
Diagnosis and Treatment of Langerhans Cell Sarcoma: A Case Report and Review of the Literature

 , , , , ,
, , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
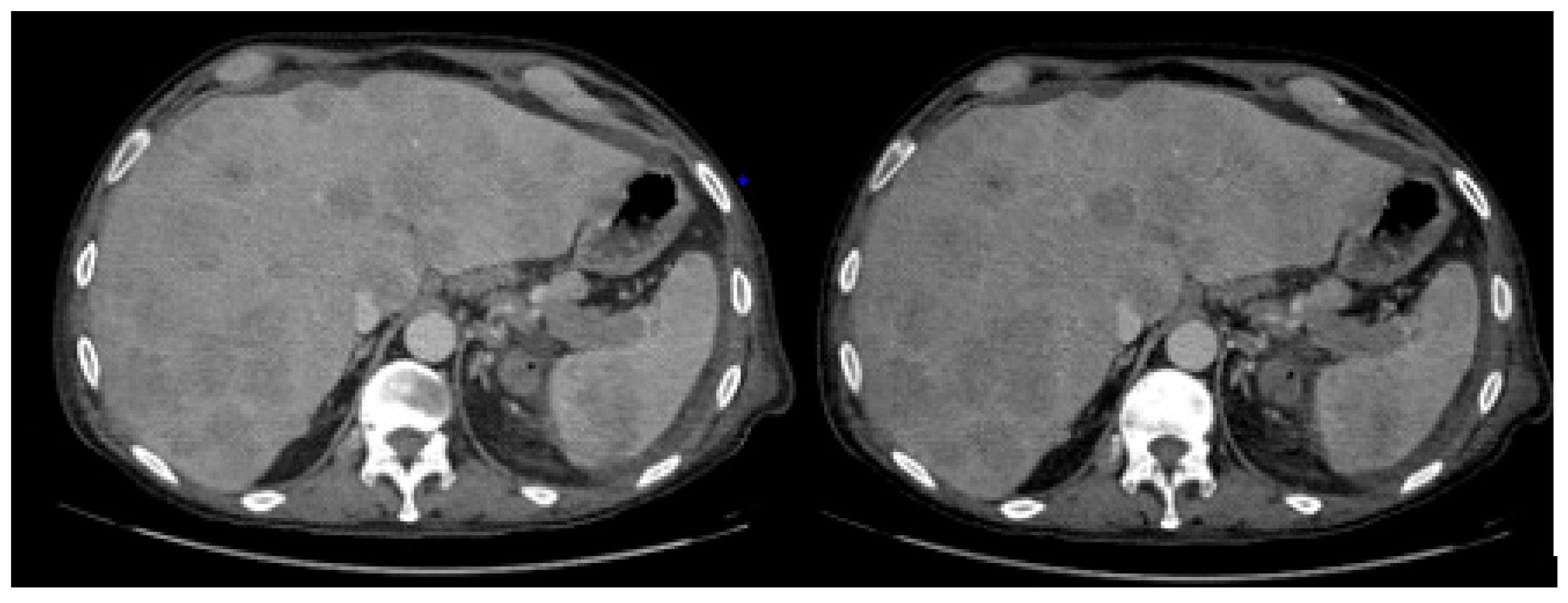
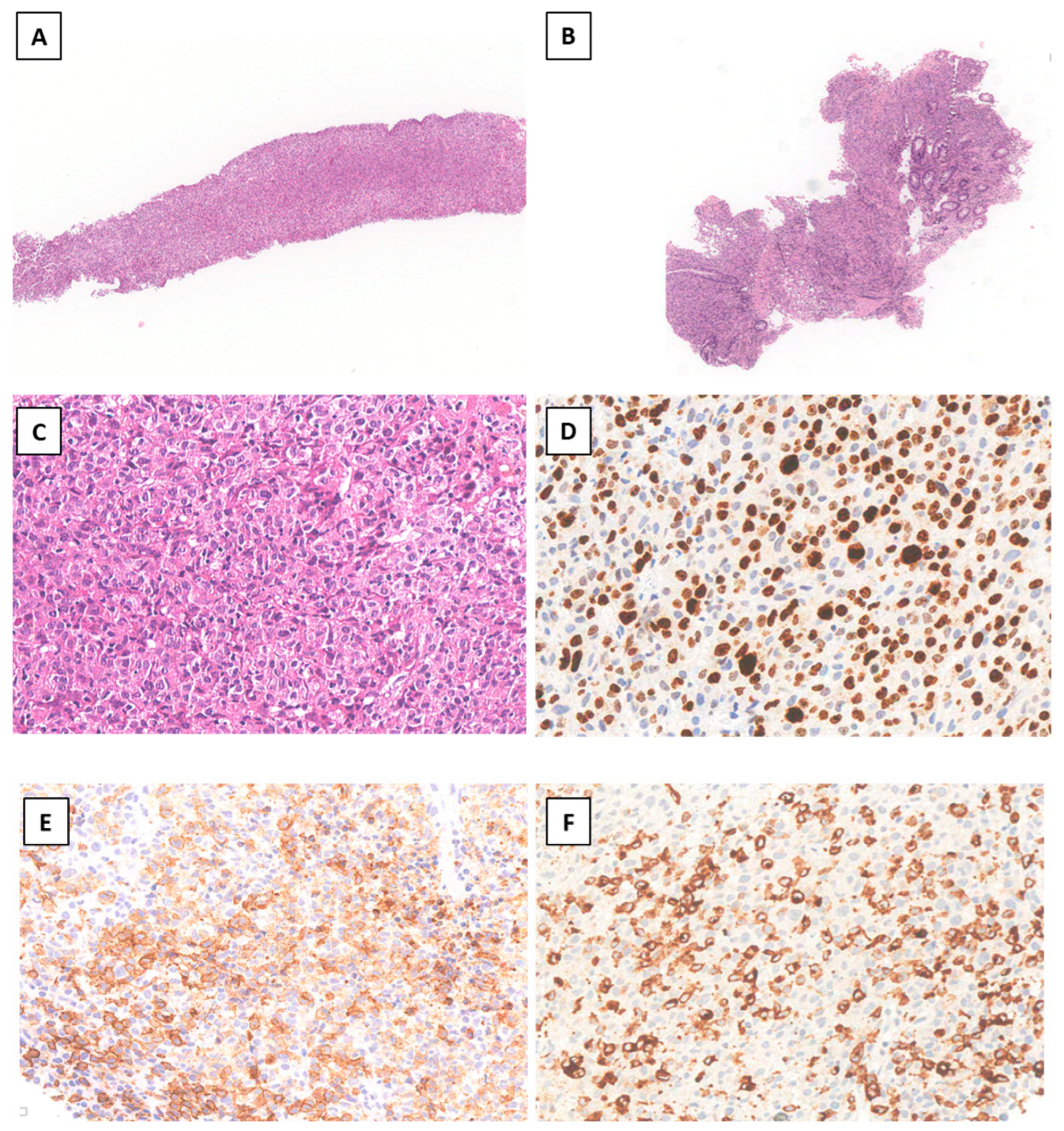
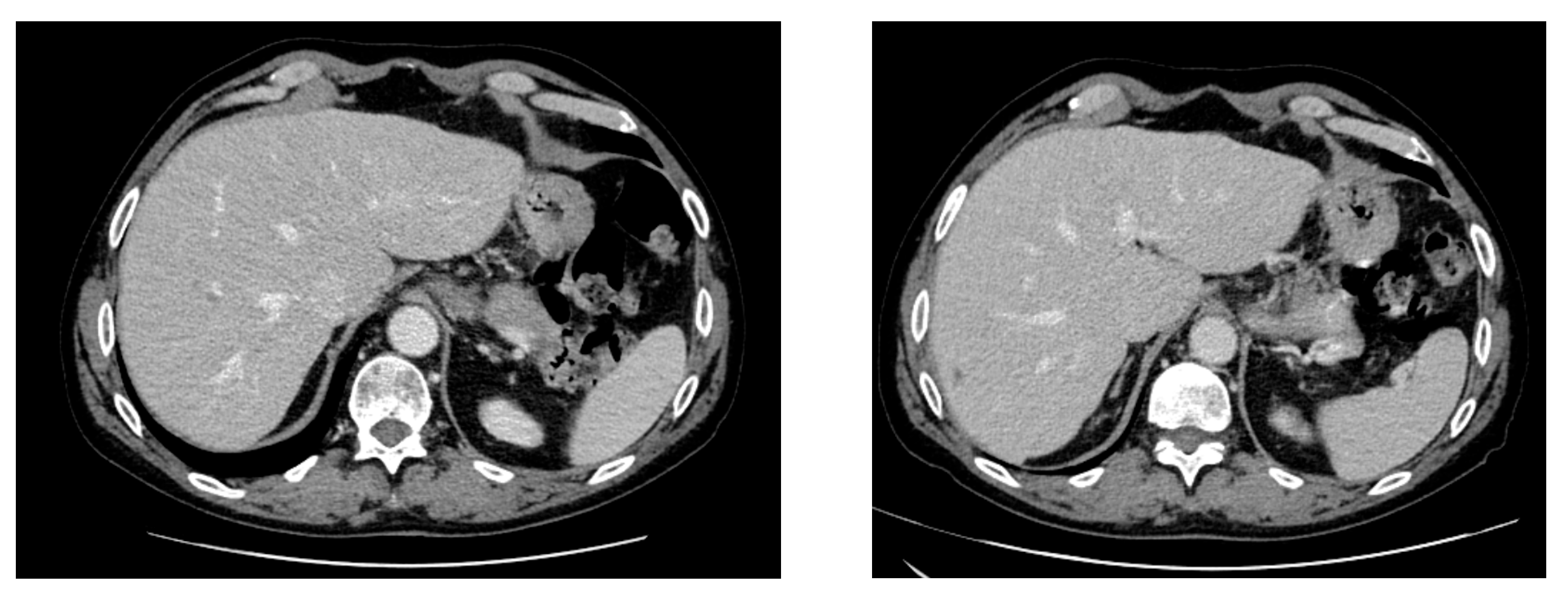
2. Case Report
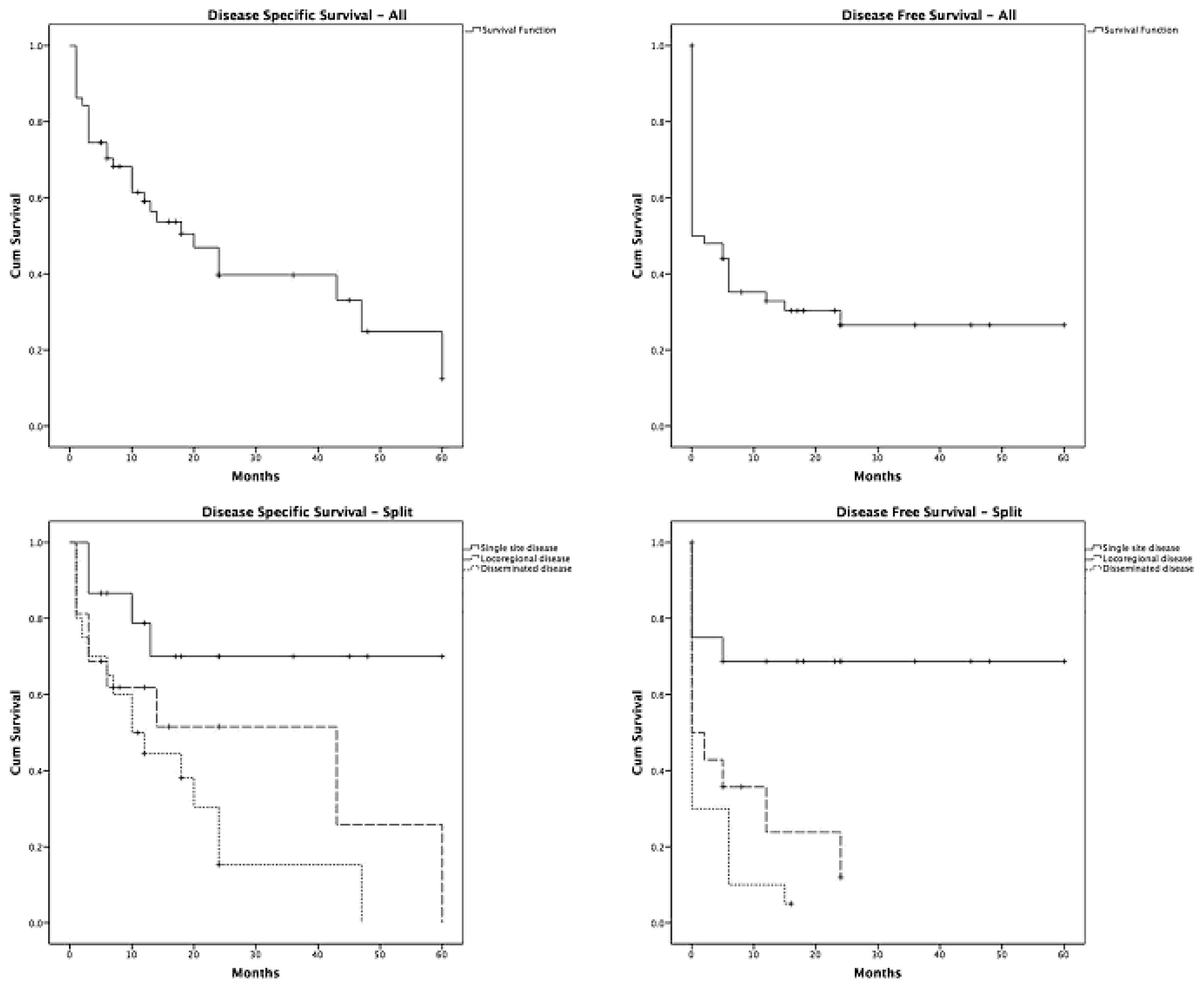
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pathology & Genetics: Tumours of Haematopoietic and Lymphoid Tissues; Jaffe, E.S., Harris, N.L., Stein, H., Vardiman, J.W., Eds.; IARC Press: Lyon, France, 2001; p. 283. [Google Scholar]
- Mali, B.; Mali, A.; Mali, A.; Abdulrazzak, M.; Jobran, A.W. The epidemiological and clinical characteristics of Langerhans cell sarcoma in the United States: A population study based on SEER data from 2000 to 2019. Medicine 2024, 103, e39315. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.T.; Friesenbichler, J.; Holzer, L.A.; Liegl-Atzwanger, B.; Beham-Schmid, C.; Leithner, A. Langerhans cell sarcoma: A case report and review of the literature. Pol. J. Pathol. 2016, 67, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Valladeau, J.; Clair-Moninot, V.; Dezutter-Dambuyant, C.; Pin, J.-J.; Kissenpfennig, A.; Mattéi, M.-G.; Ait-Yahia, S.; Bates, E.E.M.; Malissen, B.; Koch, F.; et al. Identification of mouse Langerin/CD207 in Langerhans cells and some dendritic cells of lymphoid tissues. J. Immunol. 2002, 168, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Ferringer, T.; Banks, P.M.; Metcalf, J.S. Langerhans cell sarcoma. Am. J. Dermatopathol. 2006, 28, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Pileri, S.A.; Grogan, T.M.; Harris, N.L.; Banks, P.; Campo, E.; Chan, J.K.C.; Favera, R.D.; Delsol, G.; De Wolf-Peeters, C.; Falini, B.; et al. Tumours of histiocytes and accessory dendritic cells: An immunohistochemical approach to classification from the International Lymphoma Study Group based on 61 cases. Histopathology 2002, 41, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Charfi, M.; Lajnaf, M.; Kallel, F.; Saguem, I.; Kassar, O.; Elloumi, M. Complexity of diagnosing and treating langerhans cell sarcoma: A case report. Tunis Med. 2023, 101, 651–653. [Google Scholar] [PubMed] [PubMed Central]
- Nakamine, H.; Yamakawa, M.; Yoshino, T.; Fukumoto, T.; Enomoto, Y.; Matsumura, I. Langerhans Cell Histiocytosis and Langerhans Cell Sarcoma: Current Understanding and Differential Diagnosis. J. Clin. Exp. Hematop. 2016, 56, 109–118. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yi, W.; Chen, W.Y.; Yang, T.X.; Lan, J.P.; Liang, W.N. Langerhans cell sarcoma arising from antecedent langerhans cell histiocytosis: A case report. Medicine 2019, 98, e14531. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.E.; Dwivedi, R.C.; Masterson, L.; Jani, P. Langerhans cell sarcoma: A systematic review. Cancer Treat. Rev. 2015, 41, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Valentín-Nogueras, S.M.; Seijo-Montes, R.; Montalván-Miró, E.; Sánchez, J.L. Langerhans cell sarcoma: A case report. J. Cutan. Pathol. 2013, 40, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Saven, A.; Burian, C. Cladribine activity in adult langerhans-cell histiocytosis. Blood 1999, 93, 4125–4130. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Takahashi, K.; Hori, M.; Okumura, T.; Saito, M.; Yamakawa, M.; Tabuchi, K.; Hara, A. Langerhans cell sarcoma of the cervical lymph node: A case report and literature review. Auris Nasus Larynx 2010, 37, 7503. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.; Hampton, O.A.; Shen, X.; Simko, S.J.; Shih, A.; Abhyankar, H.; Lim, K.P.H.; Covington, K.R.; Trevino, L.; Dewal, N.; et al. Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood 2014, 124, 3007–3015. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.E.; Li, L.; Peters, T.L.; Leung, H.C.; Yu, A.; Man, T.K.; Gurusiddappa, S.; Phillips, M.T.; Hicks, M.J.; Gaikwad, A.; et al. Cell-specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct profile compared with epidermal Langerhans cells. J. Immunol. 2010, 184, 4557–4567. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.N.; Zhou, X.G.; Wang, Z. Langerhans cell sarcoma in the cervical lymph node: A case report and literature review. Acta Haematol. 2013, 129, 114–120. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pileggi, G.; Mariani, S.; De Santis, V.; Maiorana, G.; Lubrano Lobianco, F.; Togni, C.; Piedimonte, M.; Tatarelli, C.; Conte, E.; di Napoli, A.; et al. Diagnosis and Treatment of Langerhans Cell Sarcoma: A Case Report and Review of the Literature. Hemato 2025, 6, 18. https://doi.org/10.3390/hemato6030018
Pileggi G, Mariani S, De Santis V, Maiorana G, Lubrano Lobianco F, Togni C, Piedimonte M, Tatarelli C, Conte E, di Napoli A, et al. Diagnosis and Treatment of Langerhans Cell Sarcoma: A Case Report and Review of the Literature. Hemato. 2025; 6(3):18. https://doi.org/10.3390/hemato6030018
Chicago/Turabian StylePileggi, Giulia, Sabrina Mariani, Valentina De Santis, Gianluca Maiorana, Federica Lubrano Lobianco, Chiara Togni, Monica Piedimonte, Caterina Tatarelli, Esmeralda Conte, Arianna di Napoli, and et al. 2025. "Diagnosis and Treatment of Langerhans Cell Sarcoma: A Case Report and Review of the Literature" Hemato 6, no. 3: 18. https://doi.org/10.3390/hemato6030018
APA StylePileggi, G., Mariani, S., De Santis, V., Maiorana, G., Lubrano Lobianco, F., Togni, C., Piedimonte, M., Tatarelli, C., Conte, E., di Napoli, A., Pilozzi, E., Rogges, E., Tafuri, A., & Palumbo, G. (2025). Diagnosis and Treatment of Langerhans Cell Sarcoma: A Case Report and Review of the Literature. Hemato, 6(3), 18. https://doi.org/10.3390/hemato6030018