Abstract
Тhe adequacy of chemical property predictions strongly depends on the structure representation, including the proper treatment of the tautomeric and isomeric forms. A combination of an in-house developed open-source tool for automatic generation of tautomers, Ambit-Tautomer, based on H-atom shift rules and standard quantum chemical (DFT) calculations is used for a detailed investigation of the possible geometric isomers, conformers and tautomers of unsubstituted and para-substituted phenylhydrazones, systems with experimentally observed unusual para-substituent effects on the intramolecular hydrogen bond (IMHB) for E-isomers of the compounds. The computational results show that the energetically preferred E-isomers are characterized by stronger IMHBs than the corresponding Z-isomers. The HN–N=C–C=N molecular fragment in the E-configurations is less sensitive to the substitution effect than the HN–N=C–C=O fragment in the isomers with Z-configuration. A probable reason for this decreased sensitivity of E-isomers to phenyl ring substitution is the more efficient conjugation and charge distribution in the HN–N=C–C=N fragment.
1. Introduction
Rotary switches are hydrazone-based molecular switching systems developed initially almost 50 years ago [1,2,3,4,5] where the rotation of a β-diketone rotor occurs under the addition of catalytic amounts of acid/base or irradiation. They were further improved by Aprahamian et al. [6,7,8] who modified the rotor part by introducing a pyridyl group, which leads to substantial (but not full) stabilization of one of the isomers in solution. One of the major difficulties in the investigation of this class of compounds is the large number of possible tautomers and isomers/rotamers, often coexisting in mobile equilibria [9,10]. This is especially important in light of the fact that the interconversion between the rotamers is accompanied by proton transfer [8]. It is well known that the molecular properties of tautomers including the nature of functional groups and hydrogen-bonding pattern may differ significantly. The large number of possible structural rearrangements may affect the results and efficiency of computational studies. For instance, cheminformatics applications, such as tools for virtual screening, database searches, fingerprint generation and physical/chemical property predictions, are extremely sensitive to the 2D/3D molecular structure. Therefore, a pre-DFT stage, generating all possible tautomers, can be very useful.
A combination of an in-house developed open-source tool for automatic generation of tautomers, Ambit-Tautomer [11], based on a knowledge base with hydrogen atom shift rules and standard quantum chemical (DFT) calculations has been recently used for a detailed investigation of the possible geometric isomers, conformers and tautomers of hydrazone-based molecular switches and their E/Z isomerization [12]. The current communication presents the results of testing the same computational tools on para-substituted phenylhydrazones (a series of 10 compounds). Experimentally observed unusual para-substituent effects on the intramolecular hydrogen bond have been reported for structures with HN–N=C–C=N fragment (E-isomers) in this set of compounds [8], which needs in-depth analysis. The effects are unusual in terms of resonance-assisted hydrogen bond (RAHB) theory. The RAHB concept, introduced by Gilli et al. in 1989, concerns a kind of intramolecular H-bonding strengthened by a conjugated π-system (usually in 6-, 8- or 10-membered conjugated hydrocarbon rings) [13]. For E-isomers of para-substituted phenylhydrazones studied by Su et al., it is observed that both EDGs and EWGs are making the H-bond stronger. This effect is not consistent with RAHB theory. The observations for Z-isomers (with HN–N=C–C=O molecular fragment) of the same compounds are consistent with the theory [8]. The present contribution is concerned with the chemical landscape study of the investigated molecular switches by means of exhaustive tautomer generation and selection of energetically and chemically relevant structures and a computational (DFT) study of the structures and properties of the possible rotamers of the main E- and Z-isomers of para-substituted phenylhydrazones.
2. Computational Details
2.1. Chemical Landscape Analysis: Tautomer Generation
All tautomeric structures for the studied molecules were generated with Ambit-Tautomer software tool [11], which is a part (separate module) of the chemoinformatics platform Ambit [14], written in Java programming language and implemented on top of Chemistry Development Kit (CDK) [15]. Ambit-Tautomer facilitates three algorithms for exhaustive tautomer generation: pure combinatorial method, improved combinatorial method (CMI) and incremental approach based on depth-first search algorithm (IA-DFS). Ambit-Tautomer is configured with a list of predefined tautomeric rules covering prototropic tautomerism with H-shifts of types 1–3, 1–5 and 1–7 (e.g., O=C–C=C–C–H ↔ H–O–C=C–C=C is a 1–5 H-shift) and a set of structure filtration rules (e.g., removal of artefact structures with allene atoms and detection of structure duplications by isomorphism check). In our study, tautomer generation algorithms CMI and IA-DFS were used.
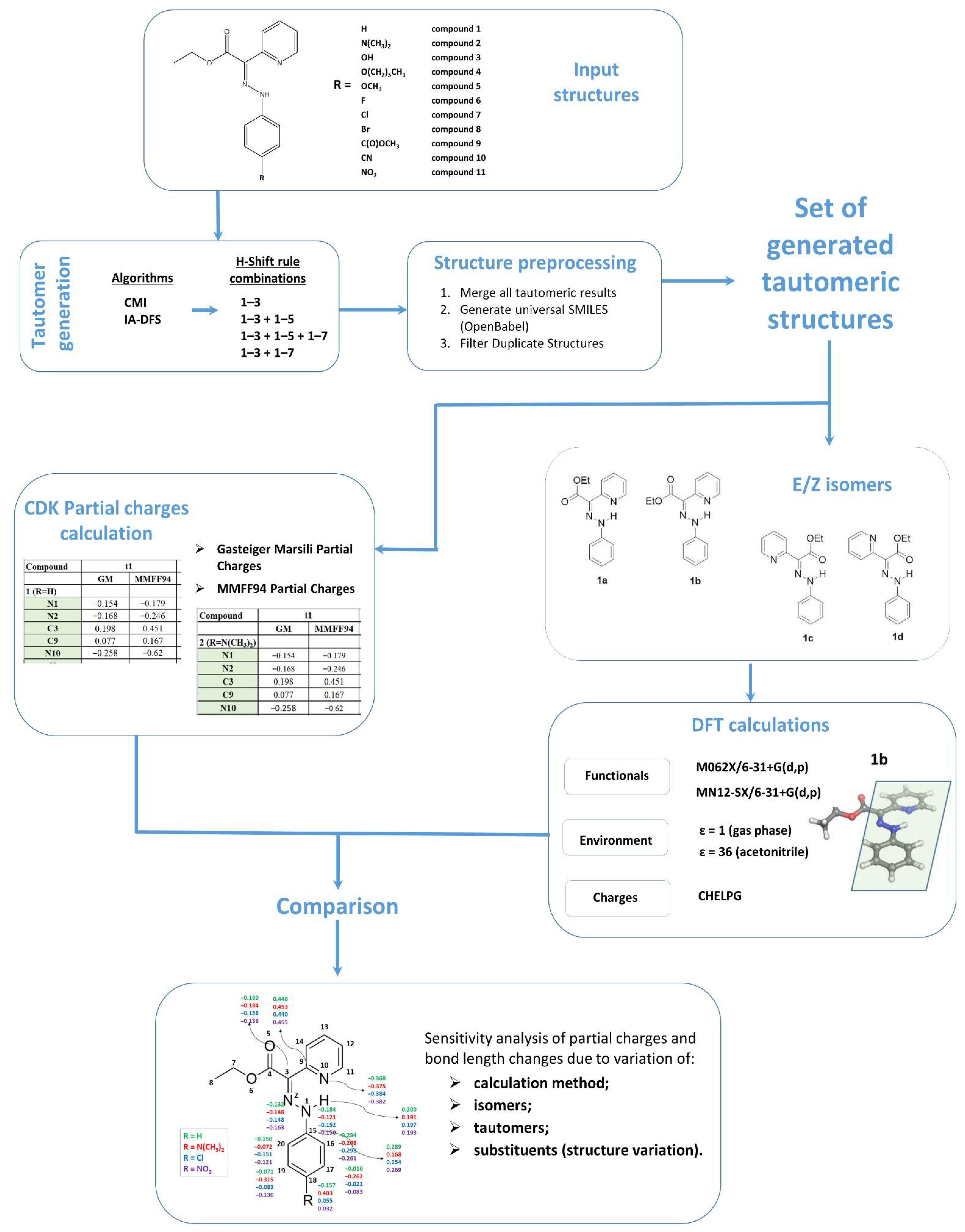
For the purpose of most exhaustive tautomer generation, we applied a calculation workflow (Figure 1) that combines the results from different Ambit-Tautomer algorithms applied with the following settings (rule combinations):
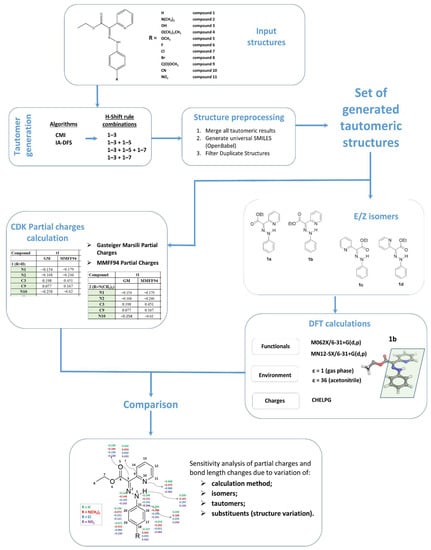
Figure 1.
Workflow of the chemoinformatics and DFT study: input, structure processing, tautomer generation, partial charge calculations, DFT calculations and parameter comparison (sensitivity analysis).
- Combination 1 (1–3 rules);
- Combination 2 (1–3 and 1–5 rules);
- Combination 3 (1–3, 1–5 and 1–7 rules);
- Combination 4 (1–3 and 1–7 rules).
The combinations listed above were applied for both CMI and IA -DFS algorithms, and consequently, all resultant tautomeric sets were merged into one final set of tautomers filtered for duplicates (duplication detection was performed on the base of generated InChI-keys).
The obtained final set of tautomers was used as input for partial charge calculations and DFT calculations. We used two fast methods for atomic partial charge calculations: Gasteiger–Marsili [16] and MMFF94 [17] partial charges. The Gasteiger–Marsili method is an iterative partial equalization of orbital electronegativity for rapid calculation of atomic charges in σ-bonded and nonconjugated π-systems. Atoms are characterized by their orbital electronegativity and only the molecule topology is considered. Another method for rapid partial charges calculation was applied based on the molecular force field, MMFF94, van der Waals and electrostatic parameters for intermolecular interactions. Both calculations were performed using the CDK software library. For this purpose, we developed small java applications available at https://github.com/PUCompChem/cdk-bits (accessed 14 April 2021).
2.2. DFT Calculations
The geometries of the studied compounds were optimized with two Minnesota functionals, M062X (global hybrid functional with 54% HF exchange) [18] and MN12-SX (screened-exchange hybrid functional with 25% HF exchange in the short range and 0% HF exchange in the long range) [19] in a combination with 6-31+G(d,p) basis set [20] using Gaussian 09 program [21]. In our previous study, M062X functional in combination with cc-pVTZ basis set was identified/selected as a computational approach, correctly predicting the distribution of the “on” and “off” forms of hydrazone-based molecular switches in solution [12]. The performance of cc-pVTZ [22] and 6-31+G(d,p) basis sets was compared in combination with M062X in the same study for the unsubstituted phenylhydrazones [12]. Diffuse functions are known to be quite important in describing weak interactions such as hydrogen bonds (and critical in describing the electron distribution of anions). The cc-pVTZ basis set (Dunning’s correlation-consistent polarized basis set) is a large basis set, and a correlation-consistent basis set augmented with diffuse functions (aug-cc-pVTZ) has versatile performance but at a high computational cost. Frequency calculations for each optimized structure were performed at the same level of theory (M062X/6-31+G(d,p) or MN12-SX/6-31+G(d,p)). All structures were confirmed as minima by frequency analysis.
Solvation effects were accounted for by employing the polarizable continuum model (PCM) [23], the default self-consistent reaction field (SCRF) method implemented in the Gaussian 09 suite of programs. The fully optimized structure of each structure/construct in the gas phase was reoptimized in an environment with dielectric constant ε = 35.688 (acetonitrile). Charges from Electrostatic Potentials Using a Grid Based Method (CHELPG) [24] atomic charges were calculated in Gaussian 09 using default settings.
PyMOL molecular graphics system was used for generation of the molecular graphics images [25].
3. Results and Discussion
It is apparent that the adequacy of chemical property predictions strongly depends on the structure representation, including the proper treatment of the tautomeric forms. In a wider context of chemoinformatics activities, chemical structure representation tackles a variety of challenges concerning representation techniques on 1D, 2D and 3D structure levels, handling of implicit and explicit hydrogen atoms, aromaticity detection and representation and many more challenges that are out of the scope of the current paper. The open-source tool for automatic generation of tautomers of a given organic compound, Ambit-Tautomer, was used for preliminary screening of the possible constitutional isomers. The rough scoring of the tautomers generated by Ambit-Tautomer was performed with subsequent DFT calculations for selected structures (Figure S1, Supporting Information), and high-energy nonconventional tautomers were excluded from further consideration. Selected keto tautomers of compounds 1–11 were subjected to quantum chemical calculations (full geometry optimization in the gas phase and in acetonitrile environment).
3.1. Unsubstituted System (Compound 1)
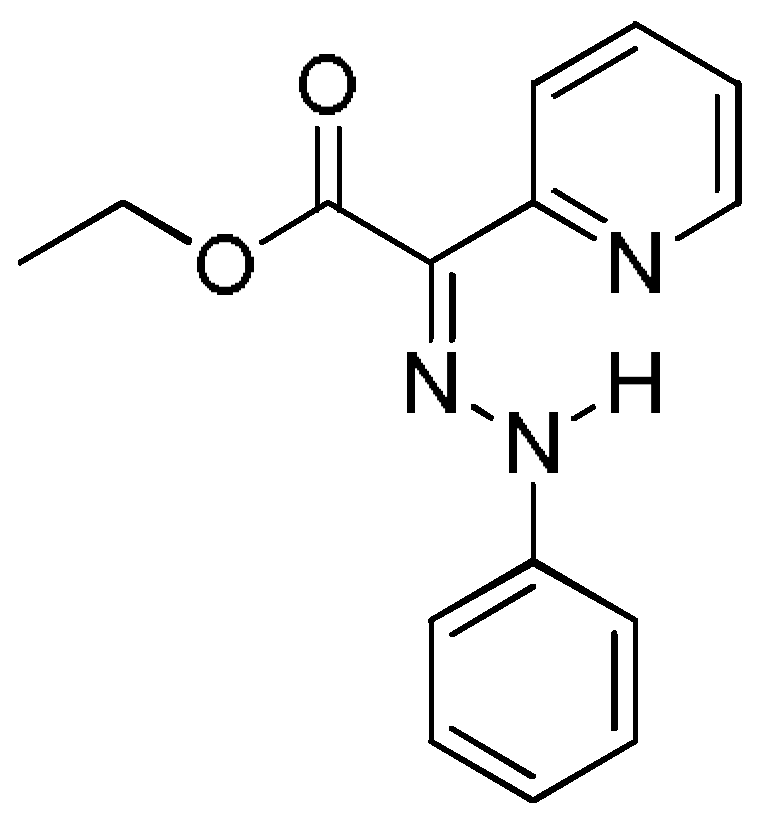
The phenylhydrazone molecular switch 1 was synthesized and a reversible, acid input, switching between the E- and Z-configurations in solution was demonstrated by Aprahamian et al. [7]. The compound was found to crystallize in E-configuration (Figure 2) and to exist predominantly in the same configuration in solution.
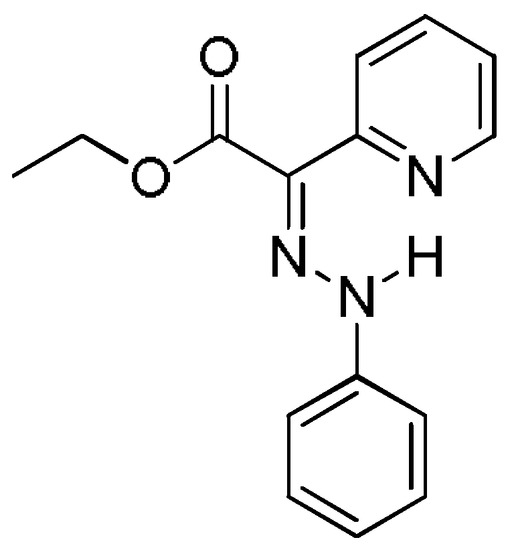
Figure 2.
E-configuration of compound 1, found in solid state and predominantly existing in solution.
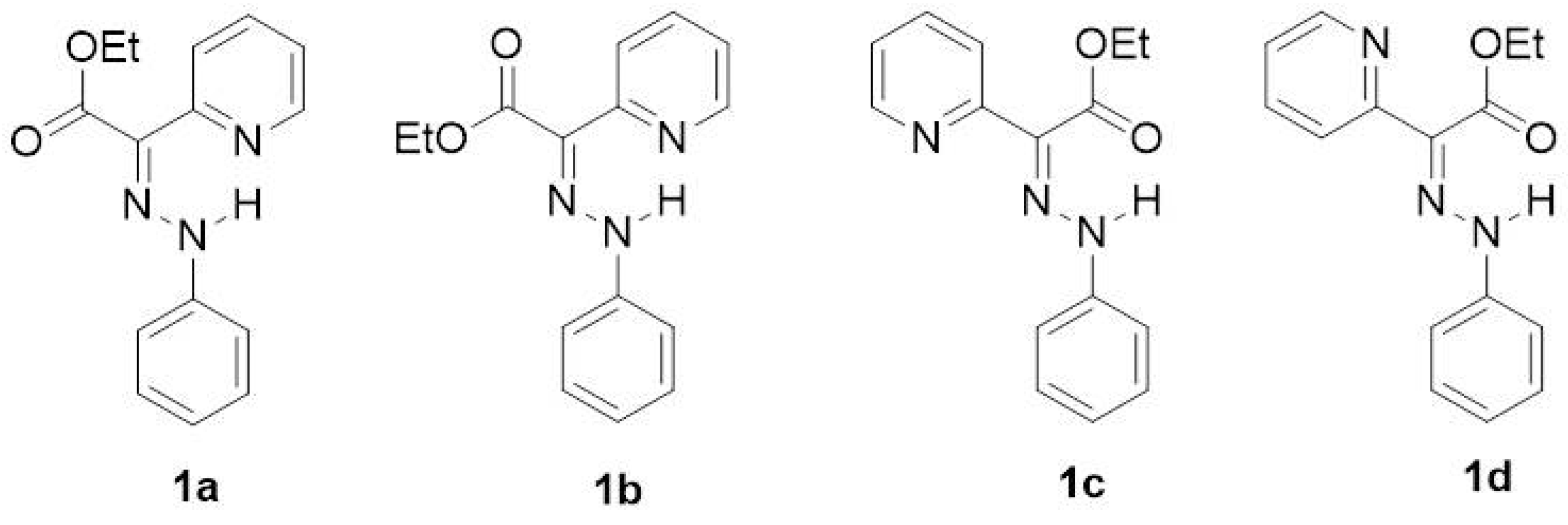
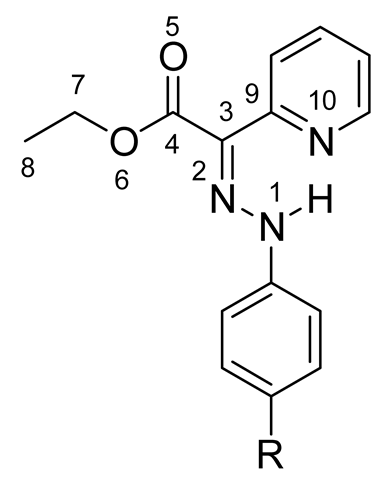
According to the DFT calculations performed by Aprahamian et al. to complement the X-ray structural analysis, the E-isomer was found to be more stable than the Z-isomer in both acetonitrile and toluene solvents [7]. The possible rotamers of the main isomers were not taken into account in the description of the E→Z isomerization process. Therefore, in our previous investigation [12] of the arylhydrazone (aryl = phenyl or naphthyl) and quinolinylhydrazone molecular switches, two E-isomers (1a and 1b) and two Z-isomers (1c and 1d) were considered (Figure 3).
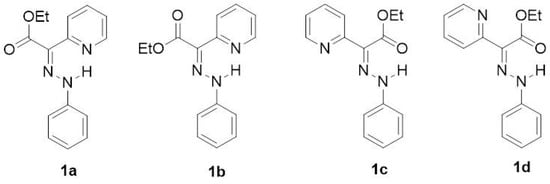
Figure 3.
Selected E-isomers (1a and 1b) and Z-isomers (1c and 1d) of compound 1.
The theoretical calculations imply that E-configurations are energetically preferred in acetonitrile over the Z-configurations at both M062X/cc-pVTZ and M062X/6-31+G(d,p) levels of theory. Here, the M062X/6-31+G(d,p) results are compared with the MN12-SX /6-31+G(d,p) ones (Table 1).

Table 1.
Gibbs energy differences in the gas phase (∆G1/dielectric constant ε = 1) and in acetonitrile (∆G36/dielectric constant ε = 36), in kcal mol−1 for compounds 1, 2, 7 and 11.
3.2. Para-Substituted Phenylhydrazones (Compounds 2–11)
The functional groups (atoms or groups of atoms) appended to the phenyl ring are shown in Table 2. The isomers of compounds 2–11 are identical to those for compound 1 (shown in Figure 3). The substituents on the phenyl ring, selected by Su et al. [8], range from typical electron-donating groups (EDGs) to typical electron-withdrawing ones (EWGs) (Table 2).
Table 2.
Para-substituted phenylhydrazones and substituent effects on the aromatic ring.
For compounds 1, 2, 7 and 11 (unsubstituted system and –N(CH3)2, –Cl and –NO2 substituted ones), the Gibbs energy differences ∆G1 and ∆G36 are given in Table 1. The Gibbs energy differences in the gas phase (∆G1) and in acetonitrile (∆G36) calculated for the rest of the compounds in the series are given in Table S1.
As expected, isomer b is found to be energetically preferred over the isomers with Z-configuration (c and d) and over isomer a (the second E-isomer) for all studied compounds (Table 1 and Table S1). The results obtained at both computational levels slightly differ, as MN12-SX functional predicts higher Gibbs energy differences, but the energy order sequence is the same. The solvation has an important effect on the equilibrium of the isomers (a–d) considered. The acetonitrile solvent reduces the preference for the most-favored isomer b over the a and c isomers.
The investigated isomers a–d of compounds 1–11 contain intramolecular H-bonded phenyl and pyridine (pyridyl) rings. The presence of an EDG in the phenyl ring is expected to enhance the electron density and to increase the intramolecular hydrogen bond (IMHB) strength, whereas EWGs have the opposite effect. On the other hand, the acidity of the proton bound to the N1 atom should decrease in presence of EDGs. The dependencies between H-bond strength and the Hammett substituent constant σ experimentally observed by Su et al. [8] are consistent with RAHB theory for the Z-configurations (isomers with HN–N=C–C=O molecular fragment) of the different derivatives but not consistent for the E-configurations (with HN–N=C–C=N fragment) of compounds 1–11. We, therefore, decided to systematically investigate the structures of a–d isomers of the 1–11 compound series. Selected bond lengths and N10⋯H distances calculated at M062X/6-31+G(d,p) level of theory for a–d isomers of compounds 1, 2, 7 and 11 are summarized in Table 3. Derivatives with EDG (2) and EWG (7 and 11) are selected as representatives. Values calculated at ε = 1 and ε = 36 are compared.
The IMHB results show that the NH⋯N unit of b isomers of compounds 1, 2, 7 and 11 is stronger than the corresponding NH⋯N or NH⋯O interactions of other forms and is consistent with the stability order. The d isomers are characterized by longer NH⋯O distances, while c isomers (with rotated pyridine ring, not involved in IMHB) have shorter NH⋯O distances, which does not support the stability order of the Z-isomers. It is interesting to differentiate trends in the NH⋯N and NH⋯O distances in presence of EDGs/EWGs. NH⋯N and NH⋯O distances calculated for 2a–d (–N(CH3)2 substituted in the phenyl ring) are slightly shorter (by ~0.02 Å) than those for the unsubstituted system. The EWG (–Cl or –NO2)-substituted compounds follow this same tendency found for EDG-substituted ones with two exceptions: 7d and 11d in acetonitrile have larger NH⋯O distances than 1d.
The single N1-N2 and double N2=C3 bonds have almost identical lengths of 1.30 Å in 1 and 2. In the EWG-substituted compounds 7 and 11, the double N2=C3 bond is slightly shorter (1.29 Å) than the single N1-N2 bond (1.31 Å–1.32 Å). The substitution effect is weaker for isomers b of compounds 1, 7 and 11; i.e., N1-N2 and N2=C3 bonds remain of almost identical length.
In this work, a popular model (CHELPG) of partial atomic charge was tested on the series of unsubstituted and p-substituted phenylhydrazones. Partial atomic charges have application in the molecular modeling of different chemically relevant areas, including the investigation of the charge transfers within a single molecule. It should be noted that the model of partial atomic charges considered here is not used to predict the ordering of possible isomers of the compounds studied. We are turning our attention only to charges on atoms in the HN–N=C–C=N and HN–N=C–C=O molecular fragments (Table 4).
As mentioned previously, the R substituents on the phenyl ring, selected by Su et al. [8], are of two types: electron-donating (EDGs) and electron-withdrawing ones (EWGs). Usually, substituents with lone pairs (e.g., –OCH3, –NH2, –N(CH3)2) are electron-donating groups (EDG)—they activate the aromatic ring by increasing the electron density on the ring through a resonance-donating effect. Substituents with π bonds to electronegative atoms (e.g., –C=O, –NO2) adjacent to the π-system are electron-withdrawing groups (EWG)—they deactivate the aromatic ring by decreasing the electron density on the ring through a resonance-withdrawing effect. Halogen substituents are not only inductive electron-withdrawing (due to their electronegativity) but also resonance-donating (lone pair donation). In summary, an EDG adds electron density to the π-system, making it more nucleophilic, and an EWG removes electron density from the π-system, making it less nucleophilic. How do these effects influence the para-positioned HN–N=C–C=N and HN–N=C–C=O molecular fragments?
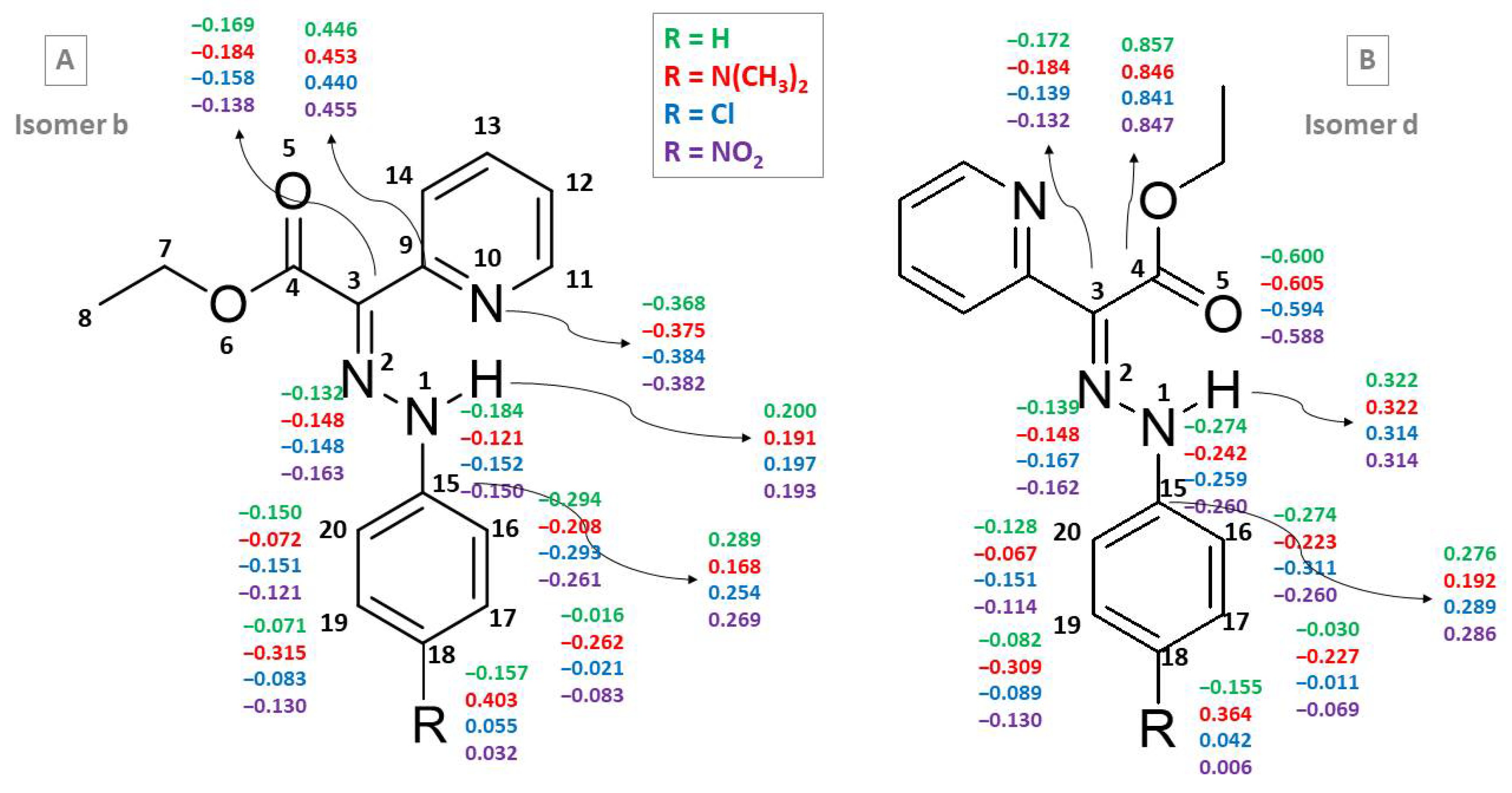
The c and d isomers of compounds 1, 2, 7 and 11 are characterized by larger partial charges of the prototropic H atom both in the gas phase and in acetonitrile (Table 4 and Figure 4). The sum of the partial charges of the atoms from the HN–N=C–C=N and HN–N=C–C=O fragments reveals different trends for compounds with these two fragments (E- and Z-isomers, respectively). Figure 4 shows the CHELPG charges for the b and d isomers of compounds 1, 2, 7 and 11 in the gas phase.
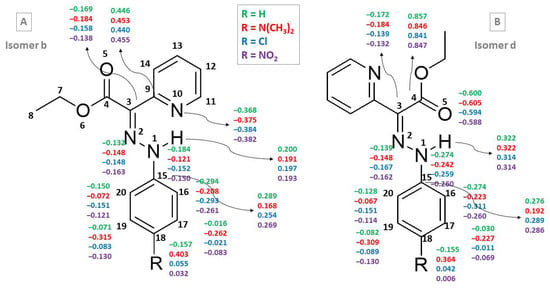
Figure 4.
CHELPG charges for selected atoms of isomers b (A) and d (B) of compounds 1, 2, 7 and 11 at ε = 1.
We performed sensitivity analysis of partial charges and bond length changes due to variation of different factors: calculation method, isomers, tautomers and substituents (structure variation). For this purpose, we calculated the root mean square deviation (RMSD) of the studied parameter, p (e.g., partial atomic charge or bond length), along the atoms/bonds (designated in the formula with index ) from the major structural fragment: N1-N2-C3-C9-N10⋯H21. For example, the RMSD for comparing isomers a and b is calculated as follows:
or the comparison between Cl substituted and unsubstituted structures is performed by calculating RMSD in the following manner:
When calculating RMSD due to a variation of a particular factor (e.g., isomer a vs. b), all other factors were fixed (e.g., method, tautomer and substituent for the case of comparing isomers a and b). A variety of tables for different combinations of the other fixed factors are given in the Supplementary Materials (see Tables S3–S18 and Raw Data Sheets 1–5).
Table 5 shows the deviations (RMSD values) between partial charges of isomers a and b and between isomers c and d. RMSD values for the gas phase are roughly twice larger than the deviation obtained in a solution environment of acetonitrile. Charge redistribution due to isomerism is less influenced by the substituent type for the calculations in the gas phase.
Table 5.
RMS deviation of the partial charges between isomers a and b (columns a/b) and between isomers c and d (columns c/d) for DFT calculation in gas phase (ε = 1) and acetonitrile (ε = 36) for different substituents.
Table 6 shows the RMSD changes of partial charges of the main structural fragment due to the substituent variation. Within the environment of acetonitrile solvent, RMSD of the charges due to substituents is about 8−10 times larger than the same RMSD in the gas phase for isomers c and d (e.g., 0.137 vs. 0.015), and it is about twice larger for isomers a and b. According to these calculations, the acetonitrile solvent is helpful for a larger charge redistribution.
Table 6.
RMS deviation of the partial charges between structures with different substituents for DFT calculation in gas phase (ε = 1) in acetonitrile (ε = 36) for isomers a–d.
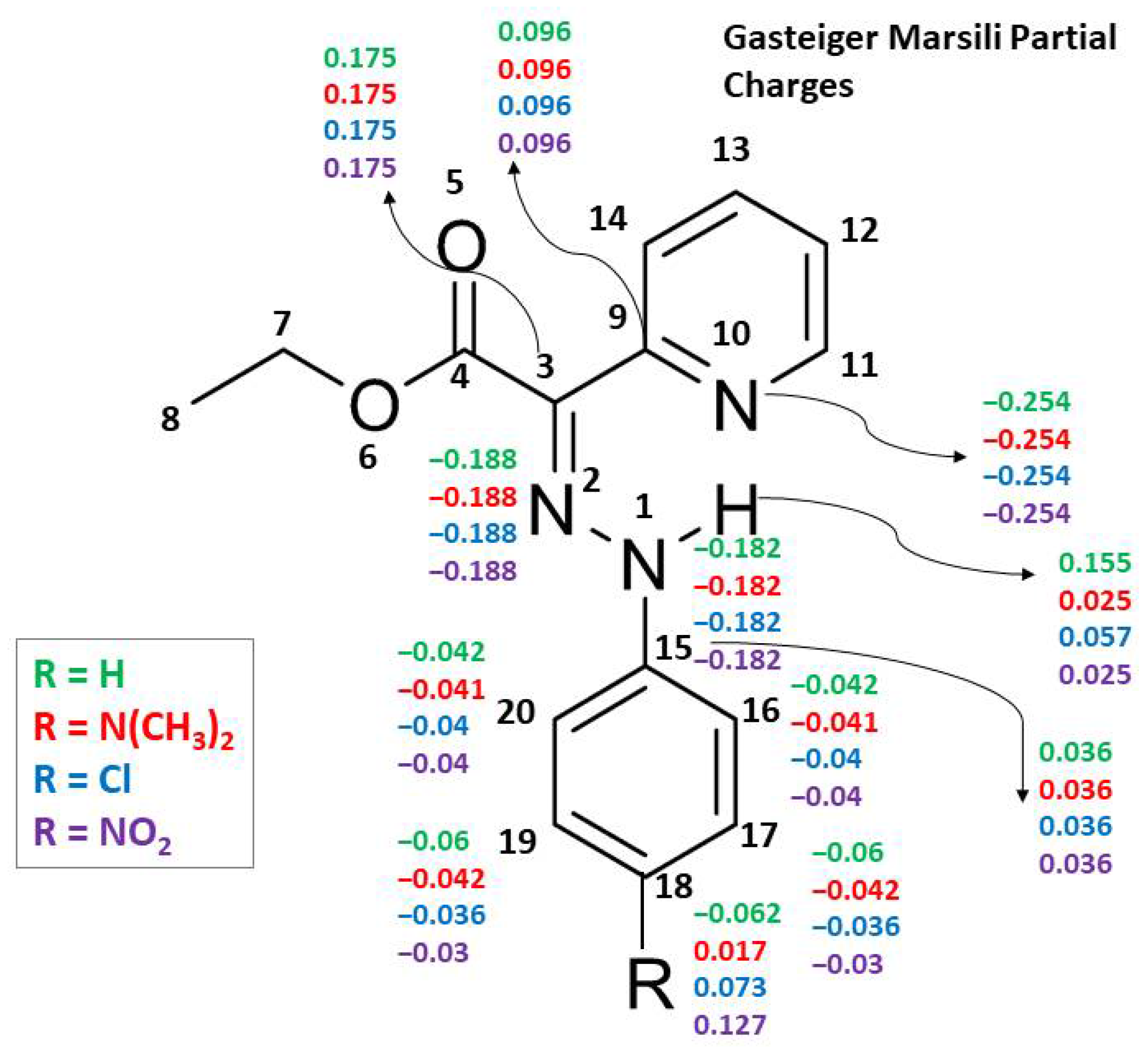
The differences between DFT partial charges and those obtained with rapid methods for partial charge calculation (GM and MMFF94) show RMSDDFT/GM values within the range 0.2–0.3 and RMSDDFT/MMFF94 within the range 0.3–0.4 (see Tables S8–S11 in the Supplementary Materials comparing tautomer 5 vs. isomer b). The sensitivity of the rapid methods of Gasteiger–Marsili and MMFF94 is not sufficient (RMSD values of about 0.05) to distinguish well the charge redistribution within tautomeric forms (see Supplementary Tables S12 and S13). The lower sensitivity of the rapid methods GM and MMFF94 is also seen by examining the partial charge changes, due to substituent variation, which are distributed up to topological distance 3 and hence not influencing the main structural fragment (see Figure 5 and the tables with detailed structures and partial charges in the Supplementary Materials). Such results are expected for rapid methods due to the application of the attenuation approach based only on topological information.
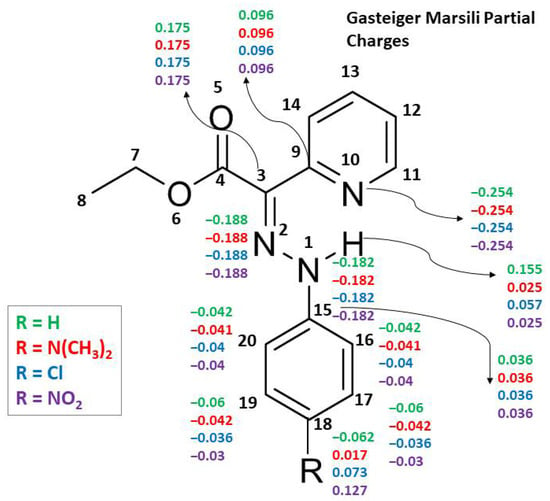
Figure 5.
Gasteiger–Marsili partial charges for selected atoms of compounds 1, 2, 7 and 11.
RMSD values for bond changes of the main structural fragment (i.e., atoms N1, N2, C3, C9, N10 and H21) show very small changes in the bond lengths (see Supplementary Tables S14–S18). The major variation is for the hydrogen bonds (N10⋯H21, O5⋯H21), which vary around 0.01–0.05 Å.
4. Conclusions
Different tautomers and isomers of unsubstituted and para-substituted phenylhydrazones were computationally studied. The effect of electron-donating and electron-withdrawing group substitution in the phenyl ring on the population and structure of E/Z-isomers was found to be divergent for the E- and Z-isomers of the compounds. The energetically preferred E-isomers are characterized by stronger intramolecular hydrogen bonds than the corresponding Z-isomers. The HN–N=C–C=N molecular fragment in the E-configurations appears to be less sensitive to the substitution effect than the HN–N=C–C=O fragment in the isomers with Z-configuration. A more efficient conjugation and charge distribution is observed in the HN-N=C–C=N fragment, leading to a decreased sensitivity of E-isomers to phenyl ring substitution. It can be concluded that differences in the structures of the interconverting E- and Z-isomers can be observed by means of DFT calculations. These differences are consistent with the experimentally observed unusual para-substituent effects on the intramolecular hydrogen bond (both EDGs and EWGs making the H-bond stronger) that have been reported by Su et al. [8] for structures with HN–N=C–C=N fragment (E-isomers). Our findings have implications for the study of the structure and stability of systems with intramolecular hydrogen bonds, especially where these systems are stabilized by intramolecular hydrogen bonds between different structural fragments.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/physchem1020013/s1, Figure S1: Relative energies of selected conventional and ring-chain tautomers of compound 1, Figure S2: Optimized structures of compounds 1–11, Table S1: Gibbs energy differences in the gas phase (∆G1) and in acetonitrile (∆G36) calculated for compounds 1–11, Table S2: List of generated tautomers for unsubstituted and substituted (substitutients H, N(CH3)2, NO2) structures and their calculated rapid partial charges with Gasteiger Marsili and MMFF94 methods; Tables S3–S18: Comparison of charges (RMS deviation) and Raw Data Sheets 1–5.
Author Contributions
Conceptualization, N.K. and S.A.; Formal analysis, V.P. and S.D.; Investigation, V.P., S.D. and N.K.; Methodology, V.P., N.K., S.A. and L.A.; Project administration, S.A. and L.A.; Visualization, V.P. and S.D.; Writing—original draft, S.A. and N.K.; Writing—review & editing, N.K., S.A. and L.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Bulgarian National Science Fund, grant number DN09/10/2016.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors acknowledge the financial support from the Bulgarian National Science Fund under the project “MolRobot” (DN09/10/2016) and the provided access to the e-infrastructure of the NCHDC, part of the Bulgarian National Roadmap for RIs, with financial support by Grant No. D01-387/18.12.2020.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Courtot, P.; Pichon, R.; Le Saint, J. Determination du site de chelation chez les arylhydrazones de tricetones et D’ α-dicetones substituees. Tetrahedron Lett. 1976, 17, 1177–1180. [Google Scholar] [CrossRef]
- Courtot, P.; Pichon, R.; Le Saint, J. Photochromisme par isomerisation syn-anti de phenylhydrazones-2- de tricetones-1,2,3 et de dicetones-1,2 substituees. Tetrahedron Lett. 1976, 17, 1181–1184. [Google Scholar] [CrossRef]
- Courtot, P.; Pichon, R.; Le Saint, J. Échanges inter et intra-molćulaires du proton entre deux atomes d’azote de cycles chélatés hydrazone-imine et azo-énamine. Tetrahedron Lett. 1979, 20, 1591–1594. [Google Scholar] [CrossRef]
- Pichon, R.; Le Saint, J.; Courtot, P. Photoisomerisation d’arylhydrazones-2 de dicetones-1,2 substituees en 2. Tetrahedron 1981, 37, 1517–1524. [Google Scholar] [CrossRef]
- Nedeltcheva-Antonova, D.; Antonov, L. Controlled Tautomerism: Is It Possible? Wiley-VCH: Weinheim, Germany, 2016; pp. 273–294. [Google Scholar] [CrossRef]
- Landge, S.M.; Aprahamian, I. A pH Activated Configurational Rotary Switch: Controlling the E/Z Isomerization in Hydrazones. J. Am. Chem. Soc. 2009, 131, 18269–18271. [Google Scholar] [CrossRef]
- Landge, S.M.; Tkatchouk, E.; Benítez, D.; Lanfranchi, D.A.; Elhabiri, M.; Goddard, W.A.; Aprahamian, I. Isomerization Mechanism in Hydrazone-Based Rotary Switches: Lateral Shift, Rotation, or Tautomerization? J. Am. Chem. Soc. 2011, 133, 9812–9823. [Google Scholar] [CrossRef] [Green Version]
- Su, X.; Lõkov, M.; Kütt, A.; Leito, I.; Aprahamian, I. Unusual para-substituent effects on the intramolecular hydrogen-bond in hydrazone-based switches. Chem. Commun. 2012, 48, 10490–10492. [Google Scholar] [CrossRef]
- Hristova, S.; Kamounah, F.S.; Crochet, A.; Vassilev, N.; Fromm, K.M.; Antonov, L. OH Group Effect in the Stator of β-Diketones Arylhydrazone Rotary Switches. Chemistry 2020, 2, 24. [Google Scholar] [CrossRef]
- Deneva, V.; Vassilev, N.; Hristova, S.; Yordanov, D.; Hayashi, Y.; Kawauchi, S.; Fennel, F.; Völzer, T.; Lochbrunner, S.; Antonov, L. Chercher de l’eau: The switching mechanism of the rotary switch ethyl-2-(2-(quinolin-8-yl)hydrazono)-2-(pyridin-2-yl)acetate. Comput. Mater. Sci. 2020, 177, 109570. [Google Scholar] [CrossRef]
- Kochev, N.T.; Paskaleva, V.; Jeliazkova, N. Ambit-Tautomer: An Open Source Tool for Tautomer Generation. Mol. Inform. 2013, 32, 481–504. [Google Scholar] [CrossRef]
- Angelova, S.; Paskaleva, V.; Kochev, N.; Antonov, L. DFT study of hydrazone-based molecular switches: The effect of different stators on the on/off state distribution. Mol. Phys. 2018, 117, 1604–1612. [Google Scholar] [CrossRef]
- Gilli, P.; Gilli, G. Hydrogen bond models and theories: The dual hydrogen bond model and its consequences. J. Mol. Struct. 2010, 972, 2–10. [Google Scholar] [CrossRef]
- Ambit Project. Available online: https://www.ambitprojects.co.uk (accessed on 17 June 2021).
- Steinbeck, C.; Hoppe, C.; Kuhn, S.; Floris, M.; Guha, R.; Willighagen, E. Recent Developments of the Chemistry Development Kit (CDK)—An Open-Source Java Library for Chemo- and Bioinformatics. Curr. Pharm. Des. 2006, 12, 2111–2120. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. II. MMFF94 van der Waals and electrostatic parameters for intermolecular interactions. J. Comput. Chem. 1996, 17, 520–552. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2007, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Peverati, R.; Truhlar, D. Screened-exchange density functionals with broad accuracy for chemistry and solid-state physics. Phys. Chem. Chem. Phys. 2012, 14, 16187–16191. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; A Pople, J. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Mennucci, B. Polarizable continuum model. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 386–404. [Google Scholar] [CrossRef]
- Breneman, C.M.; Wiberg, K.B. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; Version 1.7.6.6; Schrödinger: New York, NY, USA, 2015.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).