
Bioavailability Computations for Natural Phenolic Derivatives for Druglikeness Assessment †



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
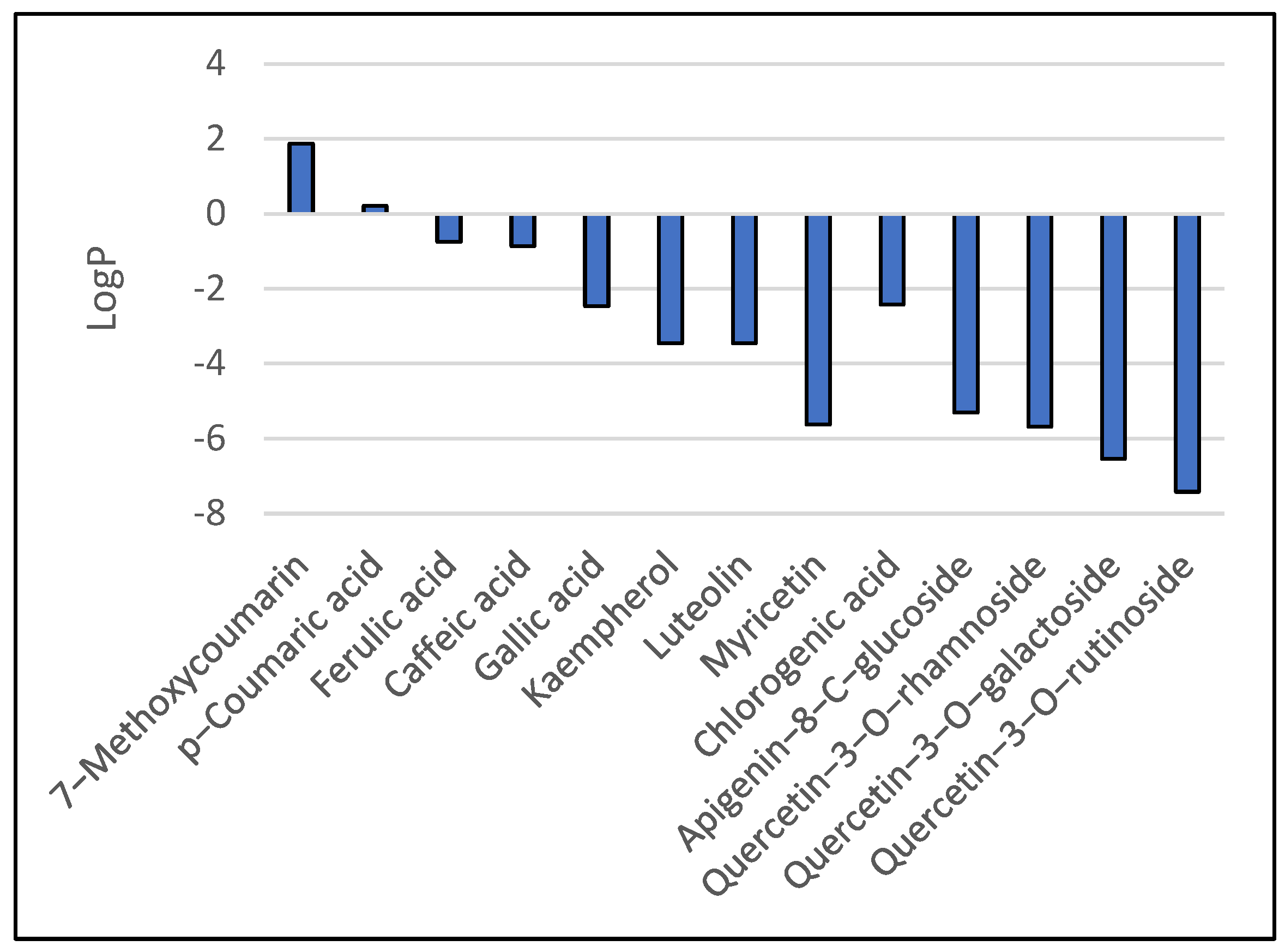
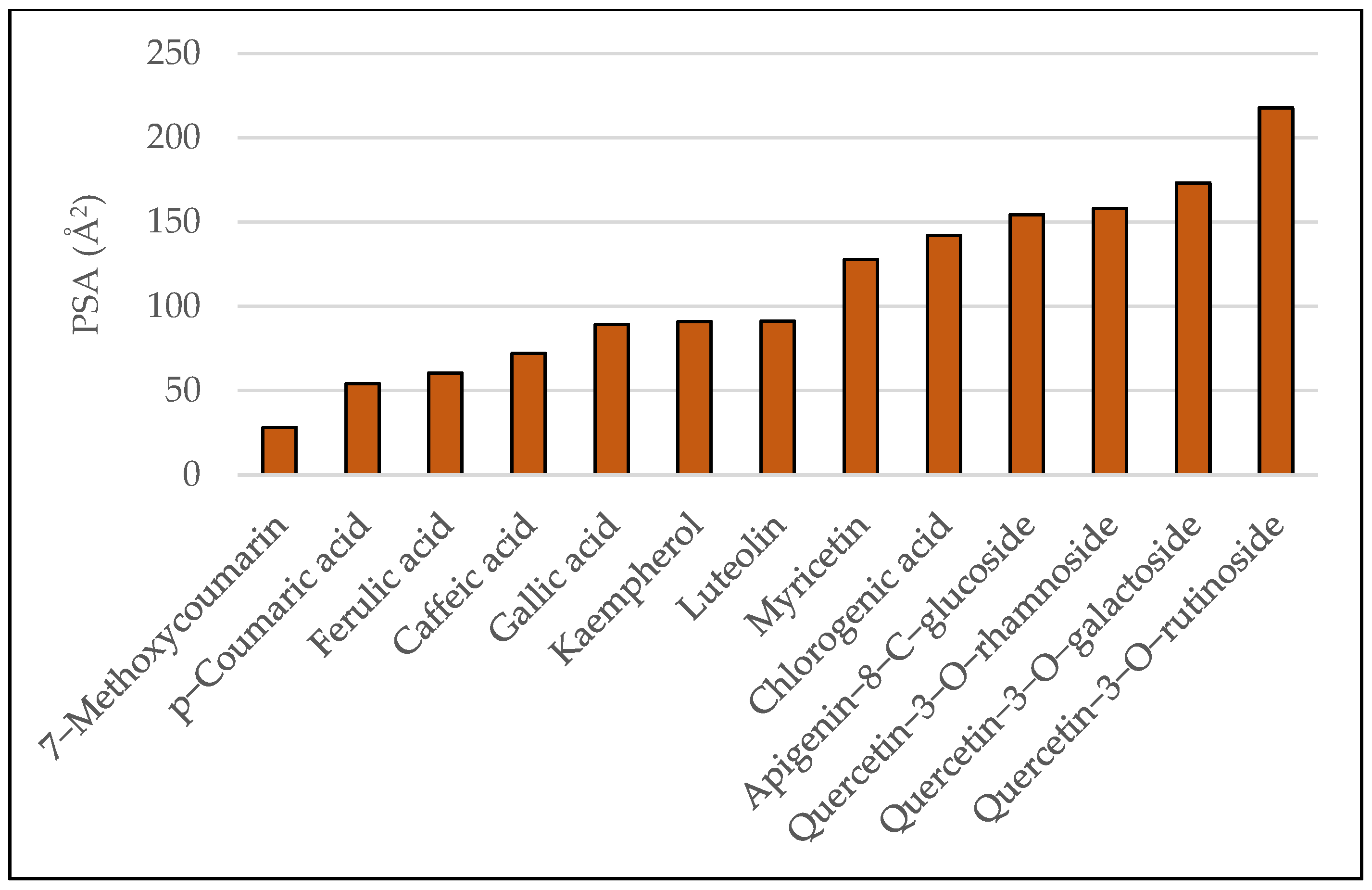
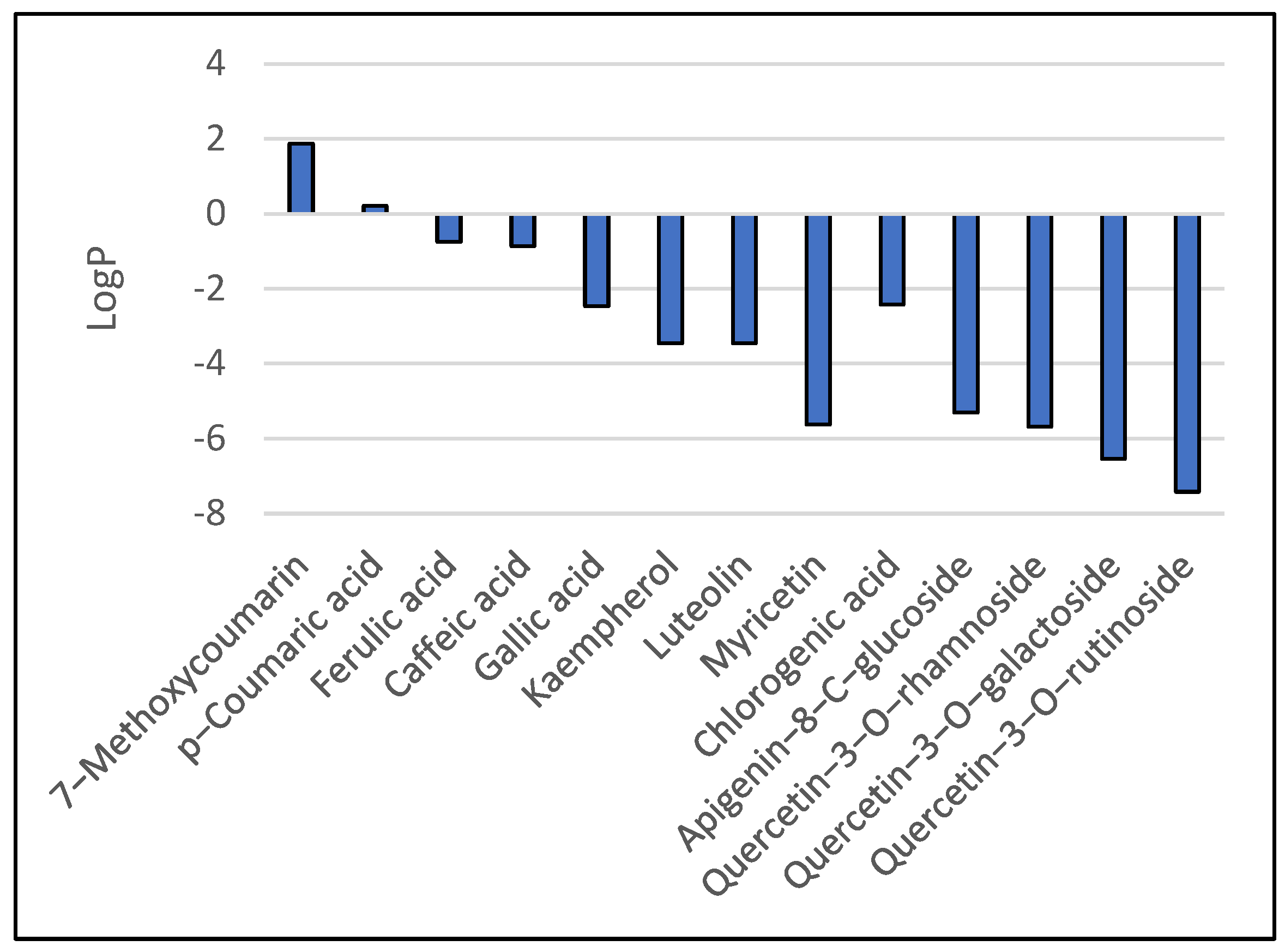
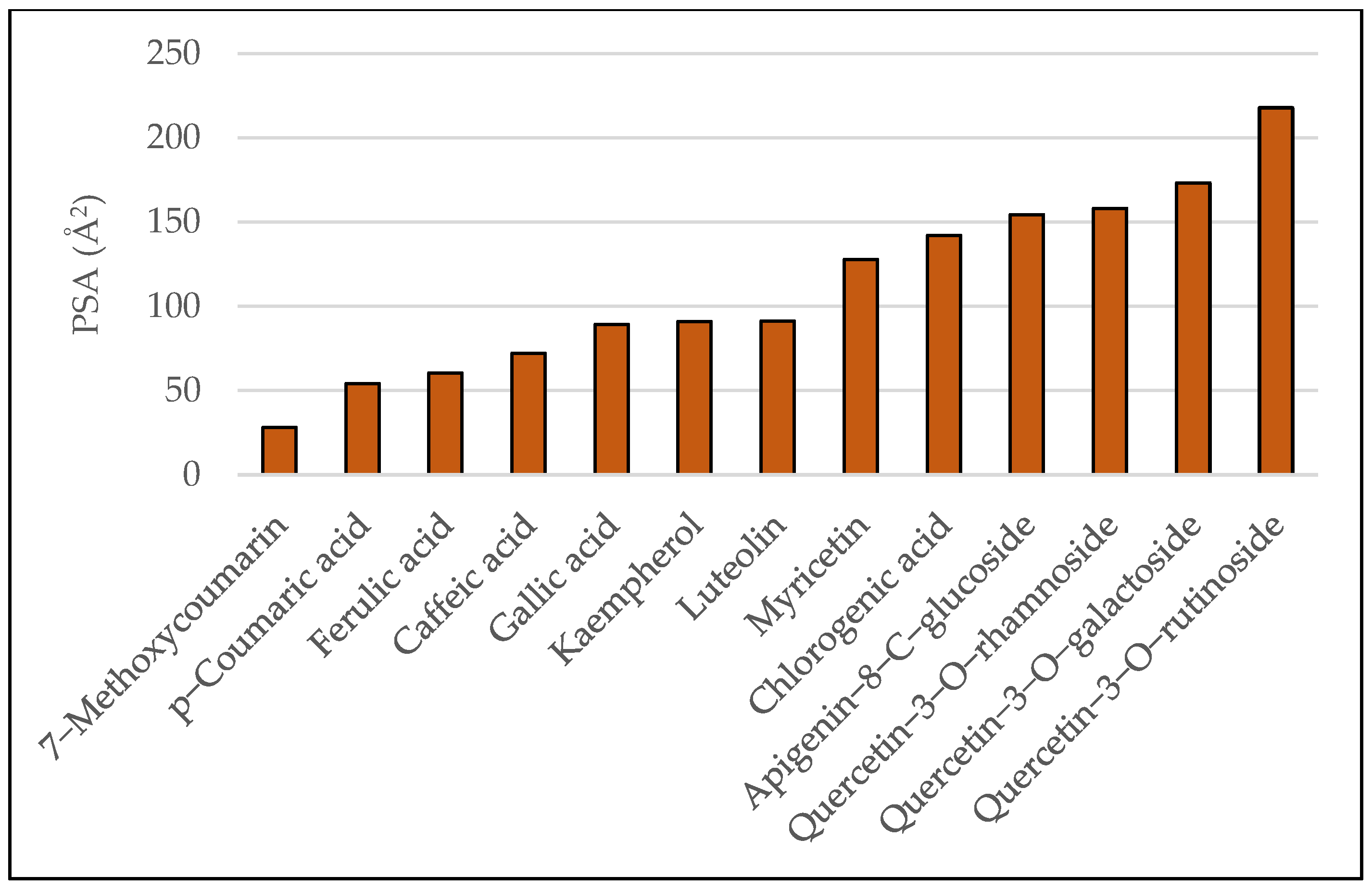
3.1. Results of Bioavailability Prediction
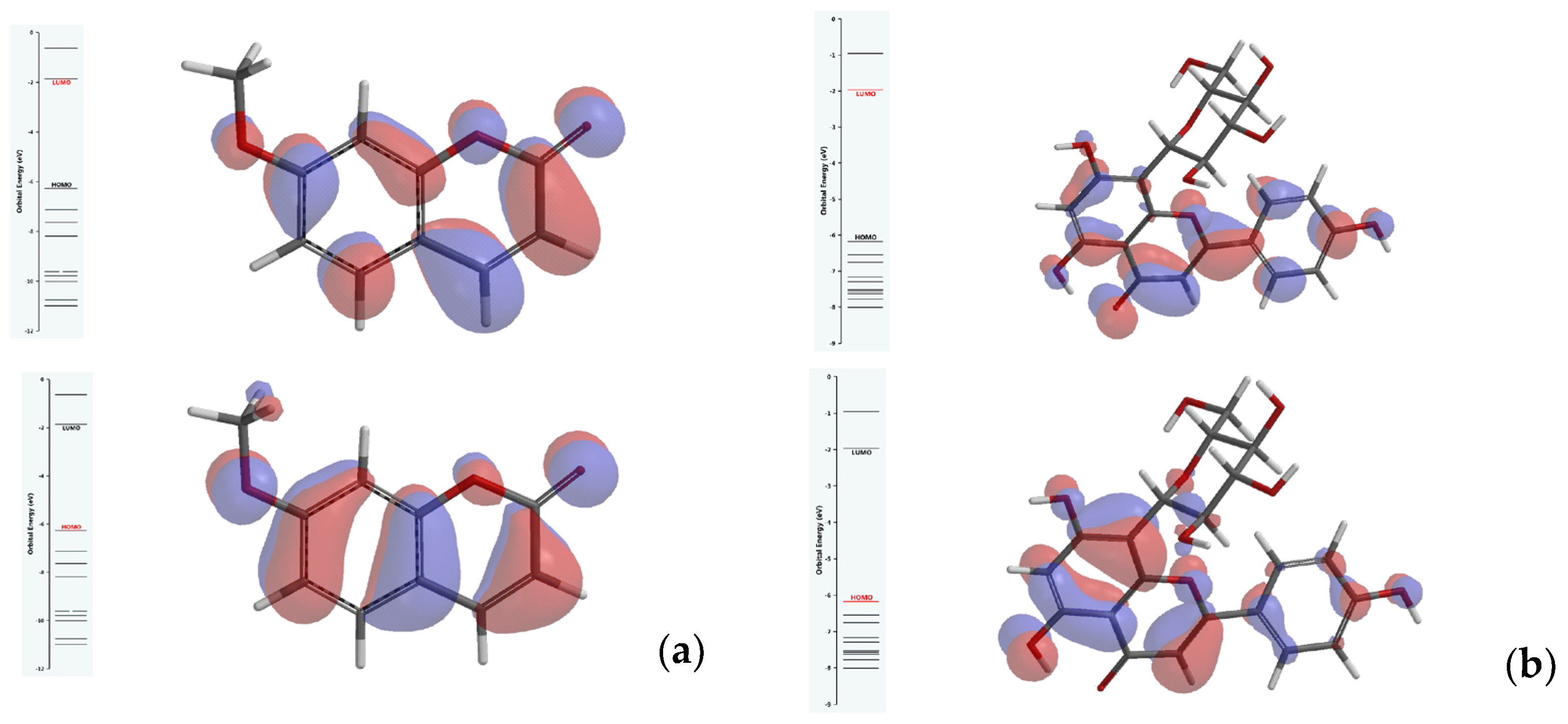
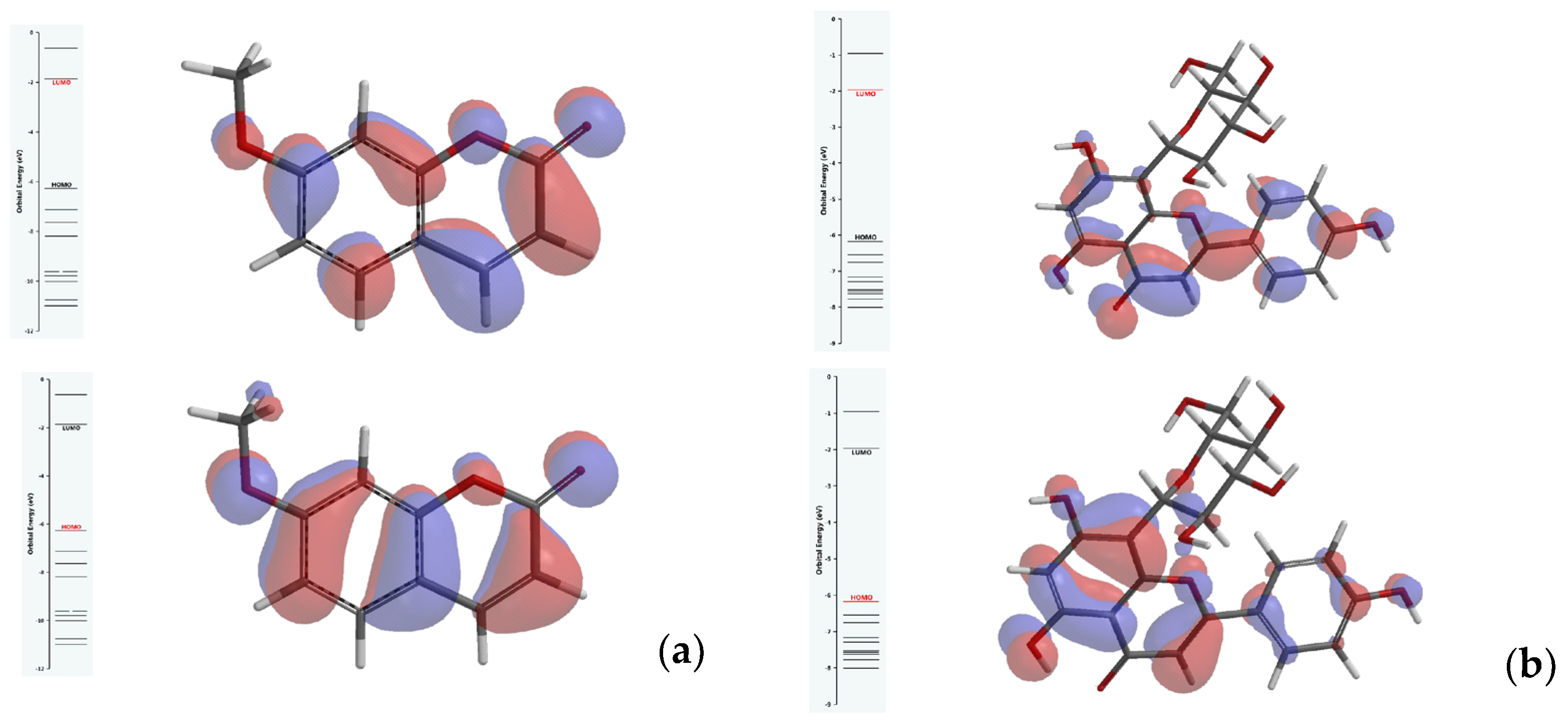
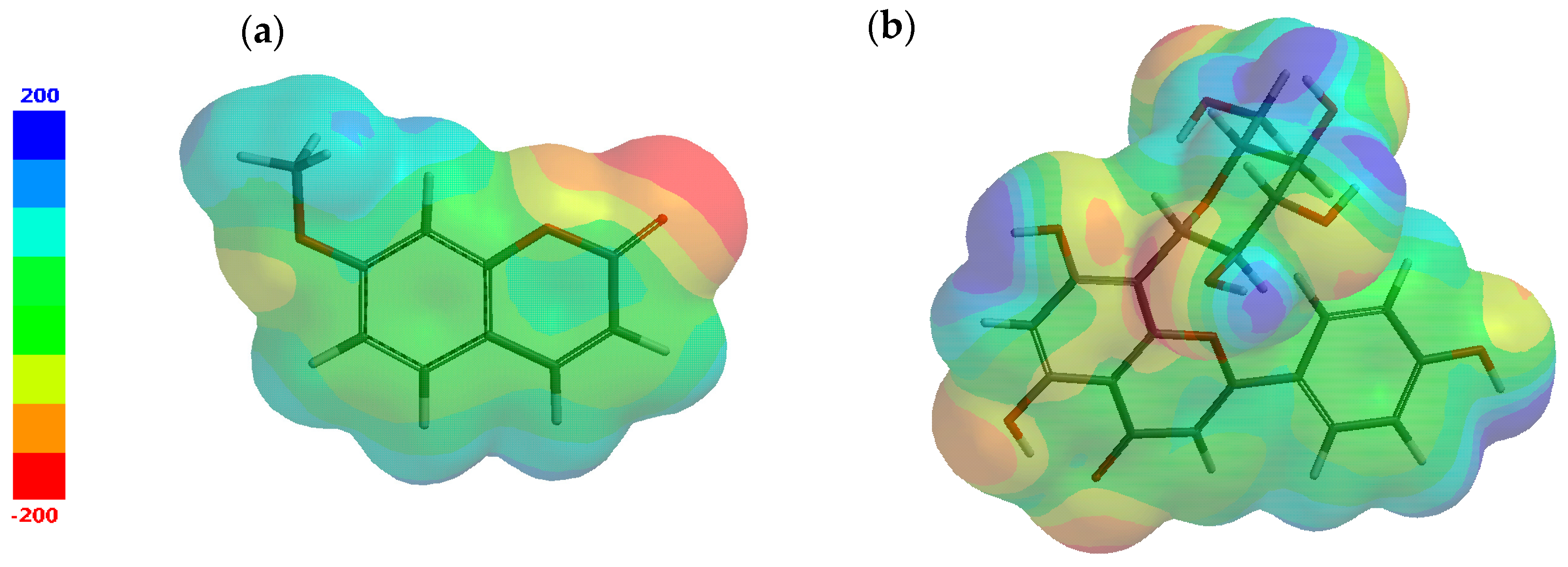
3.2. Results of Molecular Docking Simulations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Criste, A.; Urcan, A.C.; Bunea, A.; Pripon Furtuna, F.R.; Olah, N.K.; Madden, R.; Corcionivoschi, N. Phytochemical Composition and Biological Activity of Berries and Leaves from Four Romanian Sea Buckthorn (Hippophae rhamnoides L.). Varieties. Molecules 2020, 25, 1170. [Google Scholar] [CrossRef] [PubMed]
- Pirvu, L.; Hlevca, C.; Nicu, I.; Bubueanu, C. Comparative Analytical, Antioxidant and antimicrobial Activity Studies on a Series of Vegetal Extracts Prepared from Eight Plant Species Growing in Romania. JPC-J. Planar. Chromat. 2014, 27, 346–356. [Google Scholar] [CrossRef]
- Ladol, S.; Sharma, D. The effects of Hippophae rhamnoides in neuroprotection and behavioral alterations against iron-induced epilepsy. Epilepsy Res. 2021, 175, 106695. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Bala, M.; Prasad, J.; Singh, S.; Gupta, M. Leaves of Hippophae rhamnoides prevent taste aversion in gamma-irradiated rats. J. Diet. Suppl. 2011, 8, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Ji, C.; Mayfield, J.E.; Goel, A.; Xiao, J.; Dixon, J.E.; Guo, X. Ancient drug curcumin impedes 26S proteasome activity by direct inhibition of dual-specificity tyrosine-regulated kinase 2. Proc. Natl. Acad. Sci. USA 2018, 115, 8155–8160. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Shao, Y.; Molnar, L.F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S.T.; Gilbert, A.T.B.; Slipchenko, L.V.; Levchenko, S.V.; O’Neill, D.P.; et al. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Stefaniu, A.; Pirvu, L.C. In Silico Study Approach on a Series of 50 Polyphenolic Compounds in Plants; A Comparison on the Bioavailability and Bioactivity Data. Molecules 2022, 27, 1413. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.G. Absolute electronegativity and hardness: Application to inorganic chemistry. Inorg. Chem. 1988, 27, 734–740. [Google Scholar] [CrossRef]
- Geldenhuys, W.J.; Mohammad, A.S.; Adkins, C.E.; Lockman, P.R. Molecular determinants of blood–brain barrier permeation. Ther. Deliv. 2015, 6, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.P.; Price, D.A. Chapter 23—Role of Physicochemical Properties and Ligand Lipophilicity Efficiency in Addressing Drug Safety Risks. Annu. Rep. Med. Chem. 2010, 45, 380–391. [Google Scholar]
- Vasile (Corbei), A.-A.; Ștefaniu, A.; Pintilie, L.; Stanciu, G.; Ungureanu, E.-M. In silico characterization and preliminary anticancer assessment of some 1,3,4-thiadiazoles. U.P.B. Sci. Bull. Ser. B 2021, 83, 3–12. [Google Scholar]
- Schroder, V.; Radu, N.; Cornea, P.C.; Coman, O.A.; Pirvu, L.C.; Mohammed, M.S.O.; Stefaniu, A.; Pintilie, L.; Bostan, M.; Caramihai, M.D.; et al. Studies Regarding the Antimicrobial Behavior of Clotrimazole and Limonene. Antibiotics 2022, 11, 1816. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring functions for advanced protein-ligand docking with plants. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stefaniu, A.; Pirvu, L.C.; Pintilie, L.; Godeanu, S.C. Bioavailability Computations for Natural Phenolic Derivatives for Druglikeness Assessment. Chem. Proc. 2023, 13, 26. https://doi.org/10.3390/chemproc2023013026
Stefaniu A, Pirvu LC, Pintilie L, Godeanu SC. Bioavailability Computations for Natural Phenolic Derivatives for Druglikeness Assessment. Chemistry Proceedings. 2023; 13(1):26. https://doi.org/10.3390/chemproc2023013026
Chicago/Turabian StyleStefaniu, Amalia, Lucia Camelia Pirvu, Lucia Pintilie, and Sorin Constantin Godeanu. 2023. "Bioavailability Computations for Natural Phenolic Derivatives for Druglikeness Assessment" Chemistry Proceedings 13, no. 1: 26. https://doi.org/10.3390/chemproc2023013026
APA StyleStefaniu, A., Pirvu, L. C., Pintilie, L., & Godeanu, S. C. (2023). Bioavailability Computations for Natural Phenolic Derivatives for Druglikeness Assessment. Chemistry Proceedings, 13(1), 26. https://doi.org/10.3390/chemproc2023013026