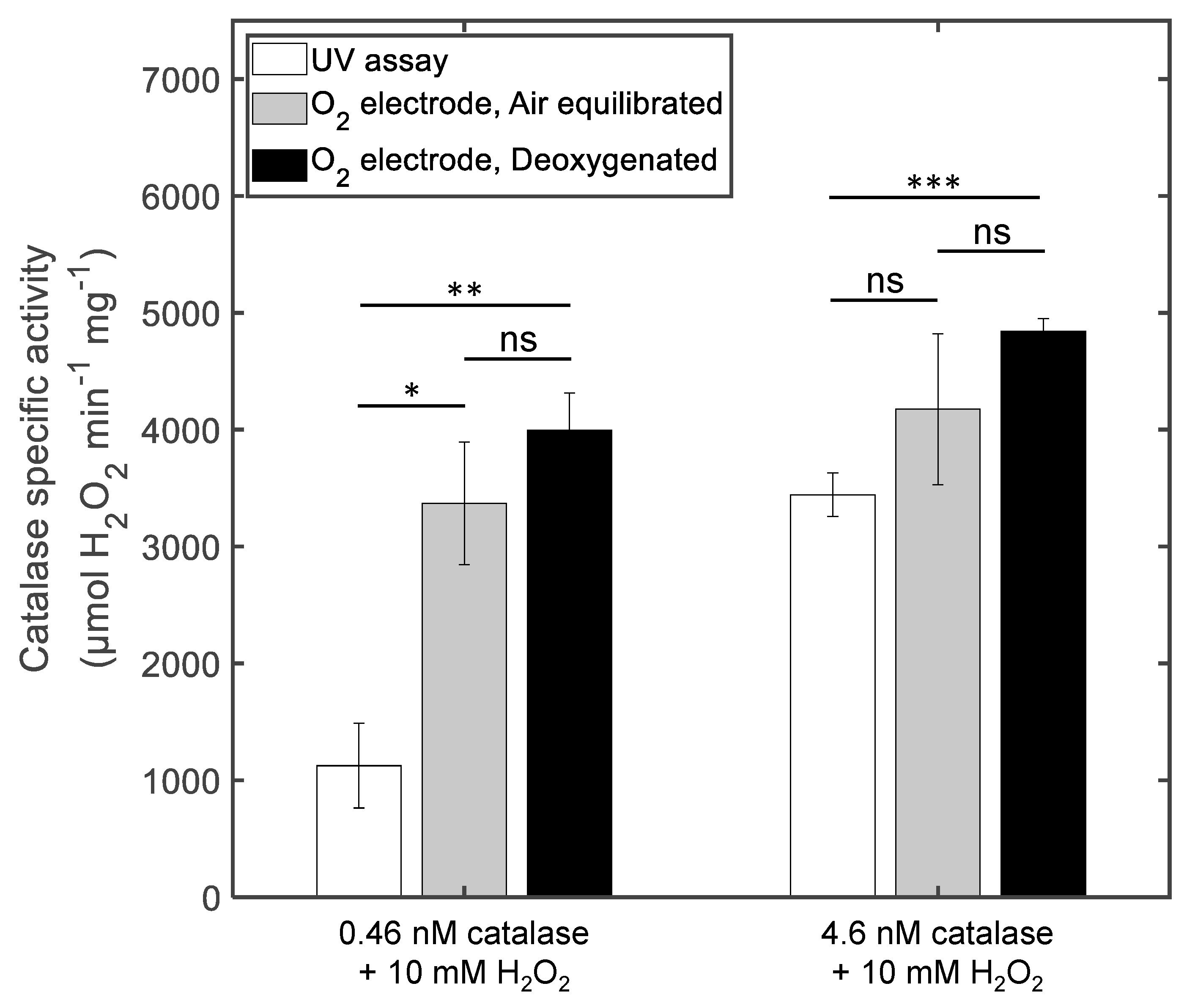
3.1. The Effect of Deoxygenation on Chronoamperometry Meaurements of Catalase Activity
3.2. Comparison between Chronoamperometry and Spectrophotometry Measuerments of Catalase Activity
4. Discussion
4.1. The Influence of Deoxygenation in the Case of Chronoamperometry Meaurements of Catalase Activity
4.2. Comparison between Chronoamperometry and Spectrophotometry Measurements of Catalase Activity
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Tirichen, H.; Yaigoub, H.; Xu, W.; Wu, C.; Li, R.; Li, Y. Mitochondrial Reactive Oxygen Species and Their Contribution in Chronic Kidney Disease Progression Through Oxidative Stress. Front. Physiol. 2021, 12, 627837. [Google Scholar] [CrossRef] [PubMed]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Herb, M.; Schramm, M. Functions of ROS in Macrophages and Antimicrobial Immunity. Antioxidants 2021, 10, 313. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxidative Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Yang, X.; Li, Y.; Li, Y.; Ren, X.; Zhang, X.; Hu, D.; Gao, Y.; Xing, Y.; Shang, H. Oxidative Stress-Mediated Atherosclerosis: Mechanisms and Therapies. Front. Physiol. 2017, 8, 600. [Google Scholar] [CrossRef]
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta 2006, 1758, 994–1003. [Google Scholar] [CrossRef]
- Patella, B.; Vincenzo, S.D.; Zanca, C.; Bollaci, L.; Ferraro, M.; Giuffrè, M.R.; Cipollina, C.; Bruno, M.G.; Aiello, G.; Russo, M.; et al. Electrochemical Quantification of H2O2 Released by Airway Cells Growing in Different Culture Media. Micromachines 2022, 13, 1762. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef]
- Mirończuk-Chodakowska, I.; Witkowska, A.M.; Zujko, M.E. Endogenous non-enzymatic antioxidants in the human body. Adv. Med. Sci. 2018, 63, 68–78. [Google Scholar] [CrossRef]
- Nimse, S.B.; Pal, D. Free radicals, natural antioxidants, and their reaction mechanisms. RSC Adv. 2015, 5, 27986–28006. [Google Scholar] [CrossRef]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef]
- Kirkman, H.N.; Gaetani, G.F. Mammalian catalase: A venerable enzyme with new mysteries. Trends Biochem. Sci. 2007, 32, 44–50. [Google Scholar] [CrossRef]
- Rojkind, M.; Domínguez-Rosales, J.-A.; Nieto, N.; Greenwel, P. Role of hydrogen peroxide and oxidative stress in healing responses. Cell. Mol. Life Sci. 2002, 59, 1872–1891. [Google Scholar] [CrossRef] [PubMed]
- Aragon, C.M.G.; Rogan, F.; Amit, Z. Ethanol metabolism in rat brain homogenates by a catalase-H2O2 system. Biochem. Pharmacol. 1992, 44, 93–98. [Google Scholar] [CrossRef]
- Middelkoop, E.; Wiemer, E.A.C.; Schoenmaker, D.E.T.; Strijland, A.; Tager, J.M. Topology of catalase assembly in human skin fibroblasts. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Res. 1993, 1220, 15–20. [Google Scholar] [CrossRef]
- Yano, S.; Arroyo, N.; Yano, N. SHP2 binds catalase and acquires a hydrogen peroxide-resistant phosphatase activity via integrin-signaling. FEBS Lett. 2004, 577, 327–332. [Google Scholar] [CrossRef]
- Hellemans, L.; Corstjens, H.; Neven, A.; Declercq, L.; Maes, D. Antioxidant enzyme activity in human stratum corneum shows seasonal variation with an age-dependent recovery. J. Investig. Dermatol. 2003, 120, 434–439. [Google Scholar] [CrossRef]
- Guarrera, M.; Ferrari, P.; Rebora, A. Catalase in the stratum corneum of patients with polymorphic light eruption. Acta Derm.-Venereol. 1998, 78, 335–336. [Google Scholar] [CrossRef]
- Szczepanczyk, M.; Ruzgas, T.; Gullfot, F.; Gustafsson, A.; Björklund, S. Catalase Activity in Keratinocytes, Stratum Corneum, and Defatted Algae Biomass as a Potential Skin Care Ingredient. Biomedicines 2021, 9, 1868. [Google Scholar] [CrossRef] [PubMed]
- Hernández, A.R.; Boutonnet, M.; Svensson, B.; Butler, E.; Lood, R.; Blom, K.; Vallejo, B.; Anderson, C.; Engblom, J.; Ruzgas, T.; et al. New concepts for transdermal delivery of oxygen based on catalase biochemical reactions studied by oxygen electrode amperometry. J. Control. Release 2019, 306, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Björklund, S.; Nowacka, A.; Bouwstra, J.A.; Sparr, E.; Topgaard, D. Characterization of stratum corneum molecular dynamics by natural-abundance 13C solid-state NMR. PLoS ONE 2013, 8, e61889. [Google Scholar] [CrossRef] [PubMed]
- Fukui, M.; Kanoh, M.; Takamatsu, Y.; Arakawa, Y. Analysis of serum catalase activities in pancreatic diseases. J. Gastroenterol. 2004, 39, 469–474. [Google Scholar] [CrossRef]
- Cullen, J.J.; Mitros, F.A.; Oberley, L.W. Expression of Antioxidant Enzymes in Diseases of the Human Pancreas: Another Link Between Chronic Pancreatitis and Pancreatic Cancer. Pancreas 2003, 26, 23–27. [Google Scholar] [CrossRef]
- Sander, C.S.; Hamm, F.; Elsner, P.; Thiele, J.J. Oxidative stress in malignant melanoma and non-melanoma skin cancer. Br. J. Dermatol. 2003, 148, 913–922. [Google Scholar] [CrossRef]
- Carvalho, L.A.C.; Queijo, R.G.; Baccaro, A.L.B.; Siena, Á.D.D.; Silva, W.A.; Rodrigues, T.; Maria-Engler, S.S. Redox-Related Proteins in Melanoma Progression. Antioxidants 2022, 11, 438. [Google Scholar] [CrossRef]
- Hwang, T.S.; Choi, H.K.; Han, H.S. Differential expression of manganese superoxide dismutase, copper/zinc superoxide dismutase, and catalase in gastric adenocarcinoma and normal gastric mucosa. Eur. J. Surg. Oncol. (EJSO) 2007, 33, 474–479. [Google Scholar] [CrossRef]
- Schallreuter, K.U.; Moore, J.; Wood, J.M.; Beazley, W.D.; Gaze, D.C.; Tobin, D.J.; Marshall, H.S.; Panske, A.; Panzig, E.; Hibberts, N.A. In vivo and in vitro evidence for hydrogen peroxide (H2O2) accumulation in the epidermis of patients with vitiligo and its successful removal by a UVB-activated pseudocatalase. J. Investig. Dermatol. Symp. Proc. 1999, 4, 91–96. [Google Scholar] [CrossRef]
- Hadwan, M.H. Simple spectrophotometric assay for measuring catalase activity in biological tissues. BMC Biochem. 2018, 19, 7. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Woollard, A.C.; Wolff, S.P. Hydrogen peroxide production during experimental protein glycation. FEBS Lett. 1990, 268, 69–71. [Google Scholar] [CrossRef]
- Ou, P.; Wolff, S.P. A discontinuous method for catalase determination at ‘near physiological’ concentrations of H2O2 and its application to the study of H2O2 fluxes within cells. J. Biochem. Biophys. Methods 1996, 31, 59–67. [Google Scholar] [CrossRef]
- Clark, L.C.J.; Lyons, C. Electrode systems for continuous monitoring in cardiovascular surgery. Ann. New York Acad. Sci. 1962, 102, 29–45. [Google Scholar] [CrossRef] [PubMed]
- del Río, L.A.; Ortega, M.G.; López, A.L.; Gorgé, J.L. A more sensitive modification of the catalase assay with the Clark oxygen electrode: Application to the kinetic study of the pea leaf enzyme. Anal. Biochem. 1977, 80, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Nocchi, S.; Björklund, S.; Svensson, B.; Engblom, J.; Ruzgas, T. Electrochemical monitoring of native catalase activity in skin using skin covered oxygen electrode. Biosens. Bioelectron. 2017, 93, 9–13. [Google Scholar] [CrossRef]
- Eskandari, M.; Rembiesa, J.; Startaite, L.; Holefors, A.; Valanciute, A.; Faridbod, F.; Ganjali, M.R.; Engblom, J.; Ruzgas, T. Polyphenol-hydrogen peroxide reactions in skin: In vitro model relevant to study ROS reactions at inflammation. Anal. Chim. Acta 2019, 1075, 91–97. [Google Scholar] [CrossRef]
- Truesdale, G.A.; Downing, A.L.; Lowden, G. The solubility of oxygen in pure water and sea-water. J. Chem. Technol. Biotechnol. 2007, 5, 53–62. [Google Scholar] [CrossRef]
- Scaglione, C.N.; Xu, Q.; Ramanujan, V.K. Direct measurement of catalase activity in living cells and tissue biopsies. Biochem. Biophys. Res. Commun. 2016, 470, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Tomassetti, M.; Pezzilli, R.; Prestopino, G.; Di Biagio, F.; Di Natale, C.; Medaglia, P.G. A New Clark-Type Layered Double Hydroxides-Enzyme Biosensor for H2O2 Determination in Highly Diluted Real Matrices: Milk and Cosmetics. Processes 2021, 9, 1878. [Google Scholar] [CrossRef]
- Beers, R.F.; Sizer, I.W. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J. Biol. Chem. 1952, 195, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Mumby, S.; Block, R.; Petros, A.J.; Gutteridge, J.M.C. Hydrogen peroxide and catalase are inversely related in adult patients undergoing cardiopulmonary bypass: Implications for antioxidant protection. Redox Rep. 1999, 4, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Ueno, S.; Iwasaka, M. Catalytic activity of catalase under strong magnetic fields of up to 8 T. J. Appl. Phys. 1996, 79, 4705. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Catalase (nM) | H2O2 (mM) | Protocol | v (µM O2 s−1) | Specific Activity (µmol H2O2 min−1 mg−1) |
|---|---|---|---|---|---|
| O2 electrode | 0.46 | 1 | Air equilibrated | 0.62 ± 0.088 | 670 ± 100 |
| 0.46 | 1 | Deoxygenated | 1.4 ± 0.26 | 1540 ± 280 | |
| 0.46 | 10 | Air equilibrated | 3.1 ± 0.48 | 3370 ± 520 | |
| 0.46 | 10 | Deoxygenated | 4.7 ± 0.29 | 4000 ± 320 | |
| 4.6 | 10 | Air equilibrated | 38 ± 5.9 | 4170 ± 650 | |
| 4.6 | 10 | Deoxygenated | 44 ± 0.10 | 4840 ± 110 | |
| UV assay | 0.46 | 10 | Air equilibrated | 1.0 ± 0.23 | 1130 ± 360 |
| 4.6 | 10 | Air equilibrated | 32 ± 1.7 | 3440 ± 190 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szczepanczyk, M.; Paul, L.; Ruzgas, T.; Björklund, S. Comparison of Oxygen Electrode Chronoamperometry and Spectrophotometry for Determination of Catalase Activity. Oxygen 2023, 3, 77-89. https://doi.org/10.3390/oxygen3010006
Szczepanczyk M, Paul L, Ruzgas T, Björklund S. Comparison of Oxygen Electrode Chronoamperometry and Spectrophotometry for Determination of Catalase Activity. Oxygen. 2023; 3(1):77-89. https://doi.org/10.3390/oxygen3010006
Chicago/Turabian StyleSzczepanczyk, Michal, Lea Paul, Tautgirdas Ruzgas, and Sebastian Björklund. 2023. "Comparison of Oxygen Electrode Chronoamperometry and Spectrophotometry for Determination of Catalase Activity" Oxygen 3, no. 1: 77-89. https://doi.org/10.3390/oxygen3010006
APA StyleSzczepanczyk, M., Paul, L., Ruzgas, T., & Björklund, S. (2023). Comparison of Oxygen Electrode Chronoamperometry and Spectrophotometry for Determination of Catalase Activity. Oxygen, 3(1), 77-89. https://doi.org/10.3390/oxygen3010006