Abstract
The main phenolic compounds in the Hippophae rhamnoides fruit with potential therapeutic activities are quercetin-3-O-rhamnoside, quercetin-3-O-galactoside, myricetin, rutin, luteolin, kaempferol, vitexin, gallic acid, chlorogenic acid, caffeic acid, 7-methoxycoumarin, p-coumaric acid, and ferulic acid. Their general features recommend them for nutritional and therapeutic purposes, exploiting their neuroprotective and radioprotective effects. This study aims to investigate the potency of polyphenol-derived structures against dual tyrosine-regulated kinase, modulating neuroblastomas and glioblastomas in humans. Structural insights from the point of view of drug-like property assessment are also provided by Density Functional Theory (DFT) predictions on the lowest energy conformers, using the B3LYP/6-311G (d,p) method.
1. Introduction
Naturally occurring polyphenolics are abundantly present in the majority of fruits and have various health benefits. Hippophae rhamnoides (common sea buckthorn) is a notorious so-called wild berry. The main phenolic compounds in the fruit part are quercetin-3-O-rhamnoside, quercetin-3-O-galactoside, myricetin, rutin, luteolin, kaempferol, vitexin, gallic acid, chlorogenic acid, caffeic acid, 7-methoxycoumarin, p-coumaric acid, and ferulic acid [1]; the products derived from the leaf part were also associated with a very similar qualitative chemical content, quercetin derivates being the dominant compounds in the polar-type extracts [2]. As for human health benefits, sea buckthorn is well known for its nutritional and therapeutic properties, including neuroprotective [3] and radioprotective effects [4]. For example, the aqueous extract from leaves of H. rhamnoides attenuates cobalt-60 γ-radiation-induced changes in behavior, oxidative stress, and serotonin levels in the jejunum and plasma of rats [4]. This study aimed to investigate the ability of the main polyphenolic compounds in H. rhamnoides (as presented above) against dual tyrosine-regulated kinase 2 (DYRK2), PDB ID: 5ZTN [5] in complex with curcumin, the native ligand. Dual tyrosine-regulated kinase 2 is a very important molecular target and tumor modulator in neuroblastomas and glioblastomas in humans; therefore, the potential effects of phenolics found in sea buckthorn-derived products could extend their use toward brain tumor adjuvant therapy.
The selection and optimization process for a given ligand is based not only on the affinity for the target kinase but also on properties that affect pharmacological activity. Thus, structure–property relationships and structure modification strategies are key steps in drug design that allow the proposal of high-quality candidates for further clinical studies. Considering these aspects, the investigation and understanding of drug-like properties such as solubility, permeability, lipophilicity balance, kinetic stability in physiological media, volume, and exposed drug area are of huge interest nowadays. The bioavailability dictates setting the dose and routes of administration of drugs.
This paper focuses on the fundamental knowledge of properties that affect the bioavailability of the most promising ligands for dual tyrosine-regulated kinase, explored using chemoinformatic and molecular modeling tools.
2. Materials and Methods
To initiate property computations and docking simulations, compounds’ structures were retrieved from the PubChem database and submitted to minimization using Molecular Mechanics Force Field (MMFF) [6] using Spartan 14 software Wavefunction, Inc. Irvine, CA, USA [7]. The lowest energy conformers of the chosen phenolic compounds were used for druglikeness property computations using Becke’s three-parameter hybrid exchange functional with the Lee–Yang–Parr correlation functional [8] with a 311G (d,p) basis set. Molecular docking studies have been performed using CLC Drug Discovery Workbench Software (QIAGEN, Aarhus, Denmark). Data related to binding affinity in terms of docking score are provided. The interaction mode is predicted; the type and count of interactions, particularly hydrogen bonding, are identified and depicted within the active binding site of DYRK2, established between ligands and the amino acid group interactions.
3. Results and Discussion
3.1. Results of Bioavailability Prediction
The calculated property data in terms of polar surface area, water-partition coefficient, molecule flexibility, electrostatic potential, and global quantum reactivity parameters related to frontier molecular orbital energies are studied, compared, and interpreted in the context of more comprehensive previously reported results [9].
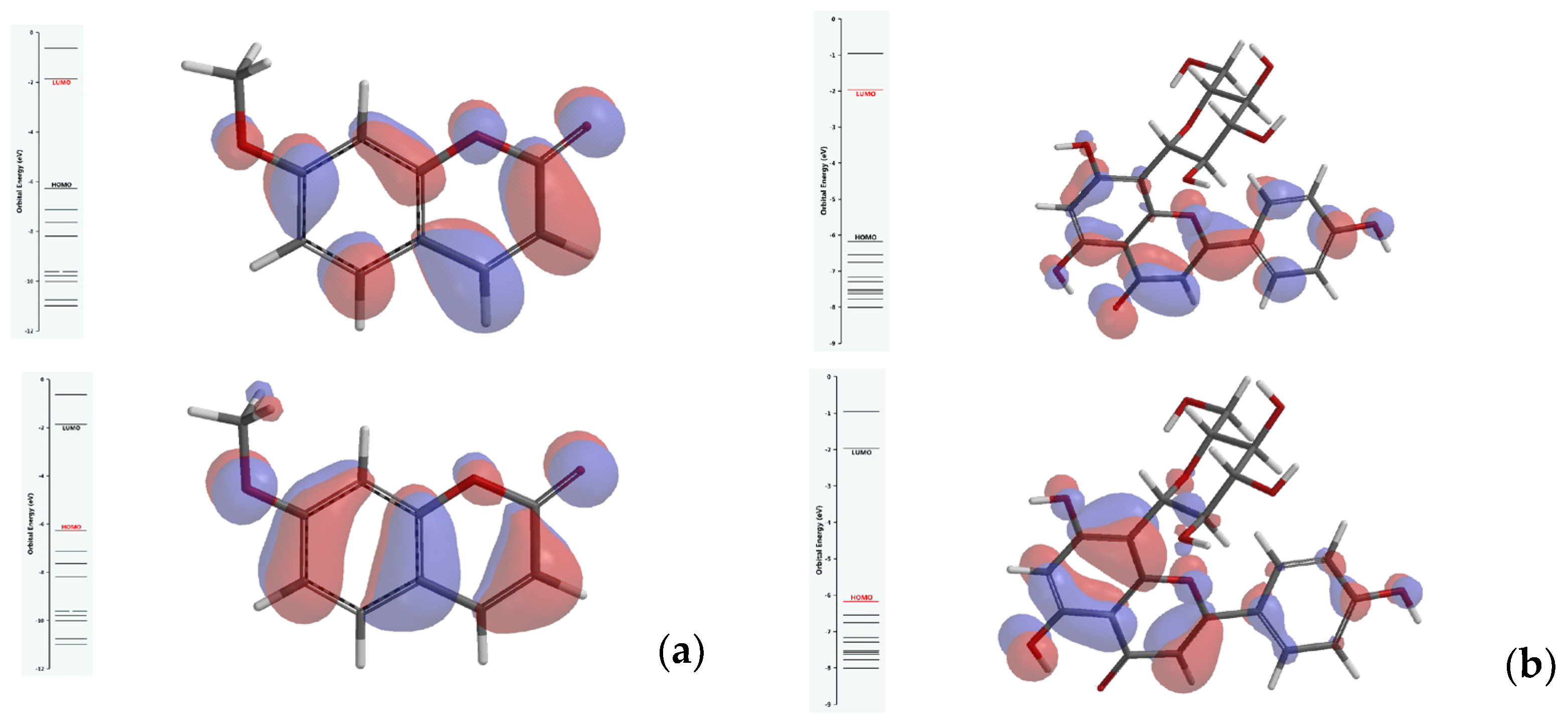
Key parameters for the evaluation of reactivity and kinetic stability of investigated ligands are calculated from the predicted energy levels of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), using equations stated by Koopmans’s theorem [10]. A larger HOMO–LUMO gap indicates more stable molecules. Contrarily, more reactivity is expected from compounds revealing a smaller HOMO–LUMO energy gap. Figure 1 illustrates a HOMO–LUMO gap of about 4.4 eV for 7-methoxycoumarin and 4.2 eV for apigenin-8-C-glucoside, respectively, predicted using B3LYP/311G (d,p) level of theory.
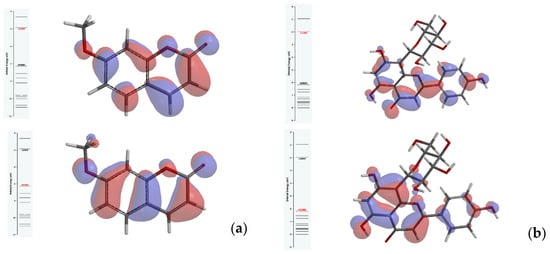
Figure 1.
Energy diagram of frontier molecular orbitals and their energy gap for (a) 7-methoxycoumarin and (b) apigenin-8-C-glucoside.
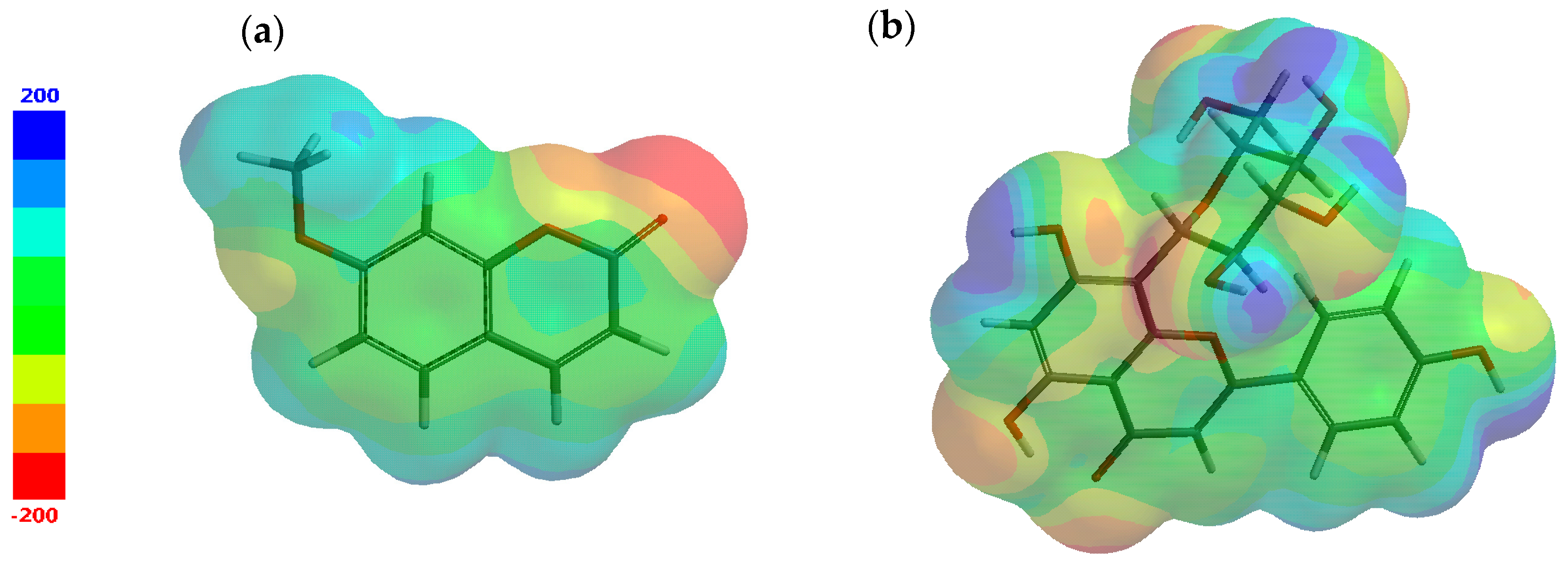
Additionally, by exploring the structures under investigation, electron-rich regions meaning negative potentials and electron-poor regions suggesting positive potentials are identified and mapped, as illustrated in Figure 2. The most negative potential is colored red and the most positive potential is colored blue. The values vary in the following order: red < orange < yellow < green < blue.
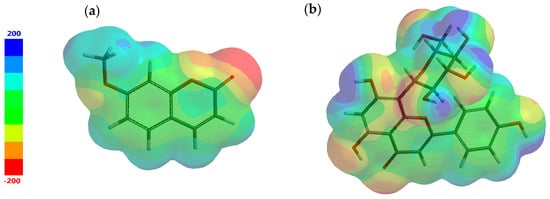
Figure 2.
Electrostatic potential map for (a) 7-methoxycoumarin and (b) apigenin-8-C-glucoside.
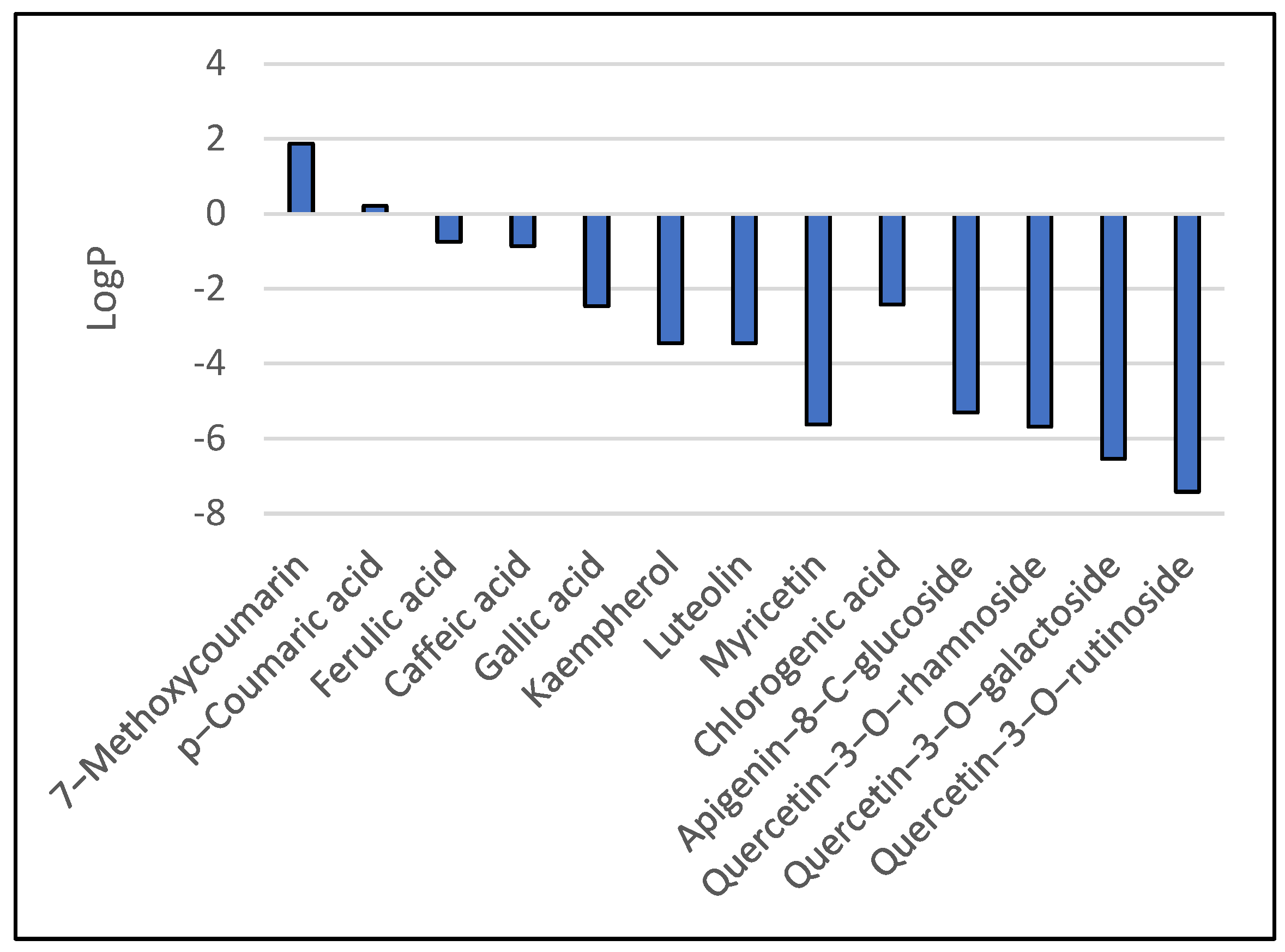
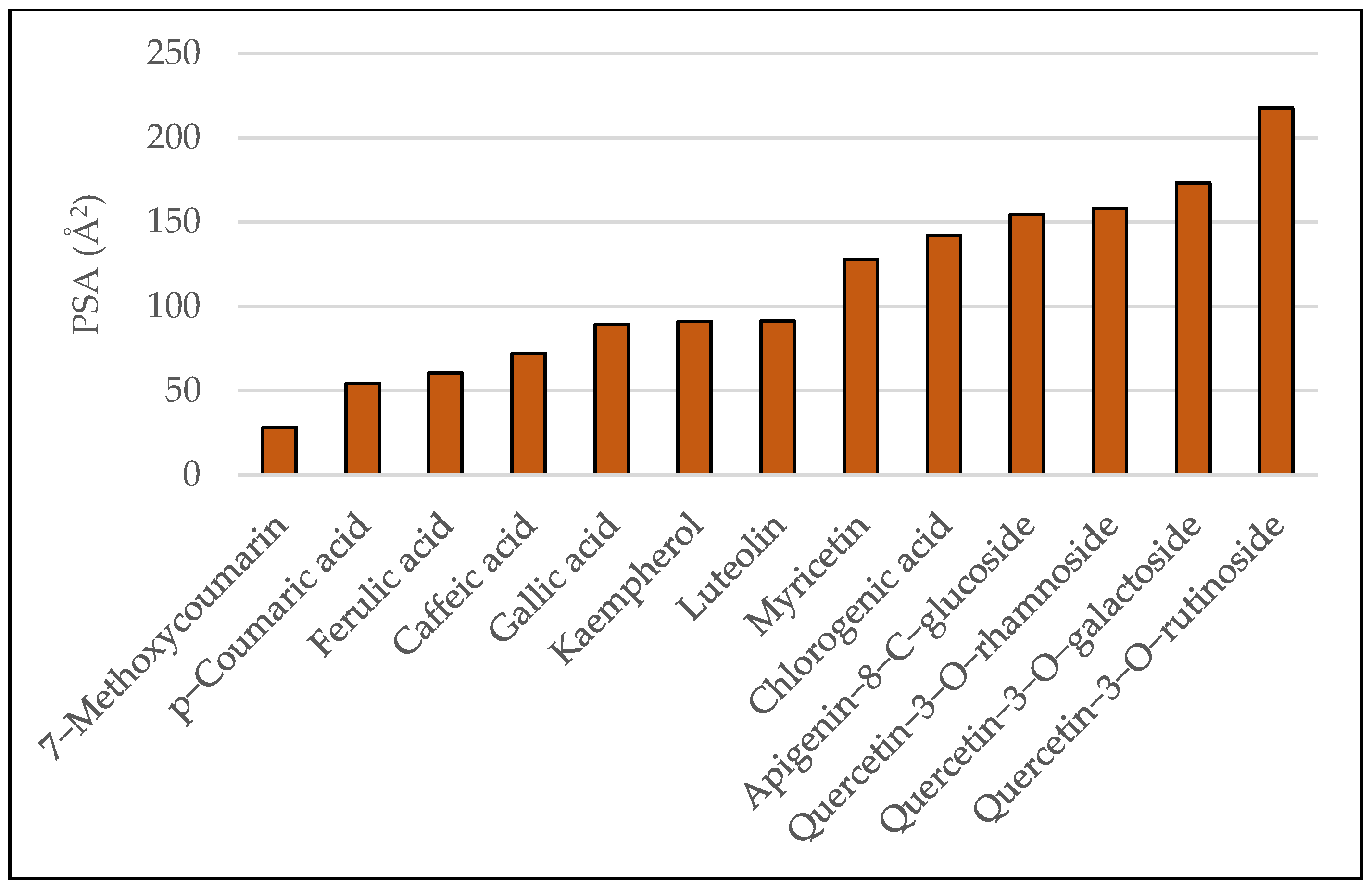
In vivo, the Blood–Brain Barrier (BBB) permeation level for drug candidates targeting the central nervous system can be easily predicted using values of the water-octanol partition coefficient (logP) and polar surface area (PSA) of the molecules, which impose restrictive transport/permeability across the brain. Equally important features to consider are molecular weight, hydrogen bond donors, and the hydrophilic–lipophilic balance [11]. Plots of polar surface area and logP for the studied ligands are given in Figure 3 and Figure 4, respectively.
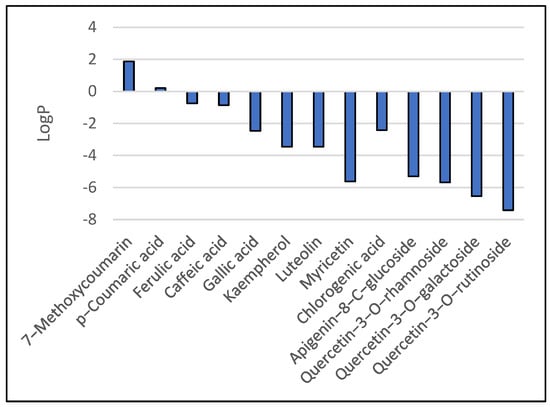
Figure 3.
Predicted logP values for the studied ligands.
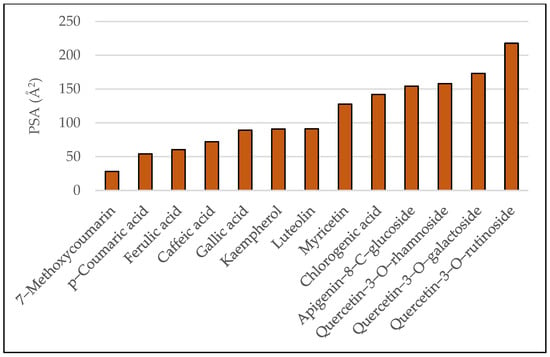
Figure 4.
Predicted PSA values for the studied ligands.
As a general rule, orally active drugs should not exceed a PSA of about 120 Å2. Values lower than 100 Å2 or even smaller (60–70 Å2) characterize drugs with good brain penetration [12]. Regarding logP, values over 5 suggest poor absorption or permeation [13].
3.2. Results of Molecular Docking Simulations
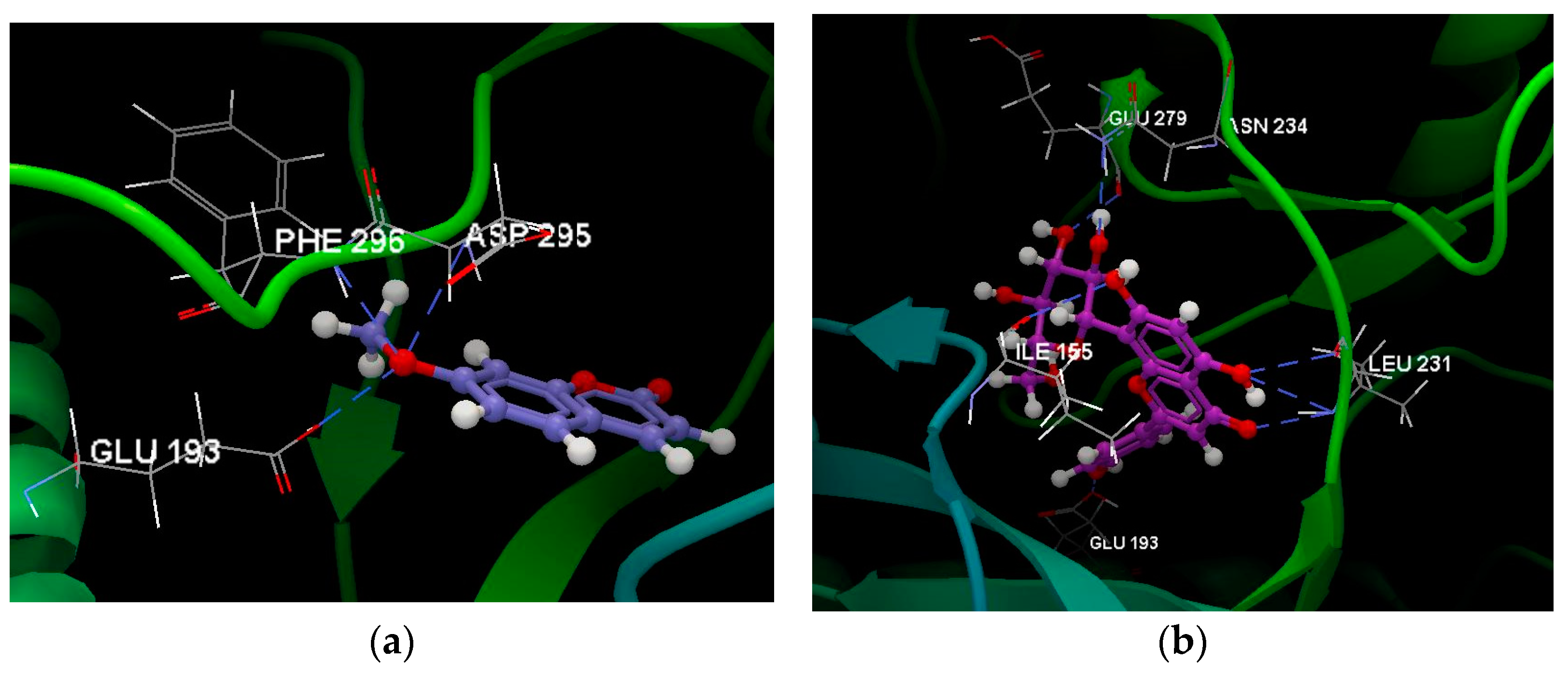
Given their drug-like potential, the most promising compounds in terms of good oral bioavailability, dictated by the water–octanol partition coefficient and the polar surface area, were chosen for molecular docking simulations to assess their ability to bind tyrosine-regulated kinase 2, PDB ID: 5ZTN [5]. An example of obtained data following the previously validated docking protocol [14] is depicted in Figure 5a, showing hydrogen-bonding interactions of the oxygen atom (sp3) of 7-methoxycoumarin with GLU193 (2.643 Å), PHE296 (3.266 Å), and ASP295 (3.044 Å) amino acid residues. Figure 5b illustrates six hydrogen bonds formed by apigenin-8-C-glucoside as follows: 3 hydrogen bonds with LEU231 (3.103 Å, 3.118 Å, and 2.890 Å, respectively), and one with ILE155 (2.860 Å), ASN234 (3.249 Å), GLU279 (2.533 Å), and GLU195 (2.964 Å), all amino acids residues belonging to the chain A of the 5ZTN fragment.
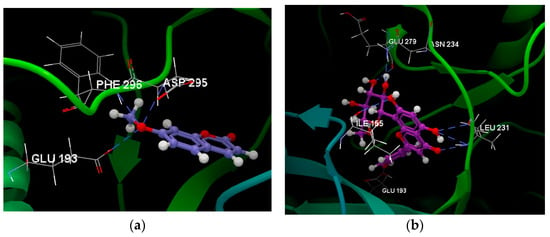
Figure 5.
Hydrogen bonding formed by (a) 7-methoxycoumarin and (b) apigenin-8-C-glucoside within the active binding site of the 5ZTN fragment.
Results obtained for the docking score as defined by Korb and co-workers [15] are negative values for all ligands, showing strong binding with the protein fragment, corresponding to the potential energy change when the protein and ligand come closer and interact. The docking score is −30.83 (RMSD: 0.05) for 7-methoxycoumarin and −53.73 (RMSD: 0.43) for apigenin-8-C-glucoside, both less than the docking score for the co-crystallized curcumin (−83.92, RMSD: 1.99), suggesting weaker binding for these investigated ligands than for the native ligand, namely curcumin.
4. Conclusions
This study reported key features and reactivity parameters for the druglikeness evaluation of several polyphenol derivatives naturally found in Hippophae rhamnoides fruits. Regarding the docking results obtained for 7-methoxycoumarin and apigenin-8-C-glucoside discussed in this paper, a less negative score than for the native ligand, curcumin, corresponds to a weaker binding within the active site of the investigated dual tyrosine-regulated kinase. Among all the investigated structures, the computation property results and binding interactions occurring in the active site of the investigated molecular target indicate the most feasible anti-neuroblastic ligands to design further functional food components with neuroprotective potential.
Author Contributions
Conceptualization, S.C.G. and L.C.P.; methodology, A.S.; software, A.S. and L.P.; validation, A.S. and L.P.; formal analysis, L.C.P. and S.C.G.; investigation, L.P.; data curation, A.S.; writing—original draft preparation, A.S.; writing—review and editing, A.S. and L.C.P.; visualization, A.S.; supervision, L.C.P.; project administration, L.C.P.; funding acquisition, L.C.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded within the NUCLEU Program, project no. PN 23-28 04 01, carried out with the support of the Ministry of Research, Innovation and Digitization, within the National Research and Innovation Strategic Plan 2023-2027.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article. Raw property data are available from the authors upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Criste, A.; Urcan, A.C.; Bunea, A.; Pripon Furtuna, F.R.; Olah, N.K.; Madden, R.; Corcionivoschi, N. Phytochemical Composition and Biological Activity of Berries and Leaves from Four Romanian Sea Buckthorn (Hippophae rhamnoides L.). Varieties. Molecules 2020, 25, 1170. [Google Scholar] [CrossRef] [PubMed]
- Pirvu, L.; Hlevca, C.; Nicu, I.; Bubueanu, C. Comparative Analytical, Antioxidant and antimicrobial Activity Studies on a Series of Vegetal Extracts Prepared from Eight Plant Species Growing in Romania. JPC-J. Planar. Chromat. 2014, 27, 346–356. [Google Scholar] [CrossRef]
- Ladol, S.; Sharma, D. The effects of Hippophae rhamnoides in neuroprotection and behavioral alterations against iron-induced epilepsy. Epilepsy Res. 2021, 175, 106695. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Bala, M.; Prasad, J.; Singh, S.; Gupta, M. Leaves of Hippophae rhamnoides prevent taste aversion in gamma-irradiated rats. J. Diet. Suppl. 2011, 8, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Ji, C.; Mayfield, J.E.; Goel, A.; Xiao, J.; Dixon, J.E.; Guo, X. Ancient drug curcumin impedes 26S proteasome activity by direct inhibition of dual-specificity tyrosine-regulated kinase 2. Proc. Natl. Acad. Sci. USA 2018, 115, 8155–8160. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Shao, Y.; Molnar, L.F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S.T.; Gilbert, A.T.B.; Slipchenko, L.V.; Levchenko, S.V.; O’Neill, D.P.; et al. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Stefaniu, A.; Pirvu, L.C. In Silico Study Approach on a Series of 50 Polyphenolic Compounds in Plants; A Comparison on the Bioavailability and Bioactivity Data. Molecules 2022, 27, 1413. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.G. Absolute electronegativity and hardness: Application to inorganic chemistry. Inorg. Chem. 1988, 27, 734–740. [Google Scholar] [CrossRef]
- Geldenhuys, W.J.; Mohammad, A.S.; Adkins, C.E.; Lockman, P.R. Molecular determinants of blood–brain barrier permeation. Ther. Deliv. 2015, 6, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.P.; Price, D.A. Chapter 23—Role of Physicochemical Properties and Ligand Lipophilicity Efficiency in Addressing Drug Safety Risks. Annu. Rep. Med. Chem. 2010, 45, 380–391. [Google Scholar]
- Vasile (Corbei), A.-A.; Ștefaniu, A.; Pintilie, L.; Stanciu, G.; Ungureanu, E.-M. In silico characterization and preliminary anticancer assessment of some 1,3,4-thiadiazoles. U.P.B. Sci. Bull. Ser. B 2021, 83, 3–12. [Google Scholar]
- Schroder, V.; Radu, N.; Cornea, P.C.; Coman, O.A.; Pirvu, L.C.; Mohammed, M.S.O.; Stefaniu, A.; Pintilie, L.; Bostan, M.; Caramihai, M.D.; et al. Studies Regarding the Antimicrobial Behavior of Clotrimazole and Limonene. Antibiotics 2022, 11, 1816. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring functions for advanced protein-ligand docking with plants. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).