Abstract
This work focuses on the investigation of ten newly synthesized spironaphthoxazines using DFT to elucidate how substituents control physicochemical behavior. Frontier-orbital analyses show substituent changes primarily shift the LUMO, controlling HOMO–LUMO gaps and electrophilicity; the open forms (MC) structures exhibit smaller gaps than closed spiro forms (SP) due to extended conjugation. Simulated IR/Raman spectra provide diagnostic markers for structural assignment. Thermodynamic parameters (S, Cp, H, G; 200–500 K) reveal higher S and Cp for MC and for longer alkyl chains, yielding lower G at elevated temperatures. Transition-state calculations indicate accessible SP↔MC isomerization barriers, confirming accessible switching. These results offer a predictive framework to position functional groups and tailor optical response, switching kinetics, and stability for responsive materials.
1. Introduction
Spironaphthoxazines belong to one of the most promising classes of organic photochromes. Their main applications include the following: optical data recording, storage, and switches; smart materials; sensors; photocontrolled drug delivery; biosensors for diagnostics; etc. Their applications, particularly in medical and related fields, stem from their high photostability, fast switching, and structural versatility [1]. Spironaphthoxazines exist in two primary forms: closed spiro (SP)—colorless with a lower dipole moment (~5 D); and open photomerocyanine (MC)—colored and having a higher dipole moment. Upon UV irradiation, temperature, solvent, or other stimuli exposure, the spironapthoxazines convert from SP to MC through the cleavage of the spiro C-O bond [1,2]. This transformation dramatically changes their molecular properties, enabling specific interactions with other molecules. The general chemical formula of SP and MC of a spironaphthoxazine derivative in a ring-opening transformation reaction is presented in Figure 1.
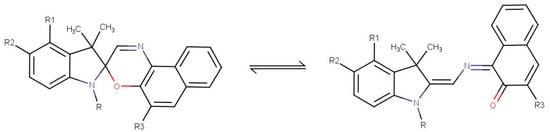
Figure 1.
The general chemical formula of SP and MC of a spironaphthoxazine derivative in a ring-opening transformation reaction. R, R1, R2, and R3 represent different fragments.
A series of new spironaphthoxazines has been synthesized, and the present work proposes the investigation of ten of the new structures using Density Functional Theory (DFT) computations. Moreover, the goal of this work is to establish a foundation for future studies and identify new applications for these compounds. Property simulations enable us to assess the advantages of specific functional groups and optimize their placement within the molecular scaffold, thereby guiding the rational synthesis of targeted derivatives for desired applications.
2. Methods
DFT calculations were used, and the molecules were described in PW91 and 6-311G(d,p) basis sets, in vacuum, for equilibrium geometry [3]. The optimized molecular structure and the energy of individual molecular orbitals were predicted for ten spironaphthoxazine derivatives with chemical structures constructed accordingly to the data provided in Table 1 for the base spironaphthoxazine presented in Figure 1. The calculations were made on the CTC conformer of the structures. HOMO-LUMO analysis with corresponding quantum global chemical reactivity descriptors of the studied molecules was obtained to predict molecular stability and reactivity of the molecules. Also, by DFT, the vibrational frequencies, thermodynamic properties, and NMR chemical shifts in molecules were predicted, and the minimum energy pathway between the two geometries of the SP and MC, with identification of the geometry of the transition state (TS) form, was calculated. The results obtained based on computations were compared and discussed in order to establish the beneficial traits of possible future applications.

Table 1.
The chemical formula of the series 0–9 of the spironaphthoxazines with corresponding substituents and the abbreviation of SP and MC used in this work according to [1].
3. Results
The analysis of the frontier orbitals, the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), indicates the shapes and distributions of these orbitals, showing where the electron density is concentrated in the considered molecules. The global reactivity descriptors as chemical potential (μ), electronegativity (χ), global hardness (η), global softness (s), and electrophilicity (ω), resulted from HOMO-LUMO analysis [4], by considering the energies determined with the GGA/PW91 density functional. The calculated values for all the structures considered are summarized in graphs presented in Figure 2.
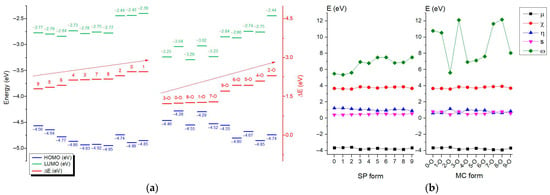
Figure 2.
(a)—The HOMO (blue horizontal lines and values) and LUMO (green horizontal lines and values) energies, energy gap values in ascending order from left to right (red horizontal lines with structures abbreviations, the red arrows mark the upward trend of the energy gap values); (b)—left panel for SP and the right panel for MC represent chemical potential (μ)—black color, electronegativity (χ)—red color, global hardness (η)—blue color, global softness (s)—magenta color, and electrophilicity (ω)—green color. The presented values are in eV.
It can be observed in Figure 2 that tuning substituents affects the acceptor part, where electron-withdrawing groups stabilize the LUMO, and electron-donating groups destabilize it. Left structures (considering the ascending order from left to right) have lower excitation energies, leading to longer wavelength absorption and increased electrophilicity, while right structures show the opposite. If these relate to closed versus open forms, MC structures generally have smaller gaps due to extended conjugation. From the results obtained, the simulated IR (Figure 3a) and Raman (Figure 3b) spectra of compounds 5, 6, 5-O, and 6-O were selected for illustration and comparison.
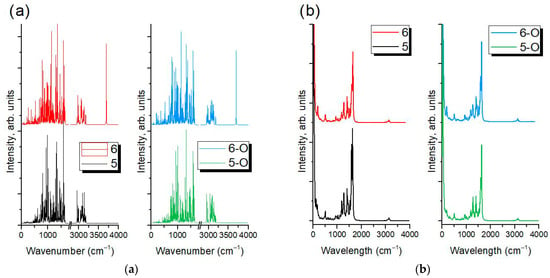
Figure 3.
(a)—IR frequencies of 5 and 6 structures (left panel) and 5-O and 6-O structures (right panel); (b)—Raman spectra at temperature of 298.0 K for 5 and 6 structures (left panel) and 5-O and 6-O structures (right panel) with Lorentzian smearing: 20 cm−1 and incident light: 514.5 nm.
Figure 4a,b shows the comparative influence of the length of the hydrocarbon radical in the R position (Figure 1) on the thermodynamic behavior of structures 3, 4, 5, and 6 and their MC. For this purpose, pairs of similar structures differing only in the R fragment (Table 1) were used.
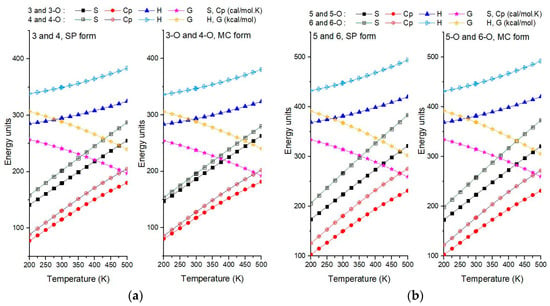
Figure 4.
The standard thermodynamic quantities (entropy (S), heat capacity (Cp), enthalpy (H), free energy (G)) for the temperature domain of 200 K to 500 K, with zero-point vibrational energy included. The calculated values are graphically presented in (a) for 3 and 4 SP (left panel) and 3-O and 4-O MC (right panel), and in (b) for 5 and 6 SP (left panel) and 5-O and 6-O MC (right panel).
Considering Figure 4a,b it was observed that, MC (3-O, 4-O, 5-O and 6-O) show higher heat capacity (Cp) and entropy (S) compared to spiro structures (3, 4, 5, and 6) probably due to greater flexibility/delocalization in the open form (more degrees of freedom in the -C4H9 chain). Enthalpy (H) changes little with fragment R length. MC has slightly higher H, but the entropy gain means its free energy (G) drops faster with temperature; thus, the open form is increasingly favored at higher temperatures, while the spiro form may be preferred at lower temperatures. Because the T·S factor (here, T·S is the change in entropy with temperature in Gibbs law) is larger for 4 and 6, the G values become relatively lower at higher temperatures, so compounds 4 and 6 are slightly more thermodynamically favored than 3 and 5 as temperature rises. Table 2 gives the computed values for the transition of state energy and the isomerization reaction parameters from the SP form to the MC form, for some of the studied structures. Therefore, Figure 5 presents the frontier orbitals HOMO and LUMO obtained after the transition state optimization and the calculated energy gap.
Table 2.
The computed values for the transition state energy (TS) and for the isomerization reaction parameters (ΔE, energy of reaction, energy of barrier) of the SP to MC transformation.
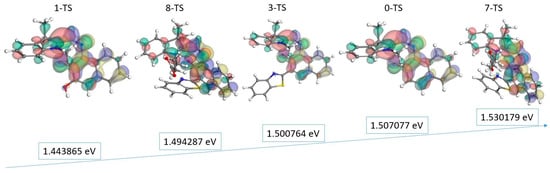
Figure 5.
The plot of energy gap (the ascending values on the blue arrow) and the frontier orbitals HOMO and LUMO (for an isovalue of 0.03) of the chemical structure of the transition state. The surfaces are drawn by yellow/blue and green/red colors for HOMO and LUMO, respectively, where the negative/positive blobs are represented by light and dark colors. The gray, red, blue, yellow, and white spheres represent the carbon, oxygen, nitrogen, sulfur, and hydrogen atoms, respectively.
4. Conclusions
In this contribution DFT study was used to set up structure–property relationships for a new series of spironaphthoxazines. Substituent engineering primarily modulates the acceptor part of the molecules, stabilizing or destabilizing the LUMO and thus tuning the HOMO–LUMO gap, electrophilicity, and expected absorption wavelengths. The obtained results have shown that MC generally display smaller gaps than the closed spiro forms, consistent with extended conjugation. Thermodynamic analyses show higher entropy and heat capacity for MC versus SP, and for longer R chains (–C4H9 > –CH3), reflecting increased flexibility; consequently, free energy for longer-chain analogs decreases more steeply with temperature, making 4 and 6 slightly more favored than 3 and 5 at elevated temperatures. Simulated IR and Raman spectra provide diagnostic fingerprints for structures 5, 6, and their MC isomers, supporting experimental assignment. Transition-state calculations confirm feasible SP→MC isomerization with modest barriers (13–21 kJ/mol) and small reaction energies (9–11 kJ/mol), indicating thermally and photochemically accessible switching.
Author Contributions
Conceptualization, A.N. and V.C.; methodology, A.N. and V.C.; software, V.C. and V.A.; validation, A.N. and S.M.; formal analysis, A.N.; investigation, A.N.; resources, V.C. and V.A.; data curation, A.N. and S.M.; writing—original draft preparation, A.N.; writing—review and editing, A.N.; visualization, S.M. and V.A.; supervision, A.N.; project administration, A.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Dataset available on request from the authors.
Acknowledgments
This research was carried out within the framework of the project “Experimental and Computational Investigations of the Adsorbed Photo-switching Molecules” through the partners: the Institute of Catalysis, Bulgarian Academy of Sciences, Sofia, Bulgaria, and the Institute of Physical Chemistry of the Romanian Academy, Bucharest, Romania.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Minkovska, S.; Hadjichristov, G.B.; Neacsu, A.; Chihaia, V.; Fedorov, Y.V. Photoswitchable Photochromic Chelating Spironaphthoxazines: Synthesis, Photophysical Properties, Quantum-Chemical Calculations, and Complexation Ability. ACS Omega 2024, 9, 4144–4161. [Google Scholar] [CrossRef] [PubMed]
- Neacsu, A.; Chihaia, V.; Alexiev, V.; Hadjichristov, G.B.; Minkovska, S. Specifics of the Molecular Conformations and Physicochemical Properties of Merocyanine Form of Spirooxazine Derivative: Insights from Experimental and Molecular Dynamics Studies. Materials 2025, 18, 2505. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787. [Google Scholar] [CrossRef] [PubMed]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. CDFT-Based Reactivity Descriptors as a Useful MEDT Chemoinformatics Tool for the Study of the Virotoxin Family of Fungal Peptides. Molecules 2019, 24, 2707. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).