Abstract
Type 2 diabetes mellitus (T2DM) and obesity affect hundreds of millions of adults worldwide and represent leading drivers of cardiovascular disease, chronic kidney disease, and escalating healthcare expenditures. Incretin-based therapies have fundamentally reshaped cardiometabolic disease management, with dual- and triple-receptor agonists extending the benefits of traditional glucagon-like peptide-1 (GLP-1) receptor agonism. By synthesizing clinical, mechanistic, and real-world data, this review examines the evolving therapeutic landscape of GLP-1-based multi-agonists. Dual GLP-1/glucose-dependent insulinotropic polypeptide (GIP) receptor agonists demonstrate superior metabolic efficacy compared with GLP-1 receptor agonists alone, achieving greater reductions in body weight and glycemic indices across diverse patient populations. Emerging triple agonists targeting GLP-1, GIP, and glucagon receptors further enhance metabolic outcomes, with weight loss approaching that observed following bariatric surgery in late-phase clinical trials. Mechanistically, multi-receptor co- agonism produces synergistic effects through complementary pathways, including appetite suppression, glucose-dependent insulin secretion, improved adipose tissue metabolism, increased energy expenditure, enhanced hepatic lipid oxidation, and reductions in hepatic steatosis. Beyond glycemic and weight endpoints, GLP-1-based therapies confer clinically meaningful cardiovascular and renal protection. Trials consistently demonstrate reductions in major adverse cardiovascular events across populations with and without T2DM, while kidney-specific trials show significant slowing of disease progression. However, gastrointestinal adverse events remain common and contribute to substantial treatment discontinuation, particularly in real-world settings. Despite their transformative efficacy, the population-level impact of these therapies is constrained by significant implementation barriers, including high drug costs, limited insurance coverage, restrictive utilization management policies, and pronounced racial and socioeconomic disparities in access. Emerging innovations including oral formulations, longer-acting injectables, and novel peptide combinations look to improve tolerability, adherence, and scalability, while therapeutic indications continue to expand to conditions such as metabolic dysfunction-associated steatohepatitis, chronic kidney disease, obstructive sleep apnea, and neurodegenerative disease. This review provides a comprehensive framework for understanding the clinical potential, mechanistic basis, and real-world challenges of GLP-1-based multi-agonists and outlines key priorities for optimizing implementation and maximizing their impact on global cardiometabolic health.
1. Introduction
1.1. The Convergent Epidemics of Type 2 Diabetes Mellitus and Obesity
Type 2 Diabetes Mellitus (T2DM) and obesity represent two of the most pressing public health challenges of the 21st century, affecting over 537 million and 890 million adults globally, respectively [1,2]. These conditions frequently coexist, with approximately 90% of individuals with T2DM classified as overweight or obese, creating a syndemic of cardiometabolic disease [3]. The economic burden is staggering, with T2DM-related healthcare expenditures exceeding $966 billion annually worldwide, with obesity expenditure at an estimated $260 billion in the United States alone [1,4]. Beyond financial costs, these conditions drive premature mortality through cardiovascular disease, chronic kidney disease, hepatic steatosis, and numerous malignancies.
Traditional management paradigms have historically addressed glycemic control and weight management as separate therapeutic targets. However, the shared pathophysiology—insulin resistance, β-cell dysfunction, chronic inflammation, and dysregulated appetite signaling—suggests that integrated pharmacological approaches targeting multiple pathways simultaneously may offer superior outcomes. The emergence of incretin-based therapies, particularly glucagon-like peptide-1 receptor agonists (GLP-1 RAs), has begun to bridge this divide, demonstrating that substantial weight reduction and glycemic improvement can be achieved through a unified mechanism.
1.2. Evolution of Incretin-Based Therapeutics
The incretin effect, whereby oral glucose elicits greater insulin secretion than intravenous glucose despite similar glycemia, was first described in 1964 [5]. This phenomenon was later attributed to gut-derived hormones, primarily GLP-1 and glucose-dependent insulinotropic polypeptide (GIP), which amplify glucose-stimulated insulin secretion. In T2DM, the incretin effect is markedly diminished, contributing to postprandial hyperglycemia and progressive β-cell failure.
The first GLP-1 RA, exenatide, was approved in 2005. It enhances glucose-dependent insulin secretion-reducing hypoglycemia risk when not used with insulin or sulfonylureas, and yields modest weight reduction of 2–3 kg [6,7]. Subsequent generations—liraglutide, dulaglutide, and semaglutide—progressively improved upon this foundation through extended half-lives enabling weekly dosing and enhanced potency. The SUSTAIN [8], LEADER [9], and REWIND [10] cardiovascular outcome trials established that GLP-1 RAs not only improve glycemic control but also significantly reduce MACE (with pooled analyses showing ~12% relative risk reduction), fundamentally repositioning these agents from purely glucose-lowering drugs to therapies with demonstrated cardioprotective benefit [11].
Despite a 10–15% mean weight reduction with the most potent GLP-1 monotherapies, many patients do not achieve clinically significantly weight reductions [12]. Furthermore, GIP—once thought to promote adiposity—has been reappraised, with GIP/GLP-1 receptors co-agonism showing synergistic metabolic benefits exceeding single-receptor agonism [13,14,15,16].
1.3. The Multi-Agonist Revolution
The approval of tirzepatide in 2022, the first dual GIP/GLP-1 receptor agonist, marked a transformative milestone in metabolic therapy. In SURMOUNT-1, tirzepatide produced up to 20.9% mean weight loss in adults with obesity, nearly doubling the effect of early GLP-1 monotherapies [16] (p. 1). The SURPASS trials further demonstrated HbA1c reductions of 2.0–2.3%, establishing tirzepatide as the most potent glucose-lowering agent to date [17]. Direct head-to-head comparison in SURMOUNT-5 (2025) confirmed tirzepatide’s superiority over semaglutide 2.4 mg, with 20.2% versus 13.7% weight loss at 72 weeks [13].
These results have accelerated development of next-generation multi-agonists incorporating glucagon receptor activity. Retatrutide, the most advanced triple GIP/GLP-1/glucagon agonist, achieved 24.2% weight loss at 48 weeks (12 mg weekly) in phase 2 trials—weight reduction previously attainable only through bariatric surgery [18]. Additional investigational agents such as survodutide (GLP-1/glucagon dual agonist) and early-stage quadruple agonists incorporating IGF-1 activity (bioglutide) aim to further enhance efficacy and preserve lean mass during substantial weight loss.
The mechanistic rationale for multi-receptor agonism is compelling. GLP-1 receptor activation suppresses appetite through hypothalamic POMC/CART neuronal pathways, slows gastric emptying, and enhances glucose-dependent insulin secretion. GIP receptor signaling complements these effects through adipose tissue regulation, supporting glucagon responses during hypoglycemia, and potentially exerting antiemetic central effects that may improve tolerability. Glucagon receptor activation uniquely increases energy expenditure and hepatic lipid oxidation, mechanisms not engaged by GLP-1 or GIP monotherapy. The convergence of these pathways produces metabolic effects exceeding simple additivity, supporting the concept of true pharmacological synergy. Table 1, provides a comprehensive overview of the incretin-based therapeutic landscape, from established GLP-1 monotherapies to investigational multi-agonists, highlighting their mechanistic diversity and expanding clinical applications.

Table 1.
Incretin-Based Therapies: Approved and Investigational Agents by Mechanism of Action.
1.4. Critical Gaps in Current Literature
Despite remarkable clinical trial successes, significant knowledge gaps limit optimal implementation of incretin-based therapies and highlight areas requiring urgent investigation.
Although clinical efficacy is well-established, the molecular mechanisms underlying multi-agonist synergy remain only partially understood. Why does co-activation of GIP and GLP-1 receptors produce effects exceeding mathematical additivity? Which signaling pathways—Gs/cAMP, β-arrestin, or biased agonism—drive therapeutic benefits versus adverse effects? Emerging pharmacogenomic evidence, including associations between treatment response and ARRB1 (β-arrestin), a key regulator of GPCR internalization and signaling duration, as well as the presence of GLP1R variants, point towards the feasibility of personalized incretin therapy, yet translation into clinical practice is still in its infancy. Addressing these unresolved mechanistic and genetic questions will be essential to fully realize the transformative potential of next-generation incretin and multi-receptor agonist treatments.
Most trials compare novel agents to placebo rather than active comparators. Head-to-head trials directly comparing tirzepatide, semaglutide, and emerging triple agonists across T2DM and obesity indications remain sparse. Network meta-analyses provide indirect comparisons, but real-world comparative effectiveness data spanning diverse patient populations are critically needed.
Clinical trials emphasize HbA1c and weight as primary endpoints, yet patients prioritize quality of life, tolerability, and treatment burden. Real-world adherence data reveal that only 32–46% of patients persist on GLP-1 therapy at 12 months, driven by gastrointestinal side effects, injection fatigue, and cost barriers [33]. Understanding factors predicting non-response, treatment discontinuation, and weight regain after cessation is essential for appropriate patient selection and counseling.
Incretin therapies exist at the intersection of efficacy and limited accessibility. Monthly costs of $1000–$1350 create substantial affordability barriers, with fewer than 1% of eligible patients able to receive treatment at current prices without exceeding healthcare budget thresholds. Insurance coverage patterns vary dramatically by indication (T2DM versus obesity), payer type (commercial versus government sponsored), and geography, creating profound health equity concerns. Prior authorization requirements consume 12 h weekly per provider and contribute to 40% of prescriptions going unfilled [5]. Cost-effectiveness analyses demonstrate favorable ICERs in selected analyses, particularly for T2DM indications ($53,000–$69,000/QALY), yet budget impact analyses reveal population-level access is financially untenable without substantial price reductions.
The pipeline of incretin-based therapies is expanding exponentially, with oral formulations (semaglutide 25/50 mg, orforglipron), monthly injectables (MariTide), triple agonists (retatrutide entering phase 3), and novel indications (MASH, Alzheimer’s disease, chronic kidney disease) rapidly advancing through clinical development, with one agent (semaglutide) recently approved by the FDA [34]. Clinicians require evidence-based guidance to navigate this evolving landscape and anticipate future treatment options.
1.5. Objectives and Scope of This Review
This narrative review addresses the identified gaps through comprehensive synthesis of recent literature (2023–2025) organized around four primary objectives. First, we elucidate the pharmacological and molecular mechanisms of GLP-1, GIP, and glucagon receptor signaling, with emphasis on synergistic effects of multi-receptor co-agonism and pharmacogenomic determinants of treatment response. Second, we systematically compare clinical efficacy and safety across incretin-based therapies, synthesizing head-to-head trials, network meta-analyses, cardiovascular and renal outcome trials, and real-world effectiveness studies. Third, we analyze practical implementation considerations including cost-effectiveness, insurance coverage patterns, patient selection strategies, drug interchangeability protocols, and adherence optimization approaches. Lastly, we evaluate emerging therapies and future directions, including oral and extended-release formulations, novel multi-agonist combinations, and expanding indications beyond T2DM and obesity.
This review is structured to provide clinicians, researchers, and policymakers with evidence-based guidance for optimizing incretin therapy in clinical practice while identifying critical research priorities to advance the field. By integrating mechanistic insights with clinical outcomes and implementation science, we aim to bridge the efficacy-effectiveness gap and maximize the population health impact of these transformative therapeutics.
2. Pharmacology and Mechanisms
The pharmacologic basis of incretin-based multi-agonist therapies rests on coordinated activation of the GLP-1, GIP, and glucagon receptors. These are class B G protein-coupled receptors that regulate glucose homeostasis, lipid metabolism, appetite, and systemic energy expenditure. The GLP-1 receptor (GLP-1R) is highly expressed in pancreatic beta cells, gastrointestinal tissues, and central appetite-regulating nuclei. Stimulation of GLP-1R primarily couples to Gs, raising intracellular cAMP, activating protein kinase A (PKA) and exchange protein activated by cAMP (EPAC) [35]. These pathways enhance glucose-stimulated insulin secretion by promoting calcium influx, granule mobilization, and exocytosis, and they function in a glucose-dependent manner that reduces risk of hypoglycemia. GLP-1R also activates PI3K/Akt and ERK1/2 pathways that support beta-cell survival, while beta-arrestin-regulated internalization influences signaling duration [35]. Within the central nervous system, GLP-1R activation reduces appetite and alters reward-based feeding circuits, while peripheral GLP-1R signaling slows gastric emptying; together these mechanisms contribute to the substantial weight loss and cardiometabolic improvements observed clinically [36]. A schematic representation of the mechanism is provided in Figure 1.
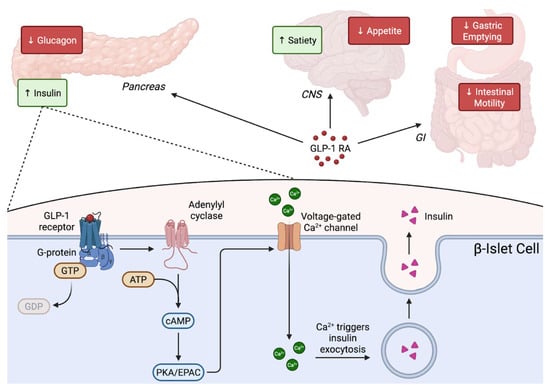
Figure 1.
Mechanism of action of GLP-1 receptor agonists. GLP-1 receptor agonists bind the GLP-1 receptor on pancreatic β-cells, activating Gs–adenylyl cyclase signaling to increase intracellular cAMP, which enhances PKA/EPAC-mediated calcium influx and stimulates insulin granule exocytosis in a glucose-dependent manner. Systemically, GLP-1 RAs slow gastric emptying and intestinal motility predominantly through peripheral vagal-enteric mechanisms, while reducing appetite and increasing satiety via central hypothalamic pathways and collectively improving postprandial and fasting glucose control.
The glucose-dependent insulinotropic polypeptide receptor (GIPR) enhances insulin secretion through similar, Gs-coupled, cAMP-dependent mechanisms but also exerts distinct effects in adipose tissue, bone, and the brain. Although individuals with obesity frequently exhibit diminished insulinotropic response to endogenous GIP, pharmacologic GIPR activation can overcome this relative resistance. When combined with GLP-1R agonism, GIPR signaling produces a supra-additive rise in cAMP and insulin secretion, improving GLP-1 tolerability [37]. Additionally, GIPR activation also modulates adipocyte lipid handling and has central actions that may reduce caloric intake while potentially mitigating nausea associated with GLP-1 agents [38]. These mechanisms help explain the enhanced glycemic and weight-loss efficacy of dual agonists such as tirzepatide compared with GLP-1 receptor agonists alone.
The glucagon receptor (GCGR), highly expressed in hepatocytes, couples to Gs to stimulate glycogenolysis and gluconeogenesis, but it also plays a prominent role in lipid and energy metabolism. GCGR activation increases hepatic mitochondrial beta-oxidation, enhances tricarboxylic acid cycle flux, and raises whole-body energy expenditure [39]. Human studies have shown that glucagon can increase hepatic mitochondrial oxidative rates by 50 to 75 percent, particularly in individuals with obesity [39]. These metabolic actions provide a compelling rationale for incorporating glucagon agonism into multi-receptor therapeutics, although such designs must balance glucagon’s glycemic effects with the glucose-lowering actions of GLP-1 agonism.
Synergistic activation of these receptors drives the superior clinical performance of multi-agonist therapies. GLP-1 and GIP co-agonism amplifies beta-cell signaling, improves appetite regulation, and enhances tolerability [40]. Adding glucagon receptor activation further extends weight-loss potential by increasing basal energy expenditure and promoting hepatic fat oxidation. Triple agonists such as retatrutide demonstrate weight-loss efficacy exceeding ~20 to 24 percent, surpassing dual agents and approaching surgical outcomes [41]. Advances in biased agonism, where ligands preferentially engage specific intracellular signaling pathways rather than uniformly activating all downstream effectors, have enabled design of agents that favor cAMP signaling while limiting beta-arrestin driven desensitization, a concept discussed in greater detail later in this section. This selective signaling contributes to more potent central and peripheral metabolic effects. Receptor trafficking behavior, signaling bias, and tissue-specific patterns of expression have become increasingly important determinants of therapeutic efficacy.
Pharmacokinetic engineering is essential for translating these mechanisms into clinical benefits. Native GLP-1 has a half-life of approximately two minutes due to rapid degradation by DPP-4 and renal clearance [42]. Therapeutic incretin analogues require structural modifications including DPP-4 resistant amino-acid substitutions, fatty-acid acylation to promote albumin binding, or fusion to IgG Fc or albumin to exploit FcRn-mediated recycling. These strategies produce half-lives of several days or longer and enable weekly dosing, with fatty-acid acylation increasing albumin binding and prolonging circulation time [43]. Tirzepatide incorporates a C20 fatty di-acid side chain that provides approximately 99 percent albumin binding and a five-day half-life [44]. Retatrutide uses similar long-acting modifications to support weekly or extended-interval administration. These pharmacokinetic designs promote sustained receptor engagement, improve adherence, and reduce peak-related gastrointestinal effects.
Inter-individual variability in response to incretin-based therapies increasingly appears linked to pharmacogenomic variation. Genome-wide studies have identified GLP1R variants that alter receptor signaling efficiency and correlate with differences in glycemic response [45]. Variants in ARRB1, which modulates beta-arrestin recruitment and receptor internalization, are also associated with enhanced HbA1c reduction among carriers [46]. These findings illustrate how genetic differences in receptor structure and downstream signaling components influence therapeutic response. Future personalization of incretin-based multi-agonist therapy may incorporate genetic markers across GLP1R, GIPR, GCGR, and key signaling regulators such as beta-arrestin and cAMP regulatory pathways.
3. Clinical Efficacy: Glycemic Control
3.1. GLP-1 Receptor Agonists
GLP-1 receptor agonists exist in both short-acting and long-acting formulations, differing in pharmacokinetics, durability of glycemic control, and clinical performance. These distinctions are clinically meaningful, as long-acting agents generally produce more sustained reductions in HbA1c and fasting glucose, whereas short-acting formulations exert stronger postprandial effects but more modest overall glycemic benefit.
Among long-acting GLP-1 receptor agonists, semaglutide demonstrates the greatest glycemic potency across randomized trials and real-world studies, producing mean HbA1c reductions of approximately 1.3–2.2% depending on dose and baseline glycemia [47] (p. 202) and [48,49,50]. In the SUSTAIN clinical trial program, semaglutide consistently outperformed placebo and active comparators, with a high proportion of patients achieving HbA1c targets below 7.0% [15,48,49]. Dulaglutide, another once-weekly GLP-1 RA, similarly produces robust glycemic improvements, typically achieving HbA1c reductions of approximately 1.1–1.7% across the AWARD trials, with incremental benefit observed at higher doses in AWARD-11 [51,52,53,54,55,56]. Liraglutide remains an effective therapy but generally demonstrates slightly lower glycemic potency than once-weekly agents in head-to-head trials and network meta-analyses [57,58,59]. Short-acting agents such as twice-daily exenatide and lixisenatide achieve more modest HbA1c reductions and are less effective for sustained glycemic control [58,60,61,62].
Comparative analyses consistently rank semaglutide as the most effective GLP-1 RA for HbA1c lowering, followed by dulaglutide and liraglutide, while short-acting agents demonstrate inferior glycemic efficacy [58,63,64,65,66]. Meta-analyses further indicate that long-acting formulations provide superior and more durable HbA1c reductions compared with short-acting agents, reinforcing a class gradient favoring sustained receptor activation [58,67]. Real-world evidence broadly confirms this hierarchy, although absolute differences between long-acting GLP-1 receptor agonists are modest relative to the larger glycemic improvements achieved with dual incretin agonism [60,68,69,70].
Taken together, clinical trial and real-world data demonstrate that long-acting GLP-1 RAs provide effective and durable glycemic control and remain a foundational com-ponent of contemporary type 2 diabetes management. However, their efficacy appears to plateau relative to emerging dual and triple incretin agonists, positioning GLP-1 monotherapy as a strong but lower-ceiling strategy and providing an important benchmark against which newer multi-receptor therapies are evaluated.
3.2. Tirzepatide
Tirzepatide, the first dual GIP/GLP-1 receptor agonist, has redefined the upper limits of glucose lowering effects achievable with incretin-based therapies. Its clinical efficacy has been characterized through the SURPASS program, a global series of phase 3 randomized controlled trials evaluating tirzepatide across diverse treatment contexts including monotherapy, add-on therapy, and comparisons with basal and prandial insulin. Collectively, these trials provide the most comprehensive assessment to date of tirzepatide’s glycemic effects, consistently demonstrating reductions that exceed those achieved with traditional GLP-1 receptor agonists and insulin-based regimens.
Across SURPASS trials, tirzepatide consistently produces larger HbA1c reductions than GLP-1 RAs and insulin-based regimens, with mean decreases typically ranging from approximately 1.9% to 2.6% depending on dose and baseline glycemia [17,71,72,73]. A substantial proportion of patients achieve stringent glycemic targets, including HbA1c levels below 7.0% and, in some cases, near-normoglycemia [71]. These effects occur alongside clinically meaningful reductions in background insulin requirements, highlighting tirzepatide’s insulin-sparing potential [14,71].
The most direct comparative evidence comes from SURPASS-2, which demonstrated superior glycemic efficacy of tirzepatide relative to semaglutide 1.0 mg across all tested doses [74]. Mechanistic analyses suggest that this advantage is mediated by greater improvements in insulin sensitivity, enhanced glucose-dependent insulin secretion, and more pronounced suppression of inappropriate glucagon signaling [75]. These findings provide biological support for the clinical superiority observed with dual incretin agonism.
Subgroup analyses indicate that tirzepatide maintains strong glycemic efficacy across a broad range of patient phenotypes, including variation in body mass index, baseline HbA1c, and duration of diabetes. While lower baseline HbA1c and shorter disease duration are associated with higher likelihood of achieving near-normoglycemia, clinically meaningful HbA1c reductions occur across all examined subgroups [71,76]. These data support tirzepatide’s applicability across heterogeneous type 2 diabetes populations rather than confinement to narrowly defined responder profiles.
Overall, the SURPASS program establishes tirzepatide as a highly potent glucose-lowering incretin therapy, consistently achieving HbA1c reductions and normoglycemia rates that surpass those of GLP-1 RAs and insulin-based regimens. These findings highlight the therapeutic potential of multi-receptor agonism and provide a clear mechanistic and clinical rationale for expanding beyond dual agonists. Building on tirzepatide’s success, emerging triple agonists targeting GIP, GLP-1, and glucagon receptors simultaneously represent the next evolution in incretin therapy and are now being investigated for even greater metabolic efficacy.
3.3. Emerging Multi-Agonist Therapies
Emerging multi-agonist therapies represent the next stage in incretin pharmacology, aiming to extend glycemic efficacy beyond dual agonism by simultaneously targeting GLP-1, GIP, and glucagon receptors. Early-phase clinical trials suggest that these agents can achieve HbA1c reductions comparable to, and in some cases exceeding, those observed with existing dual incretin agonists, although long-term comparative data remain limited.
Retatrutide, a first-in-class triple GIP/GLP-1/glucagon receptor agonist, has demonstrated substantial glycemic efficacy in phase 2 studies of adults with type 2 diabetes. In dose-ranging trials, retatrutide produced dose-dependent HbA1c reductions approaching those observed with high-potency GLP-1 RAs and tirzepatide, with a meaningful proportion of participants achieving HbA1c targets below 7.0% and near-normoglycemia [77]. These glycemic improvements were accompanied by marked reductions in fasting glucose, supporting the metabolic impact of triple-receptor engagement [77]. While higher doses produced numerically greater effects, differences between upper dose tiers were modest, suggesting a potential plateau in glycemic response at higher exposure levels. Ongoing phase 3 trials under the TRIUMPH program will be critical to defining long-term efficacy, durability, and safety [78,79].
Survodutide, a dual GLP-1/glucagon receptor agonist, has also shown promising glycemic effects in phase 2 trials. Across dose-escalation studies, survodutide produced clinically meaningful HbA1c reductions that were broadly comparable to those achieved with semaglutide 1.0 mg, with evidence of dose-responsive improvement at higher weekly exposure levels [80,81]. Beyond glycemic lowering, survodutide has demonstrated favorable effects on fasting glucose and circulating glucagon levels, although the clinical implications of these mechanistic findings remain to be clarified [81]. Ongoing phase 3 trials (SYNCHRONIZE-1 and -2) will further characterize its role in metabolic disease [28].
Taken together, available evidence indicates that emerging triple- and glucagon-containing incretin agonists can achieve glycemic efficacy comparable to leading dual-agonist therapies, potentially extending the therapeutic ceiling of incretin-based treatment. However, current data remain largely confined to early-phase studies, and direct head-to-head comparisons with tirzepatide and high-dose GLP-1 receptor agonists are lacking. As larger and longer-term trials mature, future research will need to clarify whether incremental glycemic benefits translate into meaningful advantages in cardiovascular, renal, and long-term metabolic outcomes. Table 2 summarizes the HbA1c-lowering efficacy across the spectrum of incretin-based therapies, illustrating the progressive enhancement in glycemic control from short-acting GLP-1 receptor agonists through multi-agonist formulations. This compilation highlights the dose-dependent effects within each agent class and provides a framework for understanding the relative potency of current and emerging therapies.
Table 2.
HbA1c-Lowering Efficacy of GLP-1 Receptor Agonists and Multi-Agonist Therapies.
4. Clinical Efficacy: Weight Reduction
4.1. GLP-1 Receptor Agonists
GLP-1 receptor agonists were the first incretin-based therapies shown to produce clinically meaningful weight loss, and they remain foundational agents in obesity pharmacotherapy. Among single-receptor GLP-1 RAs, semaglutide 2.4 mg consistently demonstrates the greatest weight-reducing efficacy, achieving mean weight losses of approximately 13.9–16.0% across the STEP trial program, substantially exceeding placebo and outperforming liraglutide 3.0 mg in direct comparisons [12,82,83,84,85,86,87,88,89,90]. Liraglutide produces more modest but clinically meaningful weight loss, typically in the range of 5.8–8.0% across the SCALE trials, and remains an effective option when semaglutide is unavailable or not tolerated [87,91,92]. Short-acting GLP-1 Ras and lower-dose diabetes formulations achieve smaller and less durable weight reductions [93,94].
Across agents, weight loss with GLP-1 RAs is dose-dependent and most pronounced during the first 6–12 months of therapy, with a tendency to plateau over longer treatment durations [95,96,97]. Continued therapy is generally required to maintain benefit, as discontinuation is associated with substantial weight regain and reversal of cardiometabolic improvements, as demonstrated in STEP 4 and other long-term studies [83,95]. These findings underscore obesity’s chronic, relapsing nature and position GLP-1 monotherapy as an effective but lower-ceiling strategy relative to emerging dual and triple incretin agonists.
4.2. Tirzepatide
Tirzepatide produces greater and more consistent weight loss than GLP-1 monotherapy across randomized trials and real-world studies. In the SURMOUNT program, tirzepatide achieved mean weight reductions of approximately 15.0–20.9% at 72 weeks, exceeding outcomes observed with semaglutide 2.4 mg and other single-receptor agents [13,16,83]. Direct head-to-head evidence from SURMOUNT-5 confirmed superior weight-loss efficacy compared with semaglutide, with a substantially higher proportion of participants achieving ≥20% and ≥25% weight loss [13]. Real-world analyses similarly demonstrate greater absolute and percentage weight reductions with tirzepatide relative to GLP-1 RAs [98].
Body composition studies indicate that the majority of tirzepatide-associated weight loss reflects reductions in fat mass, although lean mass loss remains clinically relevant and warrants consideration in vulnerable populations [99]. Despite superior efficacy, weight regain is common following treatment discontinuation, as demonstrated in SURMOUNT-4, reinforcing the need for sustained therapy and long-term patient engagement [100,101]. Collectively, these findings position tirzepatide as the most effective currently approved single-agent pharmacotherapy for weight reduction and establish a benchmark for next-generation multi-agonist therapies.
4.3. Emerging Multi-Agonist Therapies
Next-generation incretin therapies incorporating glucagon receptor agonism aim to extend weight-loss efficacy beyond that achieved with dual incretin agonists. Retatrutide, a triple GLP-1/GIP/glucagon receptor agonist, has demonstrated very large weight reductions in phase 2 trials, with dose-dependent mean losses ranging from approximately 17% to 24% at 48 weeks in adults with obesity [18,77]. Early phase 3 findings from the TRIUMPH program suggest that retatrutide may achieve even greater weight loss over longer treatment durations, although peer-reviewed long-term data remain pending [31]. These results establish retatrutide as a potent addition to the arsenal of available pharmacologic obesity treatments, pending full publication of late-phase outcomes.
Body composition analyses suggest that retatrutide produces substantial reductions in fat mass but is also associated with lean mass loss proportional to total weight reduction, similar to patterns observed with semaglutide and tirzepatide [102]. While the magnitude of weight loss achieved with triple agonism is unprecedented in pharmacotherapy, the long-term clinical implications such as effects on musculoskeletal health, nutritional status, and durability of benefit remain incompletely understood.
Survodutide, a dual GLP-1/glucagon receptor agonist, has also demonstrated clinically meaningful weight loss in phase 2 obesity and type 2 diabetes trials, exceeding outcomes observed with selective GLP-1 RAs but appearing less potent than tirzepatide in indirect comparisons [80,103,104]. As with other emerging multi-agonists, ongoing phase 3 trials will be essential to determine long-term efficacy, tolerability, and optimal positioning in obesity treatment algorithms [28].
This collection of early clinical evidence indicates that dual and triple incretin agonists extend the weight-loss ceiling beyond what is achievable with GLP-1 monotherapy. However, greater efficacy is accompanied by unresolved questions regarding long-term safety, tolerability, lean mass preservation, and durability following treatment discontinuation. As outcome data mature, future work will need to clarify how incremental weight-loss benefits translate into sustained cardiometabolic and functional improvements.
5. Cardiovascular Outcomes
5.1. GLP-1 Receptor Agonists and Major Adverse Cardiovascular Events
Cardiovascular disease remains the dominant driver of morbidity and mortality in both type 2 T2DM and obesity, accounting for approximately 50% of deaths in this population [105] and underscoring the central importance of cardiovascular outcomes when evaluating incretin-based therapies. Over the past decade, GLP-1 receptor agonists (GLP-1 RAs) have consistently demonstrated clinically meaningful reductions in MAC) [106], with emerging dual and triple agonists prompting investigation into whether greater metabolic effects translate into additional cardiometabolic benefit [107].
Across multiple placebo-controlled CVOTs, GLP-1 RAs have reduced the risk of 3-point MACE, typically defined as cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke. Trials including LEADER (liraglutide), SUSTAIN-6 (semaglutide), HARMONY OUTCOMES (albiglutide), REWIND (dulaglutide), and AMPLITUDE-O (efpeglenatide) demonstrated statistically significant MACE reduction, whereas ELIXA (lixisenatide), EXSCEL (exenatide once weekly), and PIONEER-6 (oral semaglutide) established cardiovascular safety without superiority [105].
In LEADER, once-daily liraglutide reduced 3-point MACE by 13% compared with placebo (HR 0.87, 95% CI 0.78–0.97), driven primarily by reductions in cardiovascular and all-cause mortality, including an approximately 22% relative risk reduction in cardiovascular death (HR 0.78, 95% CI 0.66–0.93; p = 0.007) [9]. Similarly, SUSTAIN-6 demonstrated that injectable semaglutide was associated with a 26% reduction in MACE (HR 0.74, 95% CI 0.58–0.95), with a particularly strong effect on nonfatal stroke [8]. In HARMONY Outcomes, once-weekly albiglutide was superior to placebo, reducing MACE by 22% (HR 0.78, 95% CI 0.68–0.90) in patients with established cardiovascular disease [108]. The cardioprotective benefit extended to broader and lower-risk populations in REWIND, which showed a 12% MACE reduction with dulaglutide (HR 0.88, 95% CI 0.79–0.99) among patients largely without established cardiovascular disease [10,105].
At the class level, a large meta-analysis of randomized placebo-controlled trials involving over 80,000 participants confirmed that GLP-1 RAs significantly reduced MACE (OR 0.86, 95% CI 0.80–0.94), cardiovascular mortality (OR 0.87, 95% CI 0.81–0.94), and all-cause mortality (OR 0.87, 95% CI 0.82–0.93) across subgroups defined by sex, baseline BMI, renal function, and cardiovascular disease history [106]. Importantly, post hoc analyses of LEADER and SUSTAIN-6 demonstrated that cardiovascular benefit was not proportional to the degree of weight loss, suggesting that GLP-1-mediated cardioprotection is not solely explained by weight reduction [109].
5.2. Dual Incretin Agonism: Tirzepatide and Cardiovascular Outcomes
Tirzepatide produces substantially greater reductions in HbA1c and body weight than GLP-1 RA monotherapy; however, whether these metabolic advantages translate into superior cardiovascular outcomes has been a critical question. The first active-comparator incretin CVOT, SURPASS-CVOT, demonstrated that tirzepatide was non-inferior to dulaglutide for time to first 3-point MACE (HR 0.92, 95.3% CI 0.83–1.00; p = 0.086), despite greater weight loss and glycemic improvement—consistent with the possibility that GLP-1-mediated pathways contribute substantially to the observed cardiovascular benefit [109,110]. Importantly, SURPASS-CVOT was designed to assess non-inferiority rather than superiority, and a statistically significant reduction in cardiovascular events beyond GLP-1 receptor agonist therapy was not demonstrated.
Emerging real-world evidence has raised the possibility of additional cardiovascular benefit with tirzepatide in selected high-risk populations. In a large retrospective observational study using the TriNetX network, tirzepatide use was associated with a lower risk of a composite endpoint of myocardial infarction, ischemic stroke, and all-cause mortality compared with GLP-1 receptor agonists (HR 0.60, 95% CI 0.43–0.84) among adults with T2DM, overweight or obesity, and established ischemic heart disease [111]. In a separate TriNetX observational cohort, tirzepatide was associated with lower hazards of all-cause mortality (AHR 0.58, 95% CI 0.45–0.75), MACE (AHR 0.80, 95% CI 0.71–0.91), and major adverse kidney events (AHR 0.54, 95% CI 0.44–0.67) compared with GLP-1 RAs [112]. These findings are hypothesis-generating and subject to residual confounding and do not establish causal superiority over GLP-1 receptor agonist therapy. Nevertheless, they suggest that dual GIP/GLP-1 receptor agonism may confer incremental benefit in carefully selected populations.
5.3. Obesity-Specific Cardioprotection and Heart Failure Outcomes
The SELECT trial provided definitive evidence that GLP-1 RA therapy reduces cardiovascular events in individuals with overweight or obesity without T2DM. In patients with established cardiovascular disease, semaglutide 2.4 mg reduced MACE by 20% compared with placebo (HR 0.80, 95% CI 0.72–0.90). Notably, cardiovascular risk reduction occurred early and was observed even among participants who did not achieve substantial weight loss, further reinforcing the concept that cardioprotective effects are not solely mediated by weight reduction [109,113].
Beyond atherosclerotic outcomes, incretin-based therapies have demonstrated clinically meaningful benefits in obesity-related heart failure with preserved ejection fraction (HFpEF). Randomized trials of semaglutide (STEP-HFpEF) and tirzepatide (SUMMIT) have shown improvements in health status and functional capacity, as well as reductions in worsening heart failure events; in SUMMIT, tirzepatide reduced the hierarchical composite of cardiovascular death or worsening heart failure (HR 0.62, 95% CI 0.41–0.95). These benefits were accompanied by reductions in left ventricular mass and paracardiac adiposity on cardiac imaging, as well as decreases in systemic inflammatory markers such as high-sensitivity C-reactive protein, findings observed in both patients with and without T2DM and therefore not solely attributable to glycemic improvement [114,115].
5.4. Future Trials
Despite consistent class-level benefit, heterogeneity exists across GLP-1 RA CVOTs. Meta-regression analyses suggest that higher baseline BMI and older age are associated with greater relative cardiovascular risk reduction, whereas baseline glycemic parameters are less predictive of benefit. These findings support phenotype-guided patient selection rather than exclusive reliance on glycemic thresholds when considering incretin-based therapies [105].
An overview of GLP-1RA CVOTs is summarized in Table 3.
Table 3.
Major Cardiovascular Outcomes Trials of GLP-1-Based Therapies.
6. Renal Outcomes
6.1. GLP-1 Receptor Agonists and Renal Outcomes
Chronic kidney disease (CKD) is a common and clinically consequential complication of T2DM and obesity [122], affecting an estimated ~40–50% of individuals with T2DM globally, and is strongly associated with excess cardiovascular morbidity, heart failure, and mortality. Evidence from cardiovascular outcomes trials indicates that GLP-1 RAs are associated with kidney benefits, most consistently reflected by reductions in new-onset macroalbuminuria and attenuation of eGFR decline [8,116,123].
In LEADER, liraglutide reduced the risk of a prespecified composite renal outcome by 22% compared with placebo, an effect driven primarily by a lower incidence of new-onset persistent macroalbuminuria [9,116]. Similarly, in SUSTAIN-6, semaglutide reduced new or worsening nephropathy, largely attributable to reduced albuminuria progression [8]. Renal benefit extended to broader and lower-risk populations in REWIND, in which dulaglutide reduced a composite kidney outcome—including sustained eGFR decline, end-stage kidney disease, or renal death—in post hoc analyses, with consistent effects across baseline eGFR and albuminuria strata [10,123].
The FLOW trial represents the first dedicated kidney outcomes trial of a GLP-1 RA. Among patients with T2DM and established CKD, once-weekly semaglutide reduced the risk of major kidney disease events, including kidney failure, sustained ≥50% eGFR decline, or death from kidney-related or cardiovascular causes by 24% compared with placebo (HR 0.76, 95% CI 0.66–0.88). Semaglutide also slowed the annual rate of eGFR decline relative to placebo, with benefits observed across baseline eGFR categories and among patients receiving background renin–angiotensin system blockade [124]. Collectively, secondary renal findings from cardiovascular outcomes trials and results from the FLOW trial support a kidney-protective role for GLP-1 receptor agonists in patients with T2DM and chronic kidney disease.
6.2. Dual/Triple Agonists
At present, renal outcomes data for dual and triple incretin agonists remain less mature than for GLP-1 receptor agonists, with most available evidence derived primarily from post hoc analyses and biomarker-based endpoints rather than dedicated kidney outcomes trials. Post hoc analyses of the SURPASS-4 trial, which enrolled patients with T2DM and high cardiovascular risk, demonstrated that tirzepatide was associated with favorable kidney effects compared with insulin glargine. Specifically, tirzepatide slowed the annual rate of eGFR decline and reduced progression of albuminuria, resulting in a lower risk of a composite kidney outcome driven primarily by new-onset macroalbuminuria [125]. To address potential confounding from weight-related changes in muscle mass and creatinine generation, a complementary post hoc analysis of SURPASS-4 evaluated kidney function using cystatin C-based eGFR. In this analysis, tirzepatide was again associated with a slower decline in kidney function compared with insulin glargine, supporting the presence of a kidney effect not solely attributable to changes in body composition [126].
Additional insight is provided by analyses of the SUMMIT trial in patients with obesity-related heart failure with preserved ejection fraction. Tirzepatide was associated with favorable changes in kidney-related biomarkers over longer-term follow-up, including reductions in urinary albumin-to-creatinine ratio and improvements in cystatin C-based eGFR, with consistent effects across baseline eGFR categories. Urinary albumin-to-creatinine ratio is a validated marker of kidney damage and cardiovascular risk; however, the clinical implications of these biomarker changes for long-term kidney outcomes remain to be determined. Notably, the presence of chronic kidney disease (CKD) did not attenuate cardiovascular or functional benefits, and absolute reductions in heart-failure events were numerically greater among participants with CKD [127]. Taken together, post hoc renal analyses from SURPASS-4 and secondary analyses from SUMMIT demonstrate consistent reductions in urinary albumin-to-creatinine ratio across distinct high-risk populations, supporting a reproducible effect of tirzepatide on renal risk markers, although the implications for long-term sustained reductions in kidney failure, eGFR decline or renal mortality remain to be determined.
Available data for triple incretin agonists targeting GLP-1, GIP, and glucagon receptors are limited to early-phase metabolic and weight-loss studies, and renal outcomes have not yet been systematically evaluated [107].
6.3. Clinical Implications for CKD
Evidence from cardiovascular outcomes trials and dedicated kidney outcomes studies supports the role of GLP-1 receptor agonists as adjunctive therapies in patients with T2DM and CKD, particularly among those with coexisting obesity or cardiovascular disease. Across trials, GLP-1 RA therapy has been associated with reductions in albuminuria progression, attenuation of eGFR decline, and lower risk of clinically important kidney outcomes [8,9,116,123,124].
GLP-1 RAs provide complementary kidney and cardiovascular protection alongside established therapies such as renin–angiotensin system inhibitors and sodium–glucose cotransporter-2 inhibitors [105,128]. In patients unable to tolerate SGLT2 inhibitors, GLP-1 RAs represent a reasonable alternative with demonstrated cardiorenal benefit [109]. As data from FLOW and ongoing studies continue to mature, incretin-based therapies may likely play an expanding role in integrated cardiovascular–kidney–metabolic risk management strategies.
7. Safety and Tolerability
7.1. Gastrointestinal Adverse Events
Gastrointestinal adverse events represent the most common tolerability limitation of incretin-based therapies, affecting 40–70% of patients to varying degrees but prompting discontinuation in fewer than 10% of clinical trial participants [95,129]. Nausea occurs in 28–44% of patients, diarrhea in 19–30%, vomiting in 8–24%, and constipation in 11–24% across the class [95,130]. These events are characteristically mild to moderate in severity, emerge primarily during dose escalation, and typically diminish with continued therapy at stable doses [95].
The temporal pattern of gastrointestinal symptoms follows a predictable course. Nausea typically manifests within 48 h of drug initiation or dose escalation, often presenting in the morning or after prolonged fasting periods [95]. The mechanism appears related to GLP-1 pharmacokinetics, with symptom resolution occurring once steady-state drug levels are achieved [131]. Most gastrointestinal adverse events occur within the first month of treatment initiation, with frequency and severity decreasing substantially as patients acclimate to therapy [132]. Shorter-acting formulations—exenatide twice daily more than liraglutide daily—produce nausea more frequently than longer-acting weekly preparations such as exenatide extended-release, dulaglutide, or semaglutide, likely because steady-state levels minimize the fluctuations that trigger symptoms [131].
Comparative analyses reveal variation in gastrointestinal tolerability across agents. In head-to-head comparison between tirzepatide and semaglutide for obesity (SURMOUNT-COMPARE), nausea and constipation occurred at similar rates (44% vs. 44% for nausea; 27% vs. 29% for constipation), while vomiting was less frequent with tirzepatide (15% vs. 21%) [13]. Diarrhea rates were nearly identical (24% in both groups) [13]. Critically, gastrointestinal adverse events led to treatment discontinuation more frequently with semaglutide (5.6%) than tirzepatide (2.7%) despite similar symptom incidence [13]. Network meta-analyses suggest that tirzepatide demonstrates the highest risk for nausea and diarrhea in patients with T2DM, while dulaglutide and lixisenatide show lower risks [133]. Among non-diabetic patients with obesity, orforglipron shows the highest nausea risk, followed by exenatide, tirzepatide, semaglutide, and liraglutide, with liraglutide associated with the highest risk for diarrhea, dyspepsia, and eructation [130,134].
Mitigation strategies center on gradual dose titration and dietary modifications. Starting at the lowest available dose—semaglutide 0.25 mg weekly or tirzepatide 2.5 mg weekly—with monthly escalation guided by individual tolerance rather than fixed protocols represents the cornerstone approach [95,135,136]. Extending titration schedules further improves tolerability; a randomized trial demonstrated that prolonging semaglutide titration from 8 to 16 weeks with smaller incremental increases reduced gastrointestinal-related withdrawal from 19% to 2% while maintaining equivalent efficacy [137]. During dose escalation in clinical trials, patients were permitted to remain at a given dose for up to 8 weeks to allow symptoms to dissipate before advancing [95].
Dietary counseling provides first-line symptom management. Patients should consume smaller, more frequent meals; eat slowly; and avoid high-fat, high-sugar foods during the first days after initiation or dose increases [89,95,129,136,138]. Adequate hydration is essential to prevent dehydration-related complications including acute kidney injury and cardiac palpitations [95,136]. Limiting alcohol and carbonated beverages can reduce nausea and gastroesophageal reflux [95]. For persistent nausea, ginger or peppermint tea, acupressure bands, and short-term antiemetics (prochlorperazine preferred over ondansetron to avoid worsening constipation) provide relief [95,135]. Constipation management includes gradual increases in soluble and insoluble fiber, magnesium citrate supplementation, polyethylene glycol, or bulk-forming laxatives [95,135]. For diarrhea, fiber supplements for stool bulk and anti-diarrheal medications offer acute relief [95]. Dyspepsia may respond to H2 blockers or proton pump inhibitors [135].
Proactive patient education before initiating therapy is critical. The American Association of Clinical Endocrinology and The Obesity Society recommend detailed counseling on expected side effects, early contact protocols if symptoms develop, and specific mitigation strategies [95,136]. Patients must understand that if treatment is paused, they must restart at the lowest dose and re-titrate to allow physiologic acclimation [129,136]. GLP-1 receptor agonists should be used cautiously or avoided in individuals with unexplained weight loss, undernutrition, significant or symptomatic gastroparesis, recurrent bowel obstruction, or problematic constipation. Moreover, GLP-1 receptor agonists are contraindicated in individuals with a personal or family history of medullary thyroid carcinoma or multiple endocrine neoplasia type 2 and should be discontinued in the setting of severe hypersensitivity reactions. Caution is also warranted in patients with severe gastroesophageal reflux disease, a history of intestinal pseudo-obstruction, or conditions predisposing to dehydration, as persistent nausea, vomiting, or diarrhea may precipitate acute kidney injury. These considerations are especially important in older adults, who may be more vulnerable to frailty, sarcopenia, and adverse consequences of further weight loss [139]. In older adults, careful dose titration, close monitoring of nutritional intake, and concurrent resistance exercise and adequate protein intake are recommended to mitigate risks of sarcopenia, functional decline, and falls.
7.2. Serious Adverse Events
7.2.1. Pancreatitis
Large-scale evidence indicates that GLP-1 receptor agonists and tirzepatide are not associated with increased risk of acute pancreatitis. A 2025 study comparing GLP-1 RAs to SGLT2 inhibitors in over 1.2 million patients with T2DM found similar pancreatitis risk (HR 1.01; 95% CI 0.90–1.13) [140]. A multicenter analysis of 740,370 patients demonstrated a trend toward lower risk of uncomplicated pancreatitis with GLP-1 RAs (HR 0.71; 95% CI 0.49–1.01), though this did not reach statistical significance [141]. Meta-analyses of randomized controlled trials confirm no significant increase in pancreatitis risk with GLP-1 RAs compared to placebo or active comparators [142].
For tirzepatide specifically, pancreatitis rates in clinical trials were low and comparable to placebo. In SURMOUNT-1, adjudication-confirmed pancreatitis occurred in 0.2% of participants across all tirzepatide doses and 0.2% in the placebo group [16]. The SURMOUNT-MMO safety extension reported three cases across tirzepatide groups versus one in placebo group among 1032 participants [143]. A comprehensive meta-analysis found no increased pancreatitis risk with tirzepatide compared to controls [144].
Emerging evidence suggests GLP-1 RAs may actually provide protective effects against recurrent pancreatitis in patients with prior episodes, with tirzepatide showing particularly favorable outcomes [145]. The American Association of Clinical Endocrinology recommends considering alternative therapies in patients with active or very recent pancreatitis, though history of remote pancreatitis does not constitute an absolute contraindication [146].
7.2.2. Gallbladder and Biliary Disease
GLP-1 receptor agonists and tirzepatide are associated with a modestly increased risk of gallbladder and biliary disease. A meta-analysis of 76 randomized controlled trials involving over 100,000 patients found that GLP-1 RAs increased the risk of cholelithiasis by 27% (RR 1.27; 95% CI 1.10–1.47), cholecystitis by 36% (RR 1.36; 95% CI 1.14–1.62), and overall biliary disease by 55% (RR 1.55; 95% CI 1.08–2.22), translating to an additional 27 events per 10,000 patients per year [147]. The risk of cholecystectomy was similarly elevated (RR 1.70; 95% CI 1.25–2.32) [147].
The risk demonstrates clear dose and duration dependence. Higher doses of GLP-1 RAs showed significantly increased risk (RR 1.56; 95% CI 1.36–1.78), while lower doses did not (RR 0.99; 95% CI 0.74–1.33) [147]. Treatment duration exceeding 26 weeks was associated with increased risk (RR 1.40; 95% CI 1.26–1.56), whereas shorter duration was not [147]. The risk was particularly pronounced in weight-loss trials compared to T2DM trials [147,148]. Real-world data comparing GLP-1 RAs to SGLT2 inhibitors showed a modest increase in biliary disease risk (HR 1.15; 95% CI 1.05–1.26), equivalent to fewer than one additional event per 1000 person-years [140].
For tirzepatide, SURMOUNT-MMO reported cholelithiasis in 2.1–3.6% of tirzepatide-treated participants versus 1.9% with placebo, and cholecystitis in 0.4–1.1% versus 0.4% [143]. Meta-analysis found tirzepatide associated with increased composite gallbladder/biliary disease compared to placebo or basal insulin (RR 1.97; 95% CI 1.14–3.42) [144]. Proposed mechanisms include rapid weight loss, reduced gallbladder motility, and alterations in bile composition [148,149].
7.2.3. Diabetic Retinopathy
The relationship between GLP-1 receptor agonists and diabetic retinopathy is nuanced, with the overall class not associated with increased risk except in specific high-risk populations. A comprehensive meta-analysis of 78 randomized controlled trials involving 73,640 participants found that semaglutide was not associated with increased risk of diabetic retinopathy overall (OR 1.04; 95% CI 0.92–1.17), with trial sequential analysis confirming sufficient sample size to exclude a clinically meaningful 20% increase in risk [150]. Real-world analyses found that GLP-1 RAs were associated with lower risk of sight-threatening complications including proliferative diabetic retinopathy, diabetic macular edema, and neovascular glaucoma, despite a marginally increased incidence of any diabetic retinopathy [151].
However, patients with preexisting diabetic retinopathy and poor glycemic control may experience transient worsening. The SUSTAIN-6 trial demonstrated increased rates of vitreous hemorrhage, blindness, or need for intravitreal injection/photocoagulation with semaglutide (HR 1.76; 95% CI 1.11–2.78), primarily observed in patients with established retinopathy and long-standing poor glycemic control at baseline [150,152,153]. This phenomenon likely relates to rapid glycemic improvement in patients with prolonged hyperglycemia, similar to effects observed with intensive insulin therapy [150]. The American Diabetes Association (ADA) recommends assessing retinopathy status when intensifying glucose-lowering therapies with GLP-1 RAs, as rapid HbA1c reductions can be associated with initial worsening of retinopathy [154].
7.2.4. Emerging Multi-Agonist Therapies
Serious adverse event rates with retatrutide and survodutide are low and comparable to placebo, occurring in approximately 4–8% of participants across clinical trials, with no deaths attributed to either medication in major phase 2 trials [18,77,103,155]. For retatrutide, serious adverse events occurred in 5% of participants in the T2DM trial (compared to 7% with placebo and 2% with dulaglutide) and 4% in the obesity trial (versus 4% with placebo) [18,77]. Three serious adverse events were attributed to retatrutide: one case each of cholecystitis, acute pancreatitis occurring seven days after initial dosing, and diabetic/starvation ketoacidosis [77].
For survodutide, serious adverse events occurred in 8% of participants in the MASH trial (versus 7% with placebo) and 4% in the obesity trial (compared to 7% with placebo) [103,155]. Only one serious adverse event across all survodutide trials was considered drug-related [155]. There were no deaths, life-threatening events, or cases of pancreatitis or hepatic injury in the survodutide trials [103]. Treatment discontinuation due to adverse events was notably higher—8% with retatrutide and 20–25% with survodutide—primarily driven by gastrointestinal events during dose escalation, though most were mild to moderate in severity [77,103,155].
7.3. Discontinuation Rates: Clinical Trials Versus Real-World Practice
A substantial efficacy-effectiveness gap exists between clinical trial and real-world discontinuation patterns. In clinical trials, discontinuation rates due to adverse events range from 6–8% for both semaglutide and tirzepatide [135]. The head-to-head SURMOUNT-COMPARE trial reported that 6.1% of participants discontinued tirzepatide and 8.0% discontinued semaglutide due to adverse events, with gastrointestinal events accounting for 2.7% of tirzepatide discontinuations versus 5.6% for semaglutide [13]. In STEP 1, 7.0% of participants discontinued semaglutide 2.4 mg due to adverse events compared to 3.1% in the placebo group [12]. The SURPASS-2 trial comparing tirzepatide to semaglutide 1 mg in T2DM showed discontinuation rates of 6.0–8.5% across tirzepatide doses compared to 4.1% with semaglutide [74].
In contrast, real-world discontinuation rates are substantially higher, with approximately 46–65% of patients discontinuing within the first year—though these figures reflect all reasons for stopping, not just adverse events [156,157]. A large US cohort of 125,474 adults with overweight or obesity found one-year discontinuation rates of 64.8% for patients without T2DM and 46.5% for those with T2DM. A Swedish nationwide registry study demonstrated cumulative discontinuation rates of 23.6% at 1 year and 38.5% at 3 years for GLP-1 receptor agonists [157].
Critically, more than half of patients who discontinue subsequently reinitiate treatment. The cumulative incidence of reinitiation was 41.1% at 1 year and 57.4% at 3 years after discontinuation, resulting in approximately 70–80% of patients having ongoing treatment when accounting for reinitiation patterns [157]. An academic obesity clinic reported median persistence of 10.7 months, with weight loss approximating clinical trial results among those who remained on treatment [158].
Factors associated with higher real-world discontinuation include younger age, absence of T2DM, lower socioeconomic status, higher comorbidity burden, and gastrointestinal adverse events [156,157,159]. High out-of-pocket costs and insurance instability remain among the most prominent structural barriers to persistence, particularly among individuals in lower-income areas or those with high-deductible plans [156]. Geographic and socioeconomic disparities further influence access, with markedly lower uptake in communities with limited insurance coverage, higher social vulnerability indices, and fewer obesity-specialty prescribers [156].
7.4. Special Populations
Elderly Patients
GLP-1 receptor agonists demonstrate a favorable safety profile in elderly patients, with cardiovascular and renal benefits comparable to younger populations, though specific considerations warrant attention. The most common adverse effects are gastrointestinal symptoms—nausea (25–60%), vomiting (5–15%), diarrhea, and constipation—which typically emerge during dose escalation and diminish over time [160,161]. The ADA recommends slow titration and advises particular caution when using GLP-1 receptor agonists in older adults with unexplained weight loss, undernutrition, or recurrent gastrointestinal problems, particularly those with problematic constipation, significant gastroparesis, or bowel obstruction [139]. Importantly, these clinical red flags warrant caution in younger adults as well.
Hypoglycemia risk is minimal with GLP-1 RAs alone but increases when combined with sulfonylureas or insulin [160]. Treatment deintensification of these agents or diuretics is recommended in older and frail individuals (approximately ≥65 years) to avoid hypoglycemia and hypovolemia [160]. Additional safety considerations include increased risk of gallbladder disease and, for semaglutide specifically, potential worsening of pre-existing diabetic retinopathy related to rapid glycemic improvement [94,138,160,161]. Injectable formulations require adequate visual, motor, and cognitive skills for administration, though weekly dosing schedules may facilitate adherence [139]. Patients should be monitored regularly for excessive weight loss [139].
In patients aged ≥ 80 years with T2DM, GLP-1 RAs demonstrated significant safety and efficacy, with lower risks of major adverse cardiovascular events (HR 0.86), major adverse kidney events (HR 0.86), all-cause hospitalization (HR 0.91), and all-cause mortality (HR 0.82) compared to DPP-4 inhibitors, with no significant differences in heart failure or bone fractures [162]. Meta-analyses confirm that cardiovascular and kidney benefits are consistent between older (≥65 years) and younger adults, with no evidence of age-related heterogeneity in treatment effects [139,163].
7.5. Chronic Kidney Disease
GLP-1 receptor agonists do not require dose adjustment based on renal function and can be used across the full spectrum of CKD, including in patients with eGFR as low as 15 mL/min/1.73 m2 and those on dialysis [164]. This distinguishes them from SGLT2 inhibitors, whose glucose-lowering effects decline when eGFR falls below 45 mL/min/1.73 m2 [165].
Agent-specific considerations exist. Exenatide should not be used when eGFR is below 30 mL/min/1.73 m2 due to decreased clearance and reported cases of acute renal failure [166]. Lixisenatide is contraindicated when eGFR falls below 15 mL/min/1.73 m2 [166]. In contrast, liraglutide, semaglutide (both injectable and oral), and dulaglutide require no dosage adjustments as renal function declines [166]. KDIGO recommends prioritizing liraglutide, injectable semaglutide, and dulaglutide over exenatide and lixisenatide, as the latter two did not demonstrate cardiovascular benefit in their outcome trials [167].
Recent trial data have strengthened the evidence base for GLP-1 RAs in advanced CKD. A dedicated glycemic control trial in patients with moderate-to-severe CKD (eGFR as low as 15 mL/min/1.73 m2) demonstrated that dulaglutide resulted in significantly slower eGFR decline compared with insulin glargine [168]. The FLOW trial of subcutaneous semaglutide 1 mg weekly in patients with T2DM and CKD was stopped early for clear positive efficacy on primary kidney disease outcomes [168].
Gastrointestinal side effects warrant particular attention in the CKD population. Nausea, vomiting, and diarrhea occur in 15–20% of patients with moderate-to-severe CKD but are usually tolerable with dose titration and abate over several weeks to months [164]. Caution is warranted among patients with or at risk for malnutrition due to weight loss effects [164]. No dose reduction is needed when combining GLP-1 RAs with insulin in moderate-to-severe CKD; in fact, hypoglycemia rates are reduced by one-half even with concurrent insulin therapy, though doses of insulin or insulin secretagogues may need reduction to avoid hypoglycemia when initiating GLP-1 RA therapy [164].
7.6. Pregnancy
GLP-1 receptor agonists are contraindicated in pregnancy based on animal reproductive toxicity studies and lack of adequate human safety data. Manufacturer recommend discontinuing certain long-acting GLP-1 receptor agonists-such as semaglutide and tirzepatide-at least Two months before planned conception to allow for drug washout, while all agents should be stopped prior to pregnancy [160,169]. Animal studies showed early pregnancy losses, fetal abnormalities during organogenesis, reduced fetal weight, delayed ossification, and skeletal variants—typically associated with marked maternal weight loss [169]. The Endocrine Society recommends discontinuing GLP-1 RAs before pregnancy in individuals with preexisting T2DM, emphasizing the need for effective contraception in women of childbearing age not planning pregnancy and proactive transition to appropriate glycemic management (typically insulin) upon discontinuation [169].
Human data provides initial reassurance but remains limited. A large multinational cohort study of over 50,000 pregnancies found no elevated risk of major congenital malformations with periconceptional GLP-1 RA exposure compared to insulin [170]. However, discontinuation proximal to pregnancy carries significant risks: a recent cohort study found that GLP-1 RA use with prepregnancy or early pregnancy discontinuation was associated with 3.3 kg greater gestational weight gain, higher rates of excess gestational weight gain (65% vs. 49%), and increased risk of preterm delivery, gestational diabetes, and hypertensive disorders of pregnancy [171].
Critical safety considerations include that approximately 40% of pregnancies in the United States remain unintended, making effective contraception essential for women on GLP-1 RAs [169]. GLP-1 RAs may affect oral contraceptive efficacy [169]. Abrupt discontinuation without concurrent insulin initiation risks uncontrolled hyperglycemia during organogenesis, which itself is teratogenic [172]. Coordination of care is essential: effective preconception counseling, immediate glucose monitoring upon discontinuation, and transition to insulin therapy are necessary to balance the risks of medication exposure against the well-established teratogenic effects of hyperglycemia [169].
8. Patient Selection and Personalization
8.1. Clinical Phenotyping for Optimal Drug Selection
Patient selection for incretin-based therapies has evolved from a one-size-fits-all approach to increasingly nuanced phenotyping based on baseline characteristics, treatment goals, and patient preferences. While tirzepatide demonstrates superior efficacy across most populations compared to GLP-1 receptor agonists, specific clinical phenotypes predict differential treatment responses and help guide optimal agent selection.
8.1.1. Predictors of Response Across Incretin Therapies
For both GLP-1 receptor agonists and tirzepatide, younger age, female sex, higher baseline BMI, and lower baseline HbA1c consistently predict greater weight loss and glycemic improvement [95,97,173]. However, tirzepatide achieves larger absolute reductions in HbA1c and body weight across virtually all patient subgroups, with the magnitude of benefit influenced by specific baseline characteristics [57,174,175].
Among GLP-1 receptor agonists, higher baseline BMI, younger age, and female sex represent the strongest predictors of weight loss [95,97]. In univariate analyses with semaglutide, female sex was associated with 48% greater weight loss, younger age (<55 years) with 24% greater loss, and higher baseline BMI (≥40 vs. <30 kg/m2) with 23% greater loss [95]. Additional favorable predictors include higher baseline waist circumference, hepatic steatosis indices, fat mass, and absence of prior metformin use [176]. Notably, early weight loss at 6 months strongly predicts 12-month success, with 58% of early responders maintaining ≥5% weight reduction long-term [176]. Patients without T2DM demonstrate greater weight loss than those with T2DM across all GLP-1 receptor agonists [97].
For tirzepatide, similar but more pronounced predictor patterns emerge. Female sex, younger age, White or Asian race, and higher tirzepatide doses (10–15 mg) were associated with better outcomes [165]. In multivariate analyses, female sex conferred 2.4-fold higher odds of achieving ≥20% weight reduction, while lower baseline HbA1c (1.62 higher odds), absence of hypertension (1.35 higher odds), and lower ALT (1.17 higher odds) also predicted better response [95,173]. Metformin use at baseline has been associated with greater likelihood of achieving ≥15% weight reduction with tirzepatide in exploratory analyses, though causality has not been established. Lower baseline fasting glucose and non-HDL cholesterol were also associated with higher likelihood of achieving ≥15% weight loss [173]. Early glycemic response (≥20% fasting glucose reduction at week 4) or early weight response (≥5% reduction at week 8) strongly predict superior long-term metabolic outcomes, with early responders achieving HbA1c reductions of −2.6% versus −2.0% in non-early responders at 40 weeks [177].
8.1.2. Baseline Metabolic Parameters and Treatment Outcomes
Higher baseline HbA1c predicts greater absolute HbA1c reduction with incretin-based therapies. A systematic review found that 37 of 47 DPP-4 inhibitor studies and seven of nine GLP-1 receptor agonist studies reported greater HbA1c decreases in patients with higher baseline HbA1c [178]. Meta-analyses demonstrate a negative linear correlation between baseline HbA1c and change in HbA1c (r = −0.70; p < 0.001) when incretin therapies are added to metformin [179]. Paradoxically, for tirzepatide, lower baseline HbA1c predicts higher odds of achieving ≥20% weight reduction (1.62 higher odds), suggesting that patients with better baseline glycemic control may experience superior weight loss outcomes [95].
Higher baseline BMI strongly predicts greater absolute weight loss. In univariate analyses with semaglutide, patients with BMI ≥ 40 kg/m2 achieved 23% greater weight loss compared to those with BMI < 30 kg/m2 [95]. This relationship appears consistent across incretin therapies, with higher baseline BMI, waist circumference, and fat mass all associated with enhanced weight reduction. However, the percentage of body weight lost may be similar across BMI categories, meaning absolute kilogram loss increases proportionally with baseline weight.
While T2DM duration was not extensively detailed as a predictor, longer disease duration typically correlates with reduced beta-cell function, which may attenuate glycemic response to incretin therapies. Early initiation of incretin therapy in newly diagnosed T2DM provides more durable long-term benefits than sequential monotherapy [180]. Observational modeling from real-world cohorts suggests that modest reductions in BMI and HbA1c are associated with lower long-term cardiovascular and renal event risk, emphasizing the clinical importance of sustained metabolic improvement. In an electronic health record-derived analysis, each 1% reduction in BMI was associated with ~4% lower cardiovascular disease risk and ~3% lower risk of insulin initiation, while each 1% reduction in HbA1c was associated with ~4% lower CKD risk and ~16% lower risk of insulin initiation [181].
8.1.3. Agent Selection: Tirzepatide Versus GLP-1 Receptor Agonists
The American College of Cardiology (ACC) recommends semaglutide and tirzepatide as the obesity medications of choice among incretin therapies, with agent selection primarily dictated by insurance coverage, availability, and affordability rather than specific patient characteristics [182]. However, clinical trial and real-world data provide important guidance for phenotype-based selection.
Tirzepatide produces superior weight loss across all patient populations, with mean reductions of 15–20.9% of initial body weight at 72 weeks compared to 9.6–17.4% with semaglutide 2.4 mg at 68 weeks [83,98,174,183]. In head-to-head real-world comparisons, patients receiving tirzepatide were more likely to achieve ≥5% (91% vs. 82%), ≥10% (77% vs. 67%), and ≥15% (62% vs. 42%) weight loss at 12 months [98]. This superiority persists across subgroups with and without T2DM, though patients without T2DM achieve greater absolute weight loss than those with T2DM for both agents [98,184].
For glycemic control, tirzepatide achieves HbA1c reductions of −2.10% (15 mg dose) versus −1.59% with semaglutide 2.0 mg in network meta-analyses [174,183]. Among approved incretin therapies, tirzepatide has demonstrated the greatest glycemic and weight-loss efficacy to date in head-to-head and network meta-analyses; cardiovascular outcome advantages over high-dose semaglutide remain under evaluation [185].
The dual GIP/GLP-1 receptor agonism of tirzepatide may contribute to enhanced appetite suppression and improved tolerability compared with GLP-1 agonism alone, though the specific role of GIP in central appetite regulation remains under investigation [182]. Tirzepatide has been associated with greater modeled indices of β-cell responsiveness and insulin sensitivity compared with dulaglutide [186]. Both agents share similar gastrointestinal adverse event profiles, with tirzepatide 15 mg showing comparable tolerability to high-dose GLP-1 receptor agonists [82,94,184,187].
8.1.4. Practical Selection Criteria
Tirzepatide may be prioritized for patients with obesity (especially BMI ≥ 35–40), younger patients, those without established cardiovascular disease, and when primary therapeutic goals are maximal weight loss and glycemic improvement [95,96,176,182]. The greatest advantage is observed in patients with higher baseline BMI, female sex, younger age, and when higher doses (10–15 mg) can be tolerated [95,173,176]. Some analyses report numerically larger weight reductions in White and Asian participants treated with tirzepatide, but these findings require additional validation and may reflect population characteristics rather than biologic differences [165].
Semaglutide 2.4 mg may be preferred in patients with established atherosclerotic cardiovascular disease given demonstrated MACE reduction in SELECT, whereas full cardiovascular outcome data for tirzepatide in this population are still pending. Similarly, semaglutide is favored in patients with chronic kidney disease due to dedicated renal outcome data from FLOW, while renal evidence for tirzepatide is currently limited to post hoc analyses [188,189]. For patients with T2DM requiring both glycemic control and weight management, tirzepatide achieves superior outcomes in both domains [174,183].
Liraglutide 3.0 mg represents a less efficacious but established alternative, producing mean weight loss of 5.8–7% at 26–68 weeks [82,184]. It may be considered for patients who cannot tolerate weekly injections, prefer daily dosing, or have contraindications to higher-potency agents, though its cardiovascular benefit is established only at the 1.8 mg dose approved for T2DM [190].
Both tirzepatide and semaglutide are contraindicated in patients with personal or family history of medullary thyroid carcinoma, multiple endocrine neoplasia syndrome type 2, or known hypersensitivity [182]. Beyond these absolute contraindications, agent selection should prioritize efficacy goals, cardiovascular risk profile, tolerability, practical considerations including dosing frequency, and—critically—insurance coverage and affordability [182].
8.2. Pharmacogenomic-Guided Selection
Genetic variants predict differential treatment response to incretin-based therapies, though clinical implementation of pharmacogenomic testing remains aspirational rather than standard practice. The most robust evidence supports associations between variants in the GLP-1 receptor gene (GLP1R) and β-arrestin 1 gene (ARRB1) with glycemic response to GLP-1 receptor agonists.
8.2.1. GLP1R Variants
The rs6923761 (Gly168Ser) variant in GLP1R is the most consistently replicated genetic predictor of GLP-1 receptor agonist response. Each copy of the serine allele (A allele) is associated with a smaller HbA1c reduction of approximately 0.08% (0.9 mmol/mol) following treatment [46,191,192,193]. This association has been confirmed across multiple studies and ethnicities, with individuals carrying the GG genotype achieving greater HbA1c reductions and higher rates of reaching the 7.0% HbA1c target [192,193].
However, this variant demonstrates a dual effect: while it diminishes glycemic response, it paradoxically enhances weight loss with liraglutide treatment in some cohorts [194]. The polymorphic A allele is associated with clinically relevant lower glycemic response in everyday clinical practice, suggesting tissue-specific alterations in receptor signaling [192,193].
8.2.2. ARRB1 Variants
Low-frequency coding variants in ARRB1 (β-arrestin 1) show even stronger associations with treatment response. Four specific variants—rs140226575 (Thr370Met), rs78979036 (Thr275Ile), rs58428187 (Ile158Val), and rs78052828 (Gly411Ser)—collectively contribute to differential HbA1c reduction [46]. The Thr370Met variant is particularly relevant for Hispanic and American Indian/Alaska Native populations, where its frequency reaches 6–11% compared to 0.05% in White Europeans [46]. This variant confers 0.25% (2.7 mmol/mol) greater HbA1c reduction per methionine allele, likely through enhanced β-cell mechanisms affecting GLP-1 receptor internalization and signaling [46].
Combined genotyping of GLP1R and ARRB1 identifies clinically meaningful subgroups: approximately 4% of patients show 30% greater HbA1c reduction compared to the 9% with the least response [195]. These genetic effects appear specific to GLP-1 receptor agonists and do not predict response to metformin, sulfonylureas, or other glucose-lowering medications [46].
8.2.3. GIPR Variants and Dual Agonists
Evidence for GIPR variants predicting incretin therapy efficacy remains limited. While the rs1800437 (Glu354Gln) variant is associated with obesity and insulin resistance, and the rare Arg217Leu variant shows reduced receptor function in vitro [196], no robust pharmacogenetic data demonstrate that GIPR variants predict clinical response to dual GIP/GLP-1 agonists like tirzepatide [197]. The E354Q variant does not appear to significantly alter tirzepatide signaling profiles [197].
8.2.4. Clinical Utility of Pharmacogenomic Testing
Despite the biological plausibility and statistical associations, pharmacogenomic testing for GLP1R and ARRB1 variants is not yet clinically useful for routine practice [46,192,193,195]. Current evidence supports their biological relevance but lacks the implementation framework needed for clinical application. Combined genotyping could theoretically translate to an absolute benefit of 3.2 mmol/mol HbA1c reduction and potentially three years longer before treatment failure in favorable genotype carriers [46,195].
Clinical implementation faces significant barriers. Genotyping is not routinely available at the point of prescribing, and no prospective trials have demonstrated that genotype-guided therapy improves outcomes compared to standard care [46]. Most individuals fall in overlapping regions of genetic risk distributions rather than at extremes where precision medicine approaches would be most robust [195].
Additionally, current evidence has important limitations including small sample sizes, limited ethnic diversity (particularly underrepresentation of Asian and African American populations), reliance on candidate gene approaches rather than comprehensive sequencing, and lack of data on cardiovascular and renal outcomes [46,198,199].
Other genes with preliminary associations include TCF7L2, CNR1, SORCS1, CTRB1/2, TMEM114, and CHST3, though these require replication in larger studies [191,198,199]. The field awaits prospective genotype-stratified trials to determine whether pharmacogenomic-guided GLP-1 receptor agonist selection improves patient outcomes sufficiently to justify routine implementation.
8.3. Predicting Response and Non-Response
Accurate prediction of treatment response and early identification of non-responders allows clinicians to optimize therapeutic strategies, avoid treatment inertia, and personalize management approaches for incretin-based therapies.
8.3.1. Predictors of Weight Loss Response
Dosing represents the strongest predictor of weight loss magnitude. For tirzepatide, higher doses (10 mg and 15 mg) produce significantly greater weight loss than lower doses (5 mg), with 15 mg achieving mean weight reductions of approximately 20–21% compared to 15% with 5 mg [16,83]. Similarly, semaglutide 2.4 mg produces approximately 13–16% weight loss compared to lower doses [13,200].
Demographic factors significantly influence response. Female sex, younger age, and White or Asian race are associated with higher likelihood of achieving ≥15% weight loss with tirzepatide [173]. Sex differences are notable, with men experiencing approximately six percentage points less weight reduction than women with both tirzepatide and semaglutide [13]. The presence of T2DM appears to attenuate response—patients without T2DM achieve larger weight reductions than those with T2DM for both medications [98].
Baseline metabolic parameters predict tirzepatide response. Lower baseline HbA1c, lower fasting serum glucose, and lower non-HDL cholesterol are associated with greater weight loss [173]. Metformin background therapy in patients with T2DM also predicts better response [173]. Tirzepatide consistently produces greater weight loss than semaglutide across doses, achieving approximately 15–20% weight reduction at 12 months compared to 8–14% with semaglutide, with adjusted differences of 5–7 percentage points favoring tirzepatide [13,98].
8.3.2. Early Identification of Non-Responders
Early weight loss of <5% after three months of treatment is the most reliable clinical indicator for identifying non-responders to GLP-1 receptor agonists, with this threshold consistently predicting poor long-term outcomes [189,201]. Weight loss trajectories follow a predictable pattern, with the most rapid reduction occurring during the first six months, followed by gradual slowing and relative plateauing at 18 months [95].
Patients achieving ≥5% weight loss at one month are significantly more likely to reach clinically meaningful weight reduction at three months (27% of patients), six months (45%), and 12 months (57%) [201]. Early response at six months strongly predicts 12-month outcomes, with patients achieving weight loss at six months demonstrating substantially higher likelihood of maintaining or extending their response through 12 months of therapy [176]. This early-to-late response correlation appears independent of baseline characteristics, making it a practical clinical marker.
Monthly monitoring during dose escalation and at least quarterly thereafter allows timely identification of inadequate response [135,189]. Assessment should include weight trajectories, percentage weight loss, waist circumference changes, and improvements in metabolic parameters such as blood pressure and glycemia [135]. Weight loss < 5% after 12–16 weeks warrants intervention [202].
Baseline characteristics associated with non-response include male sex, older age, longer T2DM duration, and presence of T2DM, though these factors are less predictive than early weight loss patterns [176,203,204]. Lower baseline body fat percentage, reduced insulin resistance (lower HOMA-β), and prior metformin use may also predict diminished response [176,203].
8.3.3. Management of Non-Responders
Management of identified non-responders requires avoiding treatment inertia through several strategies: (1) verifying medication adherence and assessing for missed doses that may necessitate adjusted titration; (2) continuing standard dose escalation while documenting progress, recognizing that some patients require longer than standard 17-week titration periods; (3) switching to an alternative GLP-1 receptor agonist or escalating to tirzepatide if maximum tolerated dose of a GLP-1 receptor agonist proves ineffective; and (4) intensifying treatment with additional approaches such as combination pharmacotherapy or structured lifestyle interventions [89,189,202].
Treatment adherence critically determines long-term success. Patients maintaining medication use achieve peak weight loss of approximately 12% total body weight, while medication discontinuation results in rapid weight regain at approximately 0.55% of original body weight per month [205]. Shared decision-making should guide therapeutic adjustments, balancing treatment tolerance, financial considerations, and individual preferences against the potential benefits of continued therapy [89,189].
8.3.4. Factors Predicting Discontinuation
Gastrointestinal adverse events are the primary driver of treatment discontinuation, occurring in <10% of patients in clinical trials, with nausea (25–60%), vomiting (5–15%), and diarrhea being most common [95,160]. Higher doses and specific patient populations predict increased adverse event risk. Female sex and older age are associated with higher rates of gastrointestinal events [161].
Pre-existing conditions significantly influence adverse event profiles. Patients with baseline diabetic retinopathy face increased risk of retinopathy complications, particularly when experiencing rapid glycemic improvement [94,160,206]. History of gallbladder disease predicts higher risk of cholelithiasis and cholecystitis, with GLP-1 receptor agonists increasing cholelithiasis risk by 46% overall. Concomitant medications modify safety profiles, with concurrent use of insulin or sulfonylureas substantially increasing hypoglycemia risk, particularly in older or frail patients [94,160].
8.4. Contraindications and Precautions
Understanding absolute and relative contraindications to incretin-based therapies is essential for safe and appropriate patient selection. Current evidence distinguishes between contraindications supported by mechanistic concerns versus those established through clinical outcomes data.
8.4.1. Absolute Contraindications
Personal or family history of medullary thyroid cancer or multiple endocrine neoplasia type 2 (MEN-2) are absolute contraindications [94,146,165]. This contraindication stems from preclinical rodent studies showing thyroid C-cell tumors with GLP-1 RA treatment, though cardiovascular outcomes trials have not demonstrated increased risk of medullary thyroid cancer in humans [55]. The FDA has issued black box warnings for all GLP-1 RAs based on this concern [207,208].
The contraindication persists despite reassuring human data because these are rare conditions where even theoretical risk is considered unacceptable [148]. Large-scale human studies and meta-analyses have not demonstrated a statistically significant increase in thyroid cancer risk. A large international cohort study comparing nearly 100,000 GLP-1 RA users to over 2.4 million DPP-4 inhibitor users found no evidence of increased thyroid cancer risk (pooled weighted HR: 0.81, 95% CI 0.59, 1.12) [148]. The discrepancy between rodent and human data reflects fundamental biological differences: rodents express much higher levels of GLP-1 receptors in thyroid C-cells than primates, and rodent studies used supratherapeutic doses [160,207].
GLP-1 receptor agonists should not be used in patients with MEN-2, as this represents an absolute contraindication across all agents in this class [146,160,209,210]. Clinicians must screen for personal or family history of MTC or MEN-2 before initiating any GLP-1 RA, with one case report documenting incidental detection of MEN-2 during pre-treatment evaluation [211]. Patients should be counseled regarding the contraindication and advised to report symptoms suggestive of thyroid tumors, though routine calcitonin monitoring is not recommended for patients without risk factors [146,203,212]. Several manufacturers (e.g., semaglutide, tirzepatide) recommend discontinuing therapy at least two months before planned conception to allow for drug washout [169].
8.4.2. Relative Contraindications and Precautions
ADA and other guidelines advise caution and often recommend avoiding GLP-1 RAs in patients with a history of pancreatitis [165]. However, this recommendation is increasingly difficult to justify given that meta-analyses of long-term RCTs indicate no increased risk with GLP-1 RAs [142,213]. A meta-analysis of seven placebo-controlled cardiovascular outcomes trials found no increased risk of acute pancreatitis or pancreatic cancer with GLP-1 RA treatment (Peto OR: 1.05, 95% CI 0.78, 1.40) [148,167]. These findings led the FDA and European Medicines Agency to conclude that available data do not support a causal association between incretin therapies and pancreatic cancer [146]. The recommendation to avoid GLP-1 RAs in patients with pancreatitis history appears to represent a “reconcilable divorce” between early concerns and current evidence [142,213].
Pre-existing diabetic retinopathy with poor glycemic control requires careful consideration. Semaglutide was associated with increased retinopathy complications in the SUSTAIN-6 trial (HR 1.76, 95% CI 1.11, 2.78), particularly among those with baseline retinopathy who experienced rapid HbA1c reduction [55,146]. This appears attributable to the magnitude and rapidity of glycemic improvement rather than a direct drug effect, similar to what has been observed with insulin intensification [55]. For semaglutide specifically, retinopathy screening before initiation is recommended, particularly in patients with proliferative diabetic retinopathy or HbA1c > 10% [160,209].
Gastroparesis or prior gastric surgical procedures warrant particular caution, as GLP-1 RAs delay gastric emptying and may exacerbate symptoms in patients with pre-existing gastric motility disorders [209]. The American Gastroenterological Association (AGA) recommends reviewing and potentially discontinuing GLP-1 RAs in patients with gastroparesis [214]. GLP-1 RAs are associated with increased risk of retained gastric contents during endoscopy, with long-acting formulations (semaglutide, dulaglutide, tirzepatide), high doses, procedures during dose escalation, and pre-existing gastrointestinal comorbidities all increasing this risk [129,215].
Gallbladder disease history should prompt caution. Cholelithiasis risk is significantly elevated with GLP-1 RA therapy (risk ratio 1.46, 95% CI 1.09–1.97), representing approximately two additional cases per 1000 patients treated [216]. This risk appears more pronounced in trials including individuals with overweight/obesity and with weight-loss-inducing or high-dose formulations [216]. Gastroesophageal reflux disease (GERD) risk is also increased (risk ratio 2.19, 95% CI 1.48–3.25), with approximately four additional cases per 1000 patients [216].
8.4.3. Special Considerations for Gastrointestinal Disorders
Gradual dose titration is essential when GLP-1 RAs are used to minimize gastrointestinal adverse effects [215]. For semaglutide, the AGA recommends starting at 0.25 mg weekly for 4 weeks, then increasing to 0.5 mg, 1.0 mg, and 1.7 mg weekly every four weeks until reaching the maintenance dose of 2.4 mg after 16 weeks [215]. Clinical judgment should guide adjustments based on individual patient response and tolerance [215].
For patients undergoing elective endoscopy, shared decision-making should weigh the risks of continuing versus temporarily discontinuing GLP-1 RAs [129]. In most cases, these medications can be continued periprocedurally, though a 24 h liquid diet may benefit high-risk patients [129]. For upper endoscopy specifically, point-of-care ultrasound (POCUS) examination for retained gastric content and consideration of prokinetic medications like erythromycin may enhance safety [215].
Dietary modifications represent first-line management for gastrointestinal symptoms: smaller, more frequent meals; adequate hydration; and avoidance of high-fat or high-sugar foods [129]. If symptoms persist despite dietary adjustments, antiemetics including 5-HT3 antagonists (ondansetron), H1 antagonists (promethazine), or dopamine D2 antagonists (prochlorperazine) can provide symptomatic relief [129,214]. The AGA emphasizes that dietary interventions with small-particle, low-fat, low-residue diets should be considered before or alongside pharmacologic interventions in patients with gastroparesis [214].
8.4.4. Drug Interactions and Concomitant Therapy
Concomitant use with sulfonylureas or insulin increases hypoglycemia risk and requires dose adjustment. When HbA1c is ≤7.5% or hypoglycemic episodes occur, sulfonylureas should be discontinued; basal insulin should be reduced by 20–30% if HbA1c is at or below target [209]. Treatment deintensification is particularly important in older and frail individuals (approximately ≥65 years) to avoid hypoglycemia and hypovolemia [160]. GLP-1 RAs may impact absorption of oral medications requiring rapid onset due to delayed gastric emptying, necessitating careful consideration of timing and drug–drug interactions [214,215].
Hypersensitivity to GLP-1 RAs is a recognized contraindication [209]. Beyond absolute contraindications, practical selection should consider gastrointestinal tolerability, pre-existing conditions that may be exacerbated, concomitant medications that increase adverse event risk, and patient-specific factors including age, renal function, and cardiovascular risk profile.
9. Practical Implementation
9.1. Drug Interchangeability and Switching Protocols
Successful switching between incretin-based therapies requires understanding of agent-specific pharmacology, adherence to standardized titration protocols, and proactive management of gastrointestinal adverse effects. While switching protocols are well-established for transitions among GLP-1 receptor agonists, guidance for switches involving dual GIP/GLP-1 agonists continues to evolve as clinical experience accumulates.
9.1.1. Overview and Clinical Rationale for Switching
GLP-1 RAs are classified as short-acting or long-acting based on their duration of action and mechanism [160,217]. Short-acting agents (exenatide twice daily, lixisenatide once daily) primarily delay gastric emptying and have more pronounced effects on postprandial glucose, while long-acting agents (liraglutide once daily; dulaglutide, exenatide extended-release, and semaglutide once weekly) predominantly affect fasting glucose through enhanced insulin secretion and reduced glucagon secretion [55,160,218]. Long-acting agents demonstrate greater overall glycemic efficacy, with semaglutide showing the most robust HbA1c reductions (approximately 1.2–1.5%), followed by dulaglutide and liraglutide [55,58,219]. Weight loss ranges from 1.5 to 6 kg across the class, with semaglutide and liraglutide demonstrating superior weight reduction [55,59,219].
Common reasons for switching include inadequate efficacy, gastrointestinal intolerance, cost considerations, formulary changes, and patient preference [220,221]. Importantly, comparative effectiveness studies demonstrate that both liraglutide (1.8 mg) and semaglutide have demonstrated CV benefit; renal protection is best supported by semaglutide based on FLOW, suggesting that switches driven by access or tolerability concerns are clinically reasonable [221]. Real-world data indicate that switching to semaglutide from other GLP-1 RAs can yield additional improvements in HbA1c (approximately 0.8%) and weight loss (approximately 3.4 kg), particularly in patients with suboptimal response to their initial agent [219].
9.1.2. Switching Protocols Between GLP-1 Receptor Agonists
No formal washout period is recommended when switching between GLP-1 receptor agonists; expert consensus supports initiating the new agent at its initial titration dose immediately after discontinuing the previous one [89,222]. For once-daily agents like liraglutide, the new GLP-1 RA should be initiated 1 day after the last dose, while for once-weekly agents (dulaglutide, semaglutide, exenatide extended-release), initiation occurs 1 week after the last dose [222].
Gradual dose titration of the new agent is essential to minimize gastrointestinal adverse effects, regardless of the maintenance dose of the prior agent [89,94]. For semaglutide, the AGA recommends starting at 0.25 mg weekly for four weeks, then escalating through 0.5 mg, 1.0 mg, and 1.7 mg every four weeks until reaching the maintenance dose of 2.4 mg at 16 weeks [89]. For liraglutide, initiation begins at 0.6 mg daily for seven days, followed by weekly increases through 1.2 mg, 1.8 mg, and 2.4 mg until reaching 3.0 mg daily at four weeks [89].
When switching between long-acting agents, the new agent can typically be started at its initial titration dose without a washout period, given the similar pharmacodynamic profiles within this subclass [89,219]. Clinical judgment should guide whether to resume at the same dose if 1–2 doses are missed, lower the dose if tolerance was marginal, or restart the full titration schedule if ≥3 consecutive doses are missed [89].
9.1.3. Switching Between Semaglutide and Tirzepatide
Switching between semaglutide and tirzepatide can be accomplished by initiating the new agent at its standard starting dose with gradual monthly titration, regardless of the maintenance dose of the prior agent [182]. No washout period is required, and the transition should prioritize minimizing gastrointestinal adverse effects while optimizing metabolic outcomes.
Tirzepatide demonstrates superior efficacy compared to semaglutide for both glycemic control and weight reduction. In head-to-head trials, tirzepatide 10–15 mg produced mean weight reductions of 20.9–22.9% versus 16.7% with semaglutide 2.4 mg [13,83]. For HbA1c reduction, tirzepatide 15 mg achieved reductions of approximately 2.1–2.5% compared to 1.6–1.8% with semaglutide 2.4 mg [183,223,224]. The dual agonism of GIP and GLP-1 receptors provides additional mechanisms for appetite regulation and adipocyte metabolism beyond GLP-1 receptor activation alone [13,182,225].
When switching from semaglutide to tirzepatide, initiate tirzepatide at 2.5 mg weekly and escalate monthly through 5 mg, 7.5 mg, 10 mg, 12.5 mg, and 15 mg based on individual tolerance and response [13,135]. When switching from tirzepatide to semaglutide, initiate semaglutide at 0.25 mg weekly and escalate monthly through 0.5 mg, 1.0 mg, 1.7 mg, and 2.4 mg [13,135]. In both directions, start at the initial titration dose regardless of prior maintenance dose.
Both agents share similar gastrointestinal adverse event profiles, with nausea (33–44%), diarrhea (23–31%), and vomiting (11–25%) being most common [135]. Discontinuation rates due to adverse events range from 4.3–10% in clinical trials [182,223]. Indirect comparisons suggest comparable safety profiles, with no significant differences in gastrointestinal adverse events when comparing equivalent doses [183,224,226].
9.1.4. Safety and Tolerability Considerations
Gastrointestinal adverse events—particularly nausea, vomiting, and diarrhea—are the most common side effects across all GLP-1 RAs [58,59,227]. Once-weekly formulations (exenatide extended-release, albiglutide) demonstrate lower nausea rates compared to daily agents (liraglutide) or twice-daily exenatide, though injection-site reactions may be more frequent with exenatide formulations [59]. The intrinsic risk of hypoglycemia with GLP-1 RAs is very low when used without insulin or secretagogues [58,227].
GLP-1 RAs should not be used concurrently with other GLP-1 RAs or DPP-4 inhibitors [89]. Caution is warranted when combining with insulin or sulfonylureas due to increased hypoglycemia risk, and the delayed gastric emptying effect may impact absorption of medications requiring rapid onset [89]. Some real-world cohorts report lower discontinuation with semaglutide relative to other GLP-1 RAs, though rates vary widely depending on population, indication, insurance continuity, and follow-up duration [219].
To mitigate gastrointestinal adverse effects during transition, counsel patients on eating smaller meals, avoiding high-fat foods, and limiting alcohol [135]. Short-term antiemetics (ondansetron), H2 blockers or proton pump inhibitors for dyspepsia, and osmotic laxatives (polyethylene glycol) for constipation may be considered as needed.
9.2. Dosing and Titration Strategies
Appropriate dosing and titration represent cornerstones of successful incretin-based therapy, balancing the achievement of therapeutic goals with minimization of adverse effects. Evidence-based titration protocols have been established for each agent, though individualization based on patient tolerance remains essential.
9.2.1. Standard Titration Protocols
For semaglutide (subcutaneous), initiation begins at 0.25 mg weekly and titrates monthly to minimize gastrointestinal adverse effects [89,135]. The standard escalation schedule is: Week 0–4 at 0.25 mg weekly, Week 5–8 at 0.5 mg weekly, Week 9–12 at 1.0 mg weekly, Week 13–16 at 1.7 mg weekly, and Week 17+ at 2.4 mg weekly (maintenance dose) [89,207]. The maintenance dose of 2.4 mg is typically reached after 16–17 weeks [89,207]. For patients with T2DM, a 2.0 mg dose may be used as an alternative maintenance dose [94].
Tirzepatide is initiated at 2.5 mg weekly with dose increases every four weeks in 2.5 mg increments [135,207]. The escalation schedule is: Week 0–4 at 2.5 mg weekly, Week 5–8 at 5.0 mg weekly, Week 9–12 at 7.5 mg weekly, Week 13–16 at 10.0 mg weekly, Week 17–20 at 12.5 mg weekly, and Week 21+ at 15.0 mg weekly (maximum dose) [16,207]. The recommended maintenance dose is 5 mg, 10 mg, or 15 mg weekly, depending on treatment response and tolerability [16,207].
Liraglutide is initiated at 0.6 mg daily and titrated weekly for obesity treatment [89,207]. The escalation schedule is: Week 1 at 0.6 mg daily, Week 2 at 1.2 mg daily, Week 3 at 1.8 mg daily, Week four at 2.4 mg daily, and Week 5+ at 3.0 mg daily (maintenance dose for obesity) [89,207]. For T2DM management, the maximum dose is 1.8 mg daily, reached after at least one week at 1.2 mg [9,20].
Dulaglutide is initiated at 0.75 mg weekly and may be increased to 1.5 mg after four weeks [10,23]. For patients requiring additional glycemic control, higher doses of 3.0 mg or 4.5 mg weekly (maximum) can be used, with at least four weeks at each dose before further escalation [55,228].
9.2.2. Optimizing Titration to Minimize Adverse Events
The ADA, the European Association for the Study of Diabetes recommend gradual up-titration as the optimal strategy to minimize gastrointestinal side effects when initiating GLP-1 RAs [55]. Expert consensus and real-world experience support slower or flexible dose escalations in patients experiencing gastrointestinal intolerance, allowing the titration schedule to be extended beyond standard intervals [55].
A recent randomized controlled trial demonstrated that a 16-week flexible titration regimen for semaglutide for management of obesity significantly improved adherence and reduced adverse events compared to the label-recommended 8-week regimen [137]. In this study, patients started at 0.0675 mg weekly with gradual increases of 0.0675 mg per week, with delays permitted for gastrointestinal symptoms. Only 2% of patients in the flexible arm withdrew due to gastrointestinal adverse events versus 19% in the standard arm, while achieving similar final doses and comparable HbA1c and BMI reductions [137].
Starting with the lowest available dose is essential across all GLP-1 receptor agonists—0.25 mg for semaglutide and 2.5 mg for tirzepatide—to prevent clinically significant side effects [135,136]. The American Association of Clinical Endocrinology (AACE) emphasizes that individuals naïve to these medications require this conservative initiation, and those who discontinue or pause treatment must restart at the lowest dose [136].
9.2.3. Dose Adjustments for Gastrointestinal Side Effects
Patient education is essential to distinguish between nausea (a negative sensation requiring intervention) and satiety (a positive sensation that supports weight loss) [55]. Dietary and behavioral modifications are recommended to minimize gastrointestinal symptoms, including practicing mindful eating (eating slowly, stopping when full, avoiding eating when not hungry), consuming smaller meals or snacks, decreasing intake of high-fat and spicy foods, moderating alcohol intake, and increasing water intake [55].
Maintaining patients at the lowest effective dose represents an alternative strategy supported by clinical experience, with escalation reserved for when weight reduction ceases or efficacy wanes [95]. During pivotal clinical trials, subjects were permitted to remain at a given dose for up to 8 weeks as needed to allow gastrointestinal symptoms to dissipate [95].
The timing and composition of meals relative to medication administration influences symptom severity. Nausea often occurs in the morning or after prolonged fasting, and patients benefit from eating a small breakfast followed by additional small meals every 3–4 h with adequate fluid intake [95]. Some individuals develop a cycle where nausea prevents eating, which paradoxically worsens symptoms; breaking this pattern through scheduled small meals is important [95].
Pharmacologic interventions provide symptom relief when dietary modifications prove insufficient. For nausea, prochlorperazine may be preferable to serotonergic agents like ondansetron, which can worsen constipation [95]. Short-term antiemetics remain an option for severe nausea or vomiting, while H2 blockers or proton pump inhibitors may address dyspepsia [135]. For constipation, ensuring adequate fiber and hydration is foundational, with escalation to bulk-forming laxatives (psyllium), stool softeners (docusate sodium), or osmotic/stimulant laxatives (polyethylene glycol) as needed [135].
If symptoms become intolerable despite these interventions, dose reduction to a previously tolerated level or temporary discontinuation may be necessary [95]. Approximately 6–10% of patients in clinical trials permanently discontinued GLP-1 receptor agonists due to adverse effects, though observational data suggest discontinuation rates may reach 53% at one year in real-world practice [135].
9.2.4. Management of Missed Doses
The ADA and the European Association for the Study of Diabetes emphasize that gastrointestinal side effects tend to occur during initiation and dose escalation and diminish over time, which is the rationale for their recommendation of gradual up-titration [55]. This principle suggests that when doses are missed, particularly multiple consecutive doses, the body may lose its adaptation to the medication, potentially necessitating a more cautious approach to resumption.
For once-weekly formulations like semaglutide, dulaglutide, and tirzepatide, if a dose is missed and fewer than five days have elapsed until the next scheduled dose, the missed dose should be skipped and the regular schedule resumed. If more than 5 days remain, the missed dose should be administered as soon as possible.
The American College of Cardiology provides guidance that if treatment is suspended, reinitiation should be at the lowest dose with gradual up-titration to avoid recurrent nausea and vomiting [138]. When two or more consecutive doses are missed, therapy should be restarted at a lower dose with gradual dose increase to mitigate gastrointestinal adverse effects [135]. The AACE emphasizes that individuals who discontinue or pause treatment for a length of time will need to restart at a low dose and titrate up to enable the body time to acclimate to the medication [136].
For perioperative management, the approach varies based on formulation and risk factors. Long-acting formulations (semaglutide, dulaglutide, and tirzepatide), high doses, procedures during dose escalation, and gastrointestinal comorbidities that delay gastric emptying raise the risk of retained gastric contents [129]. Per 2023 American Society of Anesthesia consensus guidance, withholding weekly GLP-1 RAs for one week prior to procedures involving general anesthesia may be considered in patients at elevated aspiration risk; recommendations continue to evolve, and shared decision-making with anesthesia services is advised. Decision should be individualized based on patient and procedure characteristics [135].
9.3. Combination Therapy Approaches
Combination therapy with incretin-based agents offers opportunities to enhance efficacy through complementary mechanisms while managing safety considerations. Understanding which combinations provide additive benefit and which are contraindicated is essential for optimizing treatment regimens.
9.3.1. Combination with Metformin
Metformin plus GLP-1 RA represents the most evidence-based combination, with 71–82% of patients in cardiovascular outcome trials receiving metformin as baseline therapy [229]. This combination provides additive glucose-lowering effects (HbA1c reduction of 1.0–2.5%) while maintaining low hypoglycemia risk and favorable weight profiles [165,180]. For patients with established atherosclerotic cardiovascular disease or high cardiovascular risk, GLP-1 RAs should be added regardless of baseline glycemic control [165,230].
Initial combination therapy may be considered when HbA1c remains ≥1.5% above target despite monotherapy, consistent with diabetes treatment guidelines rather than GLP1-specific evidence to achieve more rapid glycemic control [165]. This combination provides additive glucose-lowering effects while maintaining low hypoglycemia risk and favorable weight profiles [180]. Routine self-monitoring of blood glucose may not be required when GLP-1 RAs are paired with metformin alone, though monitoring is appropriate during initiation, titration, appetite-related dietary changes, or when hypoglycemia risk factors are present [230].
9.3.2. Combination with SGLT2 Inhibitors
GLP-1 receptor agonists and SGLT2 inhibitors can be combined, and the ACC considers this combination reasonable when clinically indicated, despite the absence of cardiovascular outcome trials specifically studying their concomitant use [138]. The DURATION-8 trial demonstrated greater reductions in blood pressure and body weight with the combination of dapagliflozin and exenatide compared to either agent alone [138]. Additionally, randomized placebo-controlled trials showed that dulaglutide, liraglutide, and semaglutide provided additive glucose-lowering benefits over placebo in patients already receiving SGLT2 inhibitors [138].
Subsequent cardiovascular outcome trials have provided robust evidence supporting the efficacy and safety of this combination. Available evidence from subgroup analyses and observational cohorts suggests that GLP-1 RAs retain cardiovascular and renal benefits when used alongside SGLT2 inhibitors, though no large RCTs have been designed to evaluate intentional co-therapy [231]. Observational data suggest lower cardiovascular and kidney risks with combination therapy versus monotherapy, although potential confounding limits causal interpretation [232,233].
Real-world observational data suggest potentially additive benefits. A 2025 meta-analysis of cohort studies found that combination therapy was associated with lower risks of major adverse cardiovascular events (risk ratio 0.56), all-cause mortality (risk ratio 0.50), and kidney composite endpoints (risk ratio 0.48) compared to monotherapy with either agent [234]. The safety profile appears favorable, with no increased risk of serious adverse events (SAEs), diabetic ketoacidosis, or urinary tract infections when GLP-1 RAs are added to SGLT2 inhibitors [231].
An important practical consideration is that out-of-pocket costs may be very high for some patients when using both drug classes simultaneously [138].
9.3.3. Combination with Insulin
When combining with insulin, GLP-1 RAs demonstrate greater efficacy and durability than insulin intensification alone, with additional benefits of weight reduction and lower hypoglycemia rates [165,235]. Prandial insulin should be reduced or discontinued first, followed by basal insulin dose reduction as the GLP-1 RA is titrated, guided by glucose monitoring [165,229]. Combination therapy often reduces required insulin doses, with pooled estimates demonstrating modest reductions and real-world reductions varying based on baseline insulin requirements [235].
The ADA recommends GLP-1 RAs as the preferred injectable therapy over insulin for persistent hyperglycemia, except when severe hyperglycemia (glucose ≥ 300 mg/dL or HbA1c > 10%) with catabolic features is present [165]. Hypoglycemia risk increases when GLP-1 RAs are combined with insulin (OR 1.28), necessitating close glucose monitoring during the first weeks after initiation and dose titration [94,209,235].
9.3.4. Combination with Sulfonylureas
Most guidelines recommend reducing or discontinuing sulfonylureas when initiating GLP-1 RAs to minimize hypoglycemia, with dose adjustments tailored to baseline HbA1c and hypoglycemia history [209,230]. For patients with HbA1c ≤ 7.5%, sulfonylureas should be stopped at GLP-1 RA initiation; if HbA1c is 7.6–8.5%, reduce the sulfonylurea dose by 50%; only continue full-dose sulfonylureas if HbA1c > 8.5% [209]. The American College of Physicians (ACP) notes that sulfonylureas and insulin are inferior to GLP-1 RAs for reducing all-cause mortality and morbidity, though they may retain limited value for glycemic control when cost is prohibitive [230].
9.3.5. Contraindicated Combination: DPP-4 Inhibitors
The ADA and ACCE guidelines do not recommend combining GLP-1 RAs with DPP-4 inhibitors due to overlapping mechanisms without additive benefit [165]. Both GLP-1 RAs and DPP-4 inhibitors work through the incretin pathway—GLP-1 RAs directly activate GLP-1 receptors with pharmacologic doses of GLP-1 analogs, while DPP-4 inhibitors prevent the degradation of endogenous GLP-1 [165].
Head-to-head trials confirm the superiority of GLP-1 RAs over DPP-4 inhibitors across multiple efficacy endpoints. A meta-analysis of 13 randomized controlled trials involving 4330 patients showed that GLP-1 RAs reduced HbA1c by an additional 0.41% and body weight by 2.15 kg compared to DPP-4 inhibitors, with no increased hypoglycemia risk despite higher rates of gastrointestinal side effects [236]. Switching from DPP-4 inhibitors to GLP-1 RAs provides additional clinical benefit, with five interventional studies showing further mean reductions in HbA1c of 0.69% and weight loss of 2.25 kg [236].
The AGA explicitly states that liraglutide and semaglutide should not be used with DPP-4 inhibitors due to overlapping mechanisms and lack of anticipated additive efficacy [89].
9.3.6. Key Safety Considerations Across Combinations
Common adverse effects include gastrointestinal symptoms (nausea 8–21%, vomiting 3–13%, diarrhea 9–13%), which typically diminish over time with dose titration [94,229]. Contraindications include personal or family history of medullary thyroid carcinoma or multiple endocrine neoplasia type 2, pregnancy and hypersensitivity to GLP-1 RAs [209,229]. Individualized HbA1c targets of 7–8% guide therapy intensification in most adults, with deintensification recommended if HbA1c falls below 6.5% [229,230]. Coordination among the multidisciplinary care team is essential, particularly when cardiologists initiate GLP-1 RAs for cardiovascular indications while primary care physicians or endocrinologists manage ongoing antihyperglycemic medication adjustments [209]. Metformin, SGLT2 inhibitors, and GLP-1 receptor agonists all require renal consideration, with metformin contraindicated in severe kidney impairment, SGLT2 inhibitors losing glycemic efficacy at low eGFR but retaining some renal benefits, and GLP-1 agonists generally safe in mild-to-moderate CKD while monitoring for dehydration-related complications.
9.4. Patient Education and Shared Decision-Making
Effective patient education and shared decision-making represent critical components of successful incretin-based therapy. Comprehensive counseling encompasses expected outcomes, potential adverse effects, lifestyle modifications, and individualized goal-setting aligned with patient values and clinical context.
9.4.1. Foundational Patient Education
The ADA recommends educating patients to distinguish between nausea and satiety when initiating GLP-1 RA therapy, as these medications promote a sense of fullness that facilitates weight loss [55]. This distinction is crucial because satiety is a positive sensation supporting therapeutic goals, while nausea is an adverse effect requiring intervention.
Dietary and eating behavior modifications should be emphasized, including mindful eating practices such as eating slowly, stopping when full, and avoiding eating when not hungry [55]. Patients should be advised to consume smaller meals or snacks, decrease intake of high-fat and spicy foods, moderate alcohol consumption, and increase water intake to help manage gastrointestinal effects [55].
Anticipatory guidance regarding the expected timeline and transient nature of gastrointestinal symptoms is critical for promoting both short- and long-term adherence [209]. Patients should be informed that early satiety and gastrointestinal upset typically emerge soon after initiation but generally improve within four weeks of continued use [209]. Frequent small meals and avoidance of fried or fatty foods may further reduce symptoms of satiety and bloating while supporting weight loss goals [209].
9.4.2. Expected Weight Loss and Outcomes
Expected weight loss varies substantially by medication and dose. Based on phase 3 clinical trial data, semaglutide 2.4 mg weekly produces mean weight loss of approximately 15% at 68 weeks, while tirzepatide 15 mg weekly achieves up to 21% weight loss at 72 weeks [135]. Liraglutide 3.0 mg daily results in more modest weight reduction of approximately 5.8% after 26 weeks [82]. At standard glucose-lowering doses, weight loss ranges from 2–4% of total body weight for dulaglutide, exenatide, and liraglutide, and 4–6 kg for semaglutide [138].
Individual responses vary considerably, and weight loss may be greater or less than typical trial findings [135]. Patients should be counseled that reasonable weight loss expectations focus on improving physical and mental health rather than achieving specific numeric targets. The AGA emphasizes uncertainty and substantial variability in how individuals weigh desirable effects against potential adverse effects, inconvenience of weekly subcutaneous administration, cost, and monitoring burden [89].
A safe weight loss rate typically ranges from 0.5 to 2.0 lb (0.23–0.91 kg) per week [135]. Weight loss exceeding 5% per month or less than 5% after three months of therapy warrants further evaluation. Monitoring should include weight trajectories, percentage and total weight loss, waist circumference changes, and improvements in health outcomes such as mobility, blood pressure, and glycemia [135].
Loss of both fat and lean mass is a normal physiological response to caloric restriction, but adults ≥65 years may be at increased risk of sarcopenia during rapid weight loss; although long term data remain limited [135].
Several trials have assessed body composition changes and shown that a meaningful proportion of weight loss with GLP-1 receptor agonists reflects lean mass reduction. In a DXA subset of STEP 1, semaglutide 2.4 mg was associated with approximately 6.9 kg of lean soft tissue loss alongside 10.4 kg of fat mass loss (~40% of total weight loss from lean tissue). In SURMOUNT-1, tirzepatide was associated with ~5.6 kg lean soft tissue loss relative to ~15.9 kg fat mass loss (~26% of total weight reduction from lean tissue). Meta-analyses indicate that potent agents such as semaglutide and tirzepatide may lose ~25% of total weight as lean mass on average, though relative lean-mass percentages often remain stable and functional outcomes such as strength can improve with continued therapy plus lifestyle support. These findings support the importance of assessing and mitigating lean mass loss, particularly in populations vulnerable to sarcopenia. Though there are associations with GLP-1 agonism and decreased muscle mass, it should be noted that the effects of GLP-1 therapies generally improve muscle function by promoting mitochondrial homeostasis and reducing inflammation, and GLP-1RAs are being researched as a potential treatment for sarcopenia in certain cases [237,238]. Adequate protein intake is essential to mitigate lean mass loss during weight reduction. Older adults and those at elevated risk for sarcopenia may benefit from protein intake of 1.2–1.5 g/kg/day, distributed evenly across meals to optimize muscle protein synthesis. Additionally, protein supplementation may be considered when dietary intake is insufficient. Clinicians should regularly assess muscle strength and function using office-based screening such as time for 5 sit-to-stand tests or grip strength testing with a dynamometer. If decline is detected, patients should ensure protein intake of at least 1.3 g/kg/day and engage in resistance training at least two days per week, with consideration of referral to a registered dietitian nutritionist and possible deescalation or discontinuation of therapy [135].
9.4.3. Safety Counseling and Warning Signs
Gastrointestinal adverse events occur in 47–84% of patients, with higher rates at obesity-targeted doses, compared to 13–63% with placebo, with nausea, vomiting, diarrhea, and constipation being most common [82]. While adverse events are frequent (80–97% vs. 63–100% with placebo), treatment discontinuation due to adverse events (0–26% vs. 0–9%) and SAEs (0–10% vs. 0–12%) remain relatively rare [82]. Additional gastrointestinal complications include cholelithiasis, cholecystitis, gastroparesis, and bowel obstruction, which may warrant caution in susceptible individuals [161].
Warning signs of rare but SAEs should be reviewed. These include severe or persistent abdominal pain, which could indicate pancreatitis, and symptoms suggestive of gallbladder disease such as fever and right upper-quadrant abdominal pain [209]. While postmarketing reports have suggested possible associations with pancreatitis, prospective randomized trials have not confirmed an increased risk [138,209].
Patients should be counseled about hypoglycemia risk and symptoms when GLP-1 receptor agonists are combined with insulin or sulfonylureas, though the medications themselves carry minimal hypoglycemia risk when used alone [94]. Contraindications must be reviewed, specifically that GLP-1 receptor agonists should not be used in patients with a personal or family history of medullary thyroid carcinoma or multiple endocrine neoplasia type 2 [55]. Patients should also be informed about the association with increased gallbladder and biliary diseases [55,94].
Additional counseling may be needed for patients with pre-existing diabetic retinopathy and high baseline HbA1C, as rapid glycemic improvement can transiently worsen retinopathy [55,94]. Monitoring for dehydration and renal impairment is warranted in patients experiencing vomiting or diarrhea [89]. For patients using injectable formulations, injection-site reactions may be prevented by rotating injection sites [209].
Medication interactions require specific attention: GLP-1 RAs are not recommended to co-administered with dipeptidyl peptidase-4 inhibitors, as both work through GLP-1 signaling [89,138]. Delayed gastric emptying may affect absorption of medications requiring rapid onset or narrow timing windows, such as oral contraceptives, analgesics, and some antibiotics [89]. Patients should also be counseled that if treatment is suspended for more than 2–3 consecutive doses, reinitiation should begin at the lowest dose with gradual up-titration to avoid recurrent nausea and vomiting [89,138].
9.4.4. Weight Regain and Long-Term Expectations
Weight regain is common upon discontinuation, necessitating counseling that these medications may require lifelong use in conjunction with lifestyle changes [83]. In the STEP 1 trial, participants who discontinued semaglutide after 68 weeks regained a mean of 11.6% of lost weight over the subsequent 52 weeks. In STEP 4, participants transitioned from semaglutide to placebo regained 6.9% of lost weight during 48 weeks of placebo administration [83].
9.4.5. Shared Decision-Making Framework
Shared decision-making for GLP-1 RA therapy requires balancing individual patient goals, values, and clinical context with evidence on efficacy, safety, and feasibility. The World Health Organization (WHO) emphasizes that obesity is a chronic, relapsing disease requiring lifelong, person-centered care that integrates behavioral, medical, and other interventions [239]. This framework applies equally to T2DM management, where treatment selection must align with patient priorities and comorbidities.
Clinicians should review FDA-approved indications specific to each agent. Semaglutide (Ozempic) is approved for glycemic control in T2DM, reduction in MACE in adults with T2DM and established cardiovascular disease, and reduction in sustained eGFR decline, end-stage kidney disease, and cardiovascular death in adults with T2DM and chronic kidney disease [188]. Tirzepatide is approved for weight reduction and maintenance in adults with obesity or overweight with weight-related comorbidities, as well as for moderate to severe obstructive sleep apnea in adults with obesity [188].
It is not clear that individuals would consistently prioritize desired outcomes over potential adverse effects, inconvenience of weekly subcutaneous administration, potentially high cost, burden of monitoring, and challenges associated with insurance authorization [209]. The WHO guideline development process included individuals with lived experiences of obesity, whose insights emphasized the need for clarity around indications, values and preferences, and equitable access [239].
Social determinants of health screening should assess food and nutrition insecurity, nutrition and culinary knowledge, and other factors that influence equitable obesity management with GLP-1 receptor agonists [95]. The WHO framework emphasizes that effective chronic obesity care requires strong health systems dependent on adequate workforce training, supply chains, and referral systems, with financial coverage under universal health care and national insurance schemes essential to ensure affordability and equity [239].
9.4.6. Integration with Lifestyle and Behavioral Therapy
The WHO recommends pairing pharmacotherapy with intensive behavioral therapy, which represents a component of comprehensive multimodal care and can amplify and sustain therapeutic benefits [239]. However, the recommendation is conditional due to low certainty evidence of intensive behavioral therapy enhancing efficacy across tirzepatide, semaglutide, and liraglutide, as well as variability in patient weight outcome priorities, potential health equity impacts, and context-specific feasibility and cost of delivering intensive behavioral therapy [239].
Baseline screening should include usual dietary habits, emotional triggers, disordered eating, and relevant medical conditions, along with comprehensive examination including muscle strength, function, and body composition assessment [95]. Priorities during GLP-1 use include nutritional and medical management of gastrointestinal side effects, navigating altered dietary preferences and intakes, preventing nutrient deficiencies, and preserving muscle and bone mass through resistance training and appropriate diet [95].
Supportive strategies include group-based visits, registered dietitian nutritionist counseling, telehealth and digital platforms, and Food is Medicine interventions [95]. Clinicians should establish patient-centered goals for weight reduction and health at initiation, monitor progress through weight trajectories and improvements in health outcomes, and provide ongoing support to address challenges around GLP-1 treatment of obesity and T2DM [95].
9.4.7. Monitoring and Long-Term Management
Real-world discontinuation varies widely by access and insurance, ranging from ~40–65% at 1 year, with rising rates over longer follow-up. Discontinuation may relate to side effects, costs, variable individual efficacy, or patient preferences [95]. Real-world challenges include gastrointestinal side effects, risk of inadequate nutrient intake from reduced food intake combined with insufficient nutritional counseling, and potential loss of significant muscle mass and bone density [95].
Strategies to improve compliance and prevent weight regain after discontinuation require ongoing study, as limited data exist on long-term efficacy and safety, titration, maintenance, and discontinuation [95,239]. The WHO notes that trials are still ongoing, and together with high current cost, inadequate health system preparedness, and potential equity implications, these factors reduced confidence that benefits clearly outweigh undesirable attributes [239].
10. Cost-Effectiveness and Access
10.1. Cost-Effectiveness Analysis
Cost-effectiveness analyses evaluate whether the health benefits of GLP-1-based therapies justify their costs, typically using incremental cost-effectiveness ratios (ICERs) expressed in cost per quality-adjusted life-year gained. Published ICERs for semaglutide and tirzepatide vary substantially depending on the indication (obesity vs. T2DM), dose, comparator, and time horizon, with most analyses finding these agents not cost-effective at current prices for obesity treatment but potentially cost-effective for T2DM management.
For obesity management, a 2025 lifetime analysis found semaglutide (2.4 mg) had an ICER of $467,676/QALY and tirzepatide an ICER of $197,023/QALY compared to lifestyle modification alone, with both falling well above the $100,000/QALY threshold [240]. To achieve cost-effectiveness at $100,000/QALY, prices would require discounts of 81.9% for semaglutide and 30.5% for tirzepatide from current net prices [240,241]. A short-term (68-week) analysis in patients without T2DM found subcutaneous tirzepatide cost-effective compared to oral semaglutide with an ICER of $34,212/QALY, and both were cost-effective compared to subcutaneous semaglutide and liraglutide at a $150,000/QALY threshold [242].
For T2DM management, tirzepatide demonstrates more favorable cost-effectiveness. A 50-year projection comparing tirzepatide doses (5, 10, 15 mg) to semaglutide 1.0 mg found ICERs of $75,803, $58,908, and $48,785 per QALY for the respective doses, all falling below typical willingness-to-pay thresholds [243]. A short-term (52-week) analysis reported tirzepatide 10 mg vs. semaglutide 1 mg had an ICER of $2247 per 1% A1c reduction and $237 per kg weight loss [244]. However, one systematic review concluded there was insufficient evidence regarding tirzepatide’s value compared to SGLT2 inhibitors or GLP-1 receptor agonists due to methodological limitations [245].
Overall, current evidence suggests that tirzepatide is more cost-effective than semaglutide across both obesity and T2DM populations, though neither agent meets conventional cost-effectiveness thresholds for obesity treatment at current list prices. These findings underscore the central role of pricing in determining the value proposition of incretin-based therapies.
10.2. Pricing Crisis
As of 2024–2025, US list prices for GLP-1-based therapies typically range from roughly $1000 to $1600 per month, depending on dose and formulation [165,189]. These are substantially higher than those in international markets, where the same medications are sold for just a fraction of U.S. prices. International list prices for these medications are substantially lower—often 5–10× lower—than in the United States, reflecting differences in pricing regulation, patent protections, limited competition, pharmacy benefit manager (PBM)-linked rebate structures, and limited federal price-setting authority in the US.
10.3. Insurance Coverage Patterns
Coverage for incretin-based obesity pharmacotherapies in the United States remains highly fragmented and restrictive, with substantial differences across Medicare, Medicaid, and private insurers. Medicare does not cover GLP-1 receptor agonists for obesity alone, although recent FDA approval of semaglutide for cardiovascular risk reduction in patients with obesity and established cardiovascular disease has created a pathway for Part D coverage under that specific indication [87,95,182,246]. Medicaid coverage varies widely, with only a minority of states covering anti-obesity medications and some states discontinuing coverage altogether because of high costs and unsuccessful negotiations with manufacturers [87,95,246]. Private insurance coverage is similarly inconsistent; earlier analyses of marketplace plans suggested that only about 10–15% covered any obesity pharmacotherapy, and even when coverage is available, insurers frequently impose clinical restrictions or lifetime caps [95,246]. Real-world prescribing data further highlight these disparities, with only 25% of patients successfully obtaining coverage for semaglutide prescribed for obesity compared with much higher approval rates for T2DM indications such as semaglutide [247].
Across insurers, coverage is typically tied to strict eligibility criteria that align with FDA labeling, requiring a BMI of at least 30 kg/m2 or at least 27 kg/m2 with weight-related comorbidities [189,248]. Prior authorization has become nearly universal, rising from 2.8–5.0% through 2023 to nearly 100% of Medicare Part D plans by 2025, creating substantial administrative burden for clinicians and health systems [249,250]. Additional restrictions such as step-therapy requirements, documentation of prior weight-loss attempts, and proof of ongoing lifestyle interventions further limit access [95]. These combined barriers including limited coverage, high annual out-of-pocket costs of $7000–$16,000, and administrative hurdles, contribute to pronounced inequities in access, with higher use among privately insured individuals and markedly lower uptake in socioeconomically disadvantaged populations [247,251].
10.4. Health Equity Disparities
Marked health equity disparities characterize the use of GLP-1 receptor agonists for obesity, with consistently lower prescribing rates among racial and ethnic minority groups, individuals from socioeconomically disadvantaged communities, and residents of rural areas. Large national analyses show that Hispanic, Asian, and Black patients receive these medications at significantly lower rates than White patients, and these differences persist even after adjusting for clinical need [95,252,253]. Notably, inequities remain evident even within health systems designed to reduce cost-related barriers; in the Veterans Health Administration, all minority groups demonstrated significantly lower odds of GLP-1 prescribing compared with White patients despite standardized access and reduced copayments [254]. Socioeconomic status further shapes access, with patients living in the most socially vulnerable areas being substantially less likely to receive treatment. Geographic disparities also contribute, as patients in rural areas show markedly reduced prescribing compared with those in metropolitan regions [253]. Together, these patterns underscore significant inequities in access to incretin-based obesity treatment across multiple social determinants of health.
These disparities reflect multiple barriers beyond medication cost, including suboptimal insurance coverage, limited accessibility, low health literacy, and physician-patient barriers such as provider bias [255]. Notably, while 51% of U.S. adults meet FDA eligibility for semaglutide, this rises to 57% among Black and 55% among Hispanic adults, yet larger proportions of these groups face barriers such as being uninsured, lacking a regular provider, having low income, or lacking higher education [95].
Insurance type remains one of the strongest determinants of access: some analyses suggest disparities are widest among privately insured patients, narrower in Medicare, and nearly absent in Medicaid, although Medicaid beneficiaries overall have the lowest rates of use and significantly lower odds of initiating therapy compared with privately insured individuals. Coverage exclusions such as Medicare’s prohibition on obesity-only indications and wide state-level variability in Medicaid benefits further exacerbate disparities, while private plans impose substantial formulary restrictions and inconsistent approval rates [95,256]. High out-of-pocket costs, often reaching thousands of dollars annually even with insurance, pose another major barrier, disproportionately affecting racial and ethnic groups with lower median wealth and contributing to clinician hesitation in prescribing these therapies [95,257]. Provider-level factors also play a role, with evidence of differential prescribing linked to patient race and ethnicity, including notably lower odds of GLP-1 use among Asian patients; such disparities persist even in healthcare systems with minimized cost barriers, highlighting the importance of clinical communication and potential implicit bias [95,257]. Additional obstacles arise after prescriptions are written, as fill rates remain low due to prior authorization requirements and pharmacy-level restrictions. Across several analyses, fill and approval rates remain low, and appear especially low among patients facing structural barriers, including some racial and ethnic minority groups and those covered by Medicaid or Medicare [251,256]. Geographic inequities compound these issues, as individuals living in rural areas remain substantially less likely to access GLP-1 therapies due to limited specialist availability and infrastructural constraints [253].
These intersecting inequities highlight that expanding access to incretin-based therapies will require more than clinical innovation alone; it will depend on structural solutions that address coverage gaps, affordability, and systemic barriers to care.
10.5. Policy Implications
Policy strategies to improve access to GLP-1 RAs center on expanding Medicare coverage, lowering prices through federal negotiation, redesigning health system delivery, and implementing equity-focused approaches to reduce disparities. Legislative reform through the Treat and Reduce Obesity Act remains the primary pathway to allow Medicare Part D coverage for obesity treatment [258], while recent FDA approval of semaglutide for cardiovascular risk reduction has created a narrower mechanism for Medicare coverage of anti-obesity medications when prescribed for that indication [246]. Modeling studies project substantial increases, as much as $47B, in Medicare spending over the next decade if broad coverage of anti-obesity GLP-1 therapies is implemented, even under modest uptake assumptions [259]. Because current prices render GLP-1 therapies far above accepted cost-effectiveness thresholds, substantial price reductions are essential; historical Medicare negotiations have produced 38–79% price cuts across other drug classes, and semaglutide may become eligible for federal negotiation as early as 2027 based on its original T2DM approval [95]. Globally, the WHO recommends generic manufacturing, tiered pricing, pooled procurement, and voluntary licensing to expand access, with semaglutide’s patent expiration in 2026 offering a critical opportunity for scale-up [239]. At the health system level, integrating GLP-1 therapy into primary-care chronic disease management platforms with uniform coverage policies and reduced cost-sharing has been shown to eliminate racial and socioeconomic disparities in prescribing [95], although administrative burdens remain substantial as prior authorization requirements have risen to 99.9–100% of Medicare Part D plans [249]. Equity-focused implementation will require identifying patients most likely to benefit, monitoring treatment outcomes, and ensuring compliance with forthcoming requirements that state Medicaid programs cover anti-obesity medications for individuals with obesity and cardiovascular disease, although step-therapy constraints may still delay access [182,246]. Finally, even under optimistic manufacturing assumptions, current global manufacturing capacity is expected to reach only a small fraction—on the order of 10%—of the global population living with obesity, even with aggressive scale-up [239].
These policy strategies highlight that expanding access to GLP-1-based obesity treatments will require coordinated legislative, economic, and health-system reforms rather than price reductions alone. Addressing insurance exclusions, affordability, administrative burden, and structural inequities is essential to ensure that the clinical benefits of these therapies translate into population-level impact.
11. Real-World Effectiveness and Adherence
11.1. Real-World Weight Loss (Tirzepatide −21.8% vs. Semaglutide −15.4%)
Real-world evidence indicates that incretin-based therapies achieve clinically meaningful weight loss in routine practice, though the magnitude of benefit is consistently attenuated relative to randomized clinical trials. This attenuation likely reflects real-world factors such as variable dosing, treatment interruptions, and early discontinuation. Nevertheless, across diverse observational datasets, dual GIP/GLP-1 receptor agonism with tirzepatide demonstrates superior real-world weight reduction compared with GLP-1 receptor agonist monotherapy [98,260].
A large comparative effectiveness analysis conducted by Rodriguez et al. used U.S. electronic health record data linked to pharmacy dispensing records between May 2022 and September 2023 [98]. This study included adults with overweight or obesity initiating tirzepatide or semaglutide and employed propensity score matching to balance baseline characteristics (18,386 matched patients; mean baseline weight approximately 110 kg; 52% with T2DM. Treatment discontinuation was common, with follow-up ending in 55.9% of tirzepatide users and 52.5% of semaglutide users within one year. Despite these real-world constraints, tirzepatide was associated with significantly greater weight loss across clinically meaningful thresholds. Compared with semaglutide, tirzepatide users were more likely to achieve at least 5%, 10%, and 15% weight loss, with hazard ratios of 1.76, 2.54, and 3.24, respectively. Absolute on-treatment weight loss also favored tirzepatide at three, six, and 12 months, with between-group differences of −2.4%, −4.3%, and −6.9%. Rates of gastrointestinal adverse events were similar between groups, suggesting that improved effectiveness was not offset by reduced tolerability [98].
Complementary findings have been reported in administrative claims analyses focused on populations with T2DM. In a large U.S. claims-based study with linked laboratory and electronic health record data, Hoog et al. evaluated adults initiating tirzepatide or injectable semaglutide and assessed weight change over 12 months among patients with available paired measurements [260]. Tirzepatide was associated with greater mean weight reductions than semaglutide in both GLP-1 receptor agonist-naïve patients (−10.2 kg vs. −6.1 kg) and GLP-1-experienced patients (−7.9 kg vs. −3.7 kg). These results reinforce that the relative advantage of dual GIP/GLP-1 receptor agonism observed in randomized trials extends into real-world clinical practice, even in populations with prior exposure to incretin therapy [260].
Collectively, these data demonstrate a consistent effectiveness gradient favoring tirzepatide over GLP-1 receptor agonist monotherapy in routine care. However, the substantial attrition observed across real-world cohorts underscores a persistent efficacy effectiveness gap. High discontinuation rates, incomplete dose escalation, and variable adherence substantially limit the population-level impact of these therapies, highlighting the importance of implementation strategies that improve persistence and optimize dosing to better translate clinical trial efficacy into real-world benefit.
11.2. Persistence Rates
Real-world persistence with GLP-1-based therapies is substantially lower than in clinical trials, with multiple population-based studies in obesity consistently demonstrating 12-month persistence of approximately 32–46%. In a large U.S. claims analysis of adults with obesity, only 32% of individuals who initiated a GLP-1 RA remained on therapy at one year [33]. Similar discontinuation patterns were observed across commercial and Medicare cohorts, where persistence at 12 months ranged from 35–46%, with higher persistence observed among those achieving consistent dose escalation [158]. European registry-based analyses corroborate these findings; in routine clinical practice, only about half of semaglutide initiators for obesity remained on treatment after one year, despite widespread prescribing and guideline uptake [261].
Individualized titration schedules—with slower escalation or temporary dose holds—reduce gastrointestinal intolerance and improved likelihood of treatment persistence. Across U.S. health-system data, fewer than 10–15% of semaglutide users reached the recommended therapeutic dose within the expected timeframe, and many remained on subtherapeutic doses for extended periods, which was associated with higher rates of discontinuation [158]. It is important to note that persistence improves when prescriptions are continuously covered without lapses. Higher out-of-pocket costs and insurance instability strongly predict early discontinuation and lower likelihood of re-initiation. Re-initiation analyses similarly show that lapses in treatment are common; approximately 30–40% of discontinued users restart therapy within 12 months, often following insurance approval changes or weight regain [156].
Population-level utilization analyses further indicate that continuous use declines sharply over the first 6–9 months, and fewer than half of individuals prescribed GLP-1-based therapies maintain therapy over 12 months [156,157,159,262]. Across datasets, persistence remains consistently low regardless of whether treatment is initiated for T2DM or obesity, underscoring a substantial gap between clinical trial efficacy and real-world treatment durability.
Finally, real-world users often report mismatched expectations regarding onset and magnitude of weight loss, leading some to discontinue prematurely when early results fall short of marketing-driven expectations. Patterns of discontinuation followed by reinitiation—frequently triggered by weight regain—highlight the need for more structured counseling around chronic therapy expectations [95,156].
11.3. Barriers to Adherence
As showcased throughout this review, multiple structural, clinical, and behavioral factors contribute to suboptimal adherence and early discontinuation of GLP-1-based therapies in routine care. Cost and insurance instability represent the most prominent barriers. High out-of-pocket costs and lapses in coverage are consistently associated with early discontinuation, particularly among individuals from lower-income zip codes or those insured through high-deductible plans [261].
Geographic and socioeconomic disparities further influence access: clusters of markedly lower GLP-1 RA uptake have been identified in communities with limited insurance coverage, higher social vulnerability indices, and fewer obesity-specialty prescribers [263].
Gastrointestinal adverse effects, including nausea, vomiting, and early satiety, represent another major driver of early discontinuation. Beyond these GI symptoms, gastroparesis-like syndromes have been reported in association with GLP-1 RA use [264], contributing to a broader spectrum of gastrointestinal intolerance. In a population-based observational study using the PharMetrics Plus for Academics database, semaglutide and liraglutide were associated with an increased relative risk of gastroparesis (adjusted HR 3.67; 95% CI 1.15–11.90) and bowel obstruction (adjusted HR 4.22; 95% CI 1.02–17.40) compared with bupropion-naltrexone, a non-GLP-1 anti-obesity therapy. However, absolute event rates were low (7.3–9.1 events per 1000 person-years) and causal inference was limited by the observational study design [265]. These effects often emerge during dose escalation, are compounded by inconsistent titration practices, with variable resolution across patients; real-world data demonstrate that many patients titrate more quickly than recommended, while others maintain low doses that compromise efficacy and reduce motivation to continue therapy [158]. The frequency of dose interruptions, delays in refilling prescriptions, and deviations from recommended uptitration schedules collectively contribute to lower adherence.
Clinical inertia and systems-level barriers also play significant roles. Limited follow-up visits, inadequate counseling on expected side effects, and variable prescribing practices contribute to inconsistent medication use across health systems. Observational studies indicate that older age, mental health comorbidities, and multiple chronic conditions further reduce the likelihood of persistent use [261].
Finally, real-world users often report mismatched expectations regarding onset and degree of weight loss, leading some to discontinue prematurely when early results fall short of marketing-driven expectations. Patterns of discontinuation followed by reinitiation—frequently triggered by weight regain—highlight the need for more structured counseling around chronic therapy expectations [156].
11.4. Adherence Improvement Strategies
Investigation of strategies that may increase or reduce patient persistence will be crucial to widespread adherence improvement efforts. Notably, reductions in common adverse GI effects have been noted to bolster patient retention [158]. Real-world titration is highly variable, with a substantial proportion of patients either escalating more quickly than recommended or remaining on subtherapeutic doses, both of which are associated with lower long-term persistence. The method most frequently employed by physicians to help mitigate these complications is dose escalation, whereby the patient begins therapy on the lowest available dose and over time escalates to the therapeutic dose. Additionally, efforts to reduce cost burden are of paramount importance to adherence. A large 2025 cohort study examined the effects of increased cost sharing on adherence to prescribed GLP-1 regimens and found an inverse relationship between patient persistence and greater out of pocket drug costs [266]. Lastly, there are currently very few large, adherence-intervention based trials to better quantify the effects of such strategies on compliance. However, smaller pilot trials on overall medication management in T2DM suggest the benefits of patient education, consistent follow-up visits, and digital coaching as possible viable ways to improve likelihood of continued use [267,268].
12. Emerging Therapies and Future Directions
12.1. Oral Formulations (Semaglutide 25/50 mg-2025, Orforglipron Approval ~2026)
Future incretin-based therapy development aims to meet gaps in adherence, side effect profiles, and metabolic effectiveness. Emerging options include oral formulations, monthly injectables, triple-agonist therapies, and novel combinations such as GLP-1/amylin receptor agonists. Oral treatments may improve patient adherence, psychological barriers to treatment entry, and global accessibility concerns. In PIONEER 1, a 26-week phase 3b trial examining the effectiveness of oral semaglutide versus placebo in T2DM patients, significant reductions in both A1C and body weight were found at the highest dose [269]. Similar findings occurred in PIONEER 3, which showed greater effectiveness on HbA1C and body weight with oral semaglutide versus sitagliptin in patients whose T2DM was uncontrolled on metformin with or without sulfonylurea [270]. In a landmark study involving patients with obesity without T2DM, the OASIS-4 trial indicated a body weight change of −13.6% on 25 mg oral semaglutide versus −2.2% on placebo after 64 weeks [271]. Oral semaglutide is currently approved for both T2DM and weight management. Together, these findings highlight the clinical utility of orally administered incretin therapy.
One limitation is that orally administered peptides require careful timing of administration relative to meals. Studies have shown limited to no systemic semaglutide exposure if consumed in the fed state, with a fasting period of at least 30 min needed to achieve adequate absorption [272,273]. Orforglipron, a small-molecule non-peptide GLP-1 RA, demonstrated a body weight change of −11.2% on the highest dose of the drug relative to −2.1% with placebo [274]. Early data from the ACHIEVE-3 trial suggests that orforglipron may achieve greater weight loss and glycemic control than oral semaglutide, although full trial data is pending publication [275]. Early data suggest orforglipron absorption may be less dependent on fasting than peptide-based orals; these findings are based on phase 2 trials, with confirmation expected from phase 3 data [276].
12.2. Monthly Injectables (MariTide Phase 3)
Other options for increasing patient adherence include adjustments to dose frequency. While most subcutaneous treatments are currently dosed once weekly, monthly dosing options are an evolving area of study. A double blind randomized controlled trial of once-monthly maridebart cafraglutide, an investigational GLP-1 agonist currently undergoing phase 3 trials, showed significant weight reduction versus placebo in patients with obesity with or without T2DM [29]. Similarly, monthly administration of efpeglenatide, another investigational incretin-based therapy, resulted in statistically significant body weight and HbA1C reductions versus placebo in patients with T2DM uncontrolled on metformin. Efpeglenatide has a unique peptide conjugation structure that allows for its longer half-life and potential for biweekly or once-monthly dosing [277]. Although its biologic properties are promising, early studies suggest monthly regimens may achieve comparable weight reduction; effects on glycemic control relative to weekly dosing require further study [278]. Other candidates for long-acting injectables are currently in development. Met097i, which has a half-life of 15 days, nearly doubling that of most existing GLP1-agonists, is in early-stage clinical development currently, with promising preliminary results for weight loss and tolerability [279]. As there are currently few studies on the efficacy and tolerability of monthly dosing strategies, future work should seek to examine which existing incretins may be suitable for extended dosing regimens as-is, or given the addition of unique conjugation structures to lengthen drug half-lives.
12.3. Triple Agonists (Retatrutide Phase 3, Approval 2027–2030)
Triple agonists are another promising frontier within incretin treatments. Retatrutide, which combines GLP-1, GIP and glucagon agonism, has demonstrated powerful weight loss results rivaling that of bariatric surgery [280]. In a 48-week phase 2 trial, approximately one fourth of participants lost 30% or more of their body weight while on the highest dose of the drug, with a mean reduction of 24.2% among all participants [18]. Safety profiles were overall similar to dual agonist GLP-1/GIP therapies [18]. As many participants continued to lose weight throughout the conclusion of the phase 2 retatrutide trial, TRIUMPH phase 3 trials are ongoing to determine the safety and maximal efficacy of retatrutide [281]. While these results are highly promising, final approval of the drug is still underway. Other GLP-1/GIP/glucagon agonist therapies such as SAR441255 and HM15211 are currently in phase 1 and 2 clinical trials, respectively [282,283]. Given the possibility of rapid weight loss due to this drug class, future studies should prioritize investigating the rate of lean muscle versus fat loss. It has been posited that glucagon signaling may increase lipid oxidation therefore leading to a greater relative fat mass loss. Phase 2 data include body-composition measures suggesting proportionate lean/fat loss similar to other incretin therapies; additional detailed analyses are ongoing to further investigate these mechanistic possibilities [102]. Preclinical models raise questions about glucagon-related effects on protein turnover, but human data are insufficient to determine clinical relevance [284,285]. As more diverse triple-agonist therapies are developed, future studies should seek to consistently include metrics such as DEXA or MRI-based body composition scans, muscle strength and functional endpoints.
12.4. Novel Combinations (Amycretin 24% Weight Loss, Cagrisema, Bioglutide)
In addition to triple agonists, other combination therapies such as CagriSema, which pairs semaglutide with cagrilintide, a long-acting amylin analogue, are being evaluated for approval in phase 3 trials. In a 20-week phase 1b trial, mean weight loss from the drug was estimated at 17.1%, compared to a 12.4% mean loss from semaglutide 2.4 mg alone in the STEP 1 trial [286]. Similarly, amycretin, which is a unimolecular agonist of GLP-1, amylin and calcitonin receptors, has demonstrated up to 24.3% weight loss at 36 weeks versus 1.1% with placebo [30]. These findings are derived from early-stage trials as the drug has not yet been approved. Durability and body-composition effects remain under investigation. A key goal of ongoing drug development is the addition of mechanisms that may be protective of lean muscle mass during periods of rapid weight loss. NA-931 (Bioglutide), an investigational quadruple GLP-1, GIP, glucagon and IGF-1 agonist is in phase 2b/3 trials currently to assess its effectiveness alone or in combination with tirzepatide [32]. Another first-in-class GLP-1 and IGF-1 dual agonist therapy, NA-941, is being evaluated in phase 2 trials currently. IGF-1 agonism is hypothesized to support muscle metabolism, but clinical evidence confirming preservation of lean mass is currently limited [287].
12.5. Expanding Indications (MASH, Alzheimer’s, CKD, OSA)
Outside of weight management, the potential future indications for GLP-1 agonists continue to expand rapidly. Many of these use cases relate to the sequelae of metabolic syndrome. In a landmark trial examining the effect of subcutaneous semaglutide on metabolic dysfunction associated steatohepatitis (MASH), significantly higher resolution rates were achieved with semaglutide 0.4 mg versus placebo [288]. Although semaglutide improved steatohepatitis resolution in trials, antifibrotic benefit has not yet been demonstrated. In chronic kidney disease (CKD), the FLOW phase 3 trial tested 1.0 mg semaglutide and found a reduced risk of clinically meaningful renal events such as transplant or initiation of long-term dialysis. A decreased risk of cardiovascular death was also observed [124]. Semaglutide is currently approved to treat both MASH and CKD. Conditions that are commonly found to co-occur with obesity, such as obstructive sleep apnea, have also shown improvement with incretin therapy. The SURMOUNT-OSA trial demonstrated clinically significant reductions in the apnea-hypoxia index, hypoxic burden, and subjective patient-reported sleep quality measures [289]. Weight loss likely mediates much of the benefit. Ongoing trials are underway, and tirzepatide has not been formally approved to treat OSA. As GLP-1 receptors are expressed throughout the brain in regions including the brainstem, hypothalamus, amygdala and hippocampus, the potential for neurological implications is broad. Small trials including a pilot study of liraglutide have demonstrated improvements in Alzheimer’s disease benchmarks, such as a preservation of brain glucose metabolism [290]. However, topline manufacturer-published results from a larger phase 3 trial EVOKE which did not meet primary cognitive endpoints versus placebo. Final publication of these larger trials’ results are pending [291]. Another brain-based implication for incretin therapy is addiction medicine. One study examining the effect of semaglutide on alcohol consumption in rats found a greater than 50% reduction in intake, with correlated declines in addiction-related brain regions such as the nucleus accumbens [292]. Human observational studies have shown correlations between GLP-1 agonist use and reduced recurrence of alcohol use disorder [293]. Randomized placebo controlled studies are warranted to further investigate these potential effects on alcohol consumption patterns. While these results are highly promising, at this time no GLP-1 RA has been formally approved to treat neurologic or psychiatric diagnoses.
12.6. Future Research Priorities
As the implications and potential benefits of GLP-1 agonist use are far reaching, future study should aim to investigate which specific therapies are ideal for each patient population based on factors such as age, comorbid conditions, health weight loss goals, and lifestyle preferences. As more data emerge on these questions, patients and clinicians will be empowered to make sound decisions on which incretin therapy offers the greatest benefit at the lowest chance of nonresponse, adverse effects or suboptimal adherence. Importantly, as 12-month discontinuation rates have been quoted as high as 50% or more in some investigations, future drug development should aim to ameliorate side effects that commonly lead to discontinuation, such as GI distress [294]. Lastly, as the incretin therapy market becomes increasingly competitive due to demand and expanding use cases, strategies to improve patient ease of use, such as longer dosing intervals, should be prioritized.
13. Conclusions
Incretin-based therapies have fundamentally redefined medical management of obesity and T2DM, establishing a clear efficacy gradient grounded in receptor biology. Among GLP-1 receptor agonists, semaglutide remains the most potent single agonist, while tirzepatide’s dual GIP/GLP-1 activation consistently achieves greater reductions in weight and HbA1c in trials and real-world cohorts. Next-generation triple agonists such as retatrutide may approach or surpass outcomes of metabolic surgery for responders, marking the next evolution in pharmacologic treatment.
The therapeutic armamentarium is rapidly expanding. The first oral GLP-1 agonist for obesity was recently approved, improving accessibility for patients who decline injections, and long-acting injectable formulations with monthly or extended dosing intervals are in development, aiming to reduce treatment burden and improve persistence. Beyond metabolic control, GLP-1-based therapies are emerging as targeted treatments for cardiometabolic disease more broadly: multiple GLP-1 receptor agonists reduce major adverse cardiovascular events and slow kidney disease progression, and early evidence shows clinically meaningful improvements in obesity-related conditions such as obstructive sleep apnea, with ongoing investigations into MASH, neurodegenerative diseases, and addiction medicine.
Safety profiles remain favorable overall, dominated by dose-dependent gastrointestinal effects that can be mitigated through gradual titration and anticipatory counseling. Early treatment response reliably identifies non-responders who may benefit from dose escalation, switching agents, or combination therapy. While genetic predictors of treatment response show promise, pharmacogenomic testing remains investigational and is not yet incorporated into routine therapeutic decision-making.
Despite transformative efficacy, translation into population-level benefit remains constrained by cost, limited coverage, administrative burden, and discontinuation rates exceeding 50% in one year. Closing this efficacy–effectiveness gap will require coordinated structural solutions: expanded insurance coverage, price reductions through negotiation and generic entry, equitable delivery models, flexible titration protocols, and structured support to sustain adherence. With broader access and improved implementation, incretin-based therapies have the potential not only to treat obesity and type 2 diabetes at scale, but also to reduce downstream diseases such as cardiovascular disease, kidney failure, and obstructive sleep apnea—shifting the trajectory of population health in the coming decade.
Author Contributions
Conceptualization, D.P., R.H., V.G. and M.E.; Methodology, D.P., O.S., A.M., A.A., M.E.M., R.H., V.G. and M.E.; Investigation, D.P., O.S., A.M., A.A., M.E.M., R.H., D.D., V.G. and M.E.; Writing—Original Draft, D.P., O.S., A.M., A.A., M.E.M., R.H., D.D., V.G. and M.E.; Writing—Review & Editing, D.P., O.S., A.M., A.A., M.E.M., R.H., D.D., V.G. and M.E.; Supervision, R.H., V.G. and M.E. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the National Institutes of Health (HL139793, HL168056 and TR004478).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef]
- Phelps, N.H.; Singleton, R.K.; Zhou, B.; Heap, R.A.; Mishra, A.; Bennett, J.E.; Paciorek, C.J.; Lhoste, V.P.; Carrillo-Larco, R.M.; Stevens, G.A.; et al. Worldwide trends in underweight and obesity from 1990 to 2022: A pooled analysis of 3663 population-representative studies with 222 million children, adolescents, and adults. Lancet 2024, 403, 1027–1050. [Google Scholar] [CrossRef]
- Grant, B.; Sandelson, M.; Agyemang-Prempeh, B.; Zalin, A. Managing obesity in people with type 2 diabetes. Clin. Med. 2021, 21, e327–e331. [Google Scholar] [CrossRef]
- Cawley, J.; Biener, A.; Meyerhoefer, C.; Ding, Y.; Zvenyach, T.; Smolarz, B.G.; Ramasamy, A. Direct medical costs of obesity in the United States and the most populous states. J. Manag. Care Spec. Pharm. 2021, 27, 354–366. [Google Scholar] [CrossRef]
- Mcintyre, N.; Holdsworth, C.D.; Turner, D.S. New Interpretation Of Oral Glucose Tolerance. Lancet 1964, 2, 20–21. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Ratner, R.E.; Han, J.; Kim, D.D.; Fineman, M.S.; Baron, A.D. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care 2005, 28, 1092–1100. [Google Scholar] [CrossRef]
- Buse, J.B.; Henry, R.R.; Han, J.; Kim, D.D.; Fineman, M.S.; Baron, A.D.; Exenatide-113 Clinical Study Group. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in sulfonylurea-treated patients with type 2 diabetes. Diabetes Care 2004, 27, 2628–2635. [Google Scholar] [CrossRef] [PubMed]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.E.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Colhoun, H.M.; Dagenais, G.R.; Diaz, R.; Lakshmanan, M.; Pais, P.; Probstfield, J.; Riesmeyer, J.S.; Riddle, M.C.; Rydén, L.; et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): A double-blind, randomised placebo-controlled trial. Lancet 2019, 394, 121–130. [Google Scholar] [CrossRef]
- Kristensen, S.L.; Rørth, R.; Jhund, P.S.; Docherty, K.F.; Sattar, N.; Preiss, D.; Køber, L.; Petrie, M.C.; McMurray, J.J.V. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol. 2019, 7, 776–785. [Google Scholar] [CrossRef] [PubMed]
- Wilding, J.P.H.; Batterham, R.L.; Calanna, S.; Davies, M.; Van Gaal, L.F.; Lingvay, I.; McGowan, B.M.; Rosenstock, J.; Tran, M.T.D.; Wadden, T.A.; et al. Once-Weekly Semaglutide in Adults with Overweight or Obesity. N. Engl. J. Med. 2021, 384, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- Aronne, L.J.; Horn, D.B.; le Roux, C.W.; Ho, W.; Falcon, B.L.; Gomez Valderas, E.; Das, S.; Lee, C.J.; Glass, L.C.; Senyucel, C.; et al. Tirzepatide as Compared with Semaglutide for the Treatment of Obesity. N. Engl. J. Med. 2025, 393, 26–36. [Google Scholar] [CrossRef]
- Dahl, D.; Onishi, Y.; Norwood, P.; Huh, R.; Bray, R.; Patel, H.; Rodríguez, Á. Effect of Subcutaneous Tirzepatide vs Placebo Added to Titrated Insulin Glargine on Glycemic Control in Patients with Type 2 Diabetes: The SURPASS-5 Randomized Clinical Trial. J. Am. Med. Assoc. 2022, 327, 534–545. [Google Scholar] [CrossRef]
- Frías, J.P.; Auerbach, P.; Bajaj, H.S.; Fukushima, Y.; Lingvay, I.; Macura, S.; Søndergaard, A.L.; Tankova, T.I.; Tentolouris, N.; Buse, J.B. Efficacy and safety of once-weekly semaglutide 2·0 mg versus 1·0 mg in patients with type 2 diabetes (SUSTAIN FORTE): A double-blind, randomised, phase 3B trial. Lancet Diabetes Endocrinol. 2021, 9, 563–574. [Google Scholar] [CrossRef]
- Jastreboff, A.M.; Aronne, L.J.; Ahmad, N.N.; Wharton, S.; Connery, L.; Alves, B.; Kiyosue, A.; Zhang, S.; Liu, B.; Bunck, M.C.; et al. Tirzepatide Once Weekly for the Treatment of Obesity. N. Engl. J. Med. 2022, 387, 205–216. [Google Scholar] [CrossRef]
- Rosenstock, J.; Wysham, C.; Frías, J.P.; Kaneko, S.; Lee, C.J.; Fernández Landó, L.; Mao, H.; Cui, X.; Karanikas, C.A.; Thieu, V.T. Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): A double-blind, randomised, phase 3 trial. Lancet 2021, 398, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Jastreboff, A.M.; Kaplan, L.M.; Frías, J.P.; Wu, Q.; Du, Y.; Gurbuz, S.; Coskun, T.; Haupt, A.; Milicevic, Z.; Hartman, M.L. Triple–Hormone-Receptor Agonist Retatrutide for Obesity—A Phase 2 Trial. N. Engl. J. Med. 2023, 389, 514–526. [Google Scholar] [CrossRef]
- Bridges, A.; Bistas, K.G.; Jacobs, T.F. Exenatide. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: http://www.ncbi.nlm.nih.gov/books/NBK518981/ (accessed on 27 December 2025).
- Cerillo, J.L.; Parmar, M. Liraglutide. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: http://www.ncbi.nlm.nih.gov/books/NBK608007/ (accessed on 27 December 2025).
- Pfeffer, M.A.; Claggett, B.; Diaz, R.; Dickstein, K.; Gerstein, H.C.; Køber, L.V.; Lawson, F.C.; Ping, L.; Wei, X.; Lewis, E.F.; et al. Lixisenatide in Patients with Type 2 Diabetes and Acute Coronary Syndrome. N. Engl. J. Med. 2015, 373, 2247–2257. [Google Scholar] [CrossRef]
- Collins, L.; Costello, R.A. Glucagon-Like Peptide-1 Receptor Agonists. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: http://www.ncbi.nlm.nih.gov/books/NBK551568/ (accessed on 27 December 2025).
- Smith, L.L.; Mosley, J.F.; Parke, C.; Brown, J.; Barris, L.S.; Phan, L.D. Dulaglutide (Trulicity): The Third Once-Weekly GLP-1 Agonist. Pharm. Ther. 2016, 41, 357–360. [Google Scholar]
- Kommu, S.; Whitfield, P. Semaglutide. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: http://www.ncbi.nlm.nih.gov/books/NBK603723/ (accessed on 27 December 2025).
- Gerstein, H.C.; Sattar, N.; Rosenstock, J.; Ramasundarahettige, C.; Pratley, R.; Lopes, R.D.; Lam, C.S.P.; Khurmi, N.S.; Heenan, L.; Del Prato, S.; et al. Cardiovascular and Renal Outcomes with Efpeglenatide in Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Farzam, K.; Patel, P. Tirzepatide. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: http://www.ncbi.nlm.nih.gov/books/NBK585056/ (accessed on 27 December 2025).
- Davies, M.J.; Bajaj, H.S.; Broholm, C.; Eliasen, A.; Garvey, W.T.; le Roux, C.W.; Lingvay, I.; Lyndgaard, C.B.; Rosenstock, J.; Pedersen, S.D. Cagrilintide–Semaglutide in Adults with Overweight or Obesity and Type 2 Diabetes. N. Engl. J. Med. 2025, 393, 648–659. [Google Scholar] [CrossRef]
- Wharton, S.; le Roux, C.W.; Kosiborod, M.N.; Platz, E.; Brueckmann, M.; Jastreboff, A.M.; Ajaz Hussain, S.; Pedersen, S.D.; Borowska, L.; Unseld, A.; et al. Survodutide for treatment of obesity: Rationale and design of two randomized phase 3 clinical trials (SYNCHRONIZETM-1 and -2). Obesity 2025, 33, 67–77. [Google Scholar] [CrossRef]
- Jastreboff, A.M.; Ryan, D.H.; Bays, H.E.; Ebeling, P.R.; Mackowski, M.G.; Philipose, N.; Ross, L.; Liu, Y.; Burns, C.E.; Abbasi, S.A.; et al. Once-Monthly Maridebart Cafraglutide for the Treatment of Obesity—A Phase 2 Trial. N. Engl. J. Med. 2025, 393, 843–857. [Google Scholar] [CrossRef]
- Dahl, K.; Toubro, S.; Dey, S.; Duque do Vale, R.; Flint, A.; Gasiorek, A.; Heydorn, A.; Jastreboff, A.M.; Key, C.; Petersen, S.B.; et al. Amycretin, a novel, unimolecular GLP-1 and amylin receptor agonist administered subcutaneously: Results from a phase 1b/2a randomised controlled study. Lancet 2025, 406, 149–162. [Google Scholar] [CrossRef]
- Lilly’s Triple Agonist, Retatrutide, Delivered Weight Loss of Up to An Average of 71.2 lbs Along with Substantial Relief from Osteoarthritis Pain in First Successful Phase 3 Trial|Eli Lilly and Company. Available online: https://investor.lilly.com/news-releases/news-release-details/lillys-triple-agonist-retatrutide-delivered-weight-loss-average (accessed on 27 December 2025).
- Biomed Industries, Inc. A Randomized, Double-Blind, Placebo-Controlled Multi-Center Study of Oral NA-931, Alone or in Addition to Open Label Subcutaneous Tirzepatide, to Investigate the Efficacy and Safety in Overweight or Obese Men and Women. Clinicaltrials.gov, Clinical Trial Registration NCT06732245. December 2024. Available online: https://clinicaltrials.gov/study/NCT06732245 (accessed on 27 December 2025).
- Gleason, P.P.; Urick, B.Y.; Marshall, L.Z.; Friedlander, N.; Qiu, Y.; Leslie, R.S. Real-world persistence and adherence to glucagon-like peptide-1 receptor agonists among obese commercially insured adults without diabetes. J. Manag. Care Spec. Pharm. 2024, 30, 860–867. [Google Scholar] [CrossRef]
- Novo Nordisk. News Details. Available online: https://www.novonordisk.com/content/nncorp/global/en/news-and-media/news-and-ir-materials/news-details.html (accessed on 27 December 2025).
- Marzook, A.; Tomas, A.; Jones, B. The Interplay of Glucagon-Like Peptide-1 Receptor Trafficking and Signalling in Pancreatic Beta Cells. Front. Endocrinol. 2021, 12, 678055. [Google Scholar] [CrossRef]
- Al Qassab, M.; Mneimneh, M.; Jradi, A.; Derbas, B.; Dabboussi, D.; Khoury Baini, J.; Katrib, N.; Chaarani, N.; Attieh, P.; Kanaan, A.; et al. The Expanding Role of GLP-1 Receptor Agonists: Advancing Clinical Outcomes in Metabolic and Mental Health. Curr. Issues Mol. Biol. 2025, 47, 285. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E. Targeting the GIPR for obesity: To agonize or antagonize? Potential mechanisms. Mol. Metab. 2021, 46, 101139. [Google Scholar] [CrossRef]
- Samms, R.J.; Sloop, K.W.; Gribble, F.M.; Reimann, F.; Adriaenssens, A.E. GIPR Function in the Central Nervous System: Implications and Novel Perspectives for GIP-Based Therapies in Treating Metabolic Disorders. Diabetes 2021, 70, 1938–1944. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Dufour, S.; Mehal, W.Z.; Shulman, G.I. Glucagon promotes increased hepatic mitochondrial oxidation and pyruvate carboxylase flux in humans with fatty liver disease. Cell Metab. 2024, 36, 2359–2366.e3. [Google Scholar] [CrossRef]
- Liu, Z.; Yu, S.; Jin, X.; Sheng, L.; YanMu, M.R.; Gao, J.; Lu, J.; Lei, T. The Clinical Application of GLP-1RAs and GLP-1/GIP Dual Receptor Agonists Based on Pharmacological Mechanisms: A Review. Drug Des. Devel Ther. 2025, 19, 10383–10409. [Google Scholar] [CrossRef] [PubMed]
- Goldney, J.; Hamza, M.; Surti, F.; Davies, M.J.; Papamargaritis, D. Triple Agonism Based Therapies for Obesity. Curr. Cardiovasc. Risk Rep. 2025, 19, 18. [Google Scholar] [CrossRef]
- Sharma, D.; Verma, S.; Vaidya, S.; Kalia, K.; Tiwari, V. Recent updates on GLP-1 agonists: Current advancements & challenges. Biomed. Pharmacother. 2018, 108, 952–962. [Google Scholar] [CrossRef]
- Tsimihodimos, V.; Elisaf, M. Effects of incretin-based therapies on renal function. Eur. J. Pharmacol. 2018, 818, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Min, T.; Bain, S.C. The Role of Tirzepatide, Dual GIP and GLP-1 Receptor Agonist, in the Management of Type 2 Diabetes: The SURPASS Clinical Trials. Diabetes Ther. 2021, 12, 143–157. [Google Scholar] [CrossRef]
- Dorsey-Trevino, E.G.; Kaur, V.; Mercader, J.M.; Florez, J.C.; Leong, A. Association of GLP1R Polymorphisms with the Incretin Response. J. Clin. Endocrinol. Metab. 2022, 107, 2580–2588. [Google Scholar] [CrossRef] [PubMed]
- Dawed, A.Y.; Mari, A.; McDonald, T.J.; Li, L.; Wang, S.; Hong, M.-G.; Sharma, S.; Robertson, N.R.; Mahajan, A.; Wang, X.; et al. Pharmacogenomics of GLP-1 Receptor Agonists: A genome wide analysis of observational data and large randomized controlled trials. medRxiv 2022. [Google Scholar] [CrossRef]
- Katz, G. Efficacy, Safety, and Future of GLP-1 Receptor Agonists: A Systematic Literature Review and Meta-Analysis. Horm. Metab. Res. 2025, 57, 326–337. [Google Scholar] [CrossRef]
- Sorli, C.; Harashima, S.-I.; Tsoukas, G.M.; Unger, J.; Karsbøl, J.D.; Hansen, T.; Bain, S.C. Efficacy and safety of once-weekly semaglutide monotherapy versus placebo in patients with type 2 diabetes (SUSTAIN 1): A double-blind, randomised, placebo-controlled, parallel-group, multinational, multicentre phase 3a trial. Lancet Diabetes Endocrinol. 2017, 5, 251–260. [Google Scholar] [CrossRef]
- DeSouza, C.; Cariou, B.; Garg, S.; Lausvig, N.; Navarria, A.; Fonseca, V. Efficacy and Safety of Semaglutide for Type 2 Diabetes by Race and Ethnicity: A Post Hoc Analysis of the SUSTAIN Trials. J. Clin. Endocrinol. Metab. 2020, 105, dgz072. [Google Scholar] [CrossRef] [PubMed]
- Aroda, V.R.; Capehorn, M.S.; Chaykin, L.; Frias, J.P.; Lausvig, N.L.; Macura, S.; Lüdemann, J.; Madsbad, S.; Rosenstock, J.; Tabak, O.; et al. Impact of baseline characteristics and beta-cell function on the efficacy and safety of subcutaneous once-weekly semaglutide: A patient-level, pooled analysis of the SUSTAIN 1-5 trials. Diabetes Obes. Metab. 2020, 22, 303–314. [Google Scholar] [CrossRef]
- Giorgino, F.; Benroubi, M.; Sun, J.-H.; Zimmermann, A.G.; Pechtner, V. Efficacy and Safety of Once-Weekly Dulaglutide Versus Insulin Glargine in Patients with Type 2 Diabetes on Metformin and Glimepiride (AWARD-2). Diabetes Care 2015, 38, 2241–2249. [Google Scholar] [CrossRef]
- Umpierrez, G.; Tofé Povedano, S.; Pérez Manghi, F.; Shurzinske, L.; Pechtner, V. Efficacy and safety of dulaglutide monotherapy versus metformin in type 2 diabetes in a randomized controlled trial (AWARD-3). Diabetes Care 2014, 37, 2168–2176. [Google Scholar] [CrossRef]
- Blonde, L.; Jendle, J.; Gross, J.; Woo, V.; Jiang, H.; Fahrbach, J.L.; Milicevic, Z. Once-weekly dulaglutide versus bedtime insulin glargine, both in combination with prandial insulin lispro, in patients with type 2 diabetes (AWARD-4): A randomised, open-label, phase 3, non-inferiority study. Lancet 2015, 385, 2057–2066. [Google Scholar] [CrossRef] [PubMed]
- Gallwitz, B.; Dagogo-Jack, S.; Thieu, V.; Garcia-Perez, L.-E.; Pavo, I.; Yu, M.; Robertson, K.E.; Zhang, N.; Giorgino, F. Effect of once-weekly dulaglutide on glycated haemoglobin (HbA1c) and fasting blood glucose in patient subpopulations by gender, duration of diabetes and baseline HbA1c. Diabetes Obes. Metab. 2018, 20, 409–418. [Google Scholar] [CrossRef]
- Davies, M.J.; Aroda, V.R.; Collins, B.S.; Gabbay, R.A.; Green, J.; Maruthur, N.M.; Rosas, S.E.; Del Prato, S.; Mathieu, C.; Mingrone, G.; et al. Management of Hyperglycemia in Type 2 Diabetes, 2022. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2022, 45, 2753–2786. [Google Scholar] [CrossRef]
- Frias, J.P.; Bonora, E.; Cox, D.A.; Bethel, M.A.; Kwan, A.Y.M.; Raha, S.; Malik, R.E. Glycaemic efficacy of an expanded dose range of dulaglutide according to baseline glycated haemoglobin (HbA1c) subgroup: Post hoc analysis of AWARD-11. Diabetes Obes. Metab. 2021, 23, 2819–2824. [Google Scholar] [CrossRef]
- Ren, X.; Hua, H.; Wu, Y.; Zhang, W.; Long, X.; Bai, Y.; Cheng, N. Efficacy and safety of GLP-1 agonists in the treatment of T2DM: A systematic review and network meta-analysis. Sci. Rep. 2025, 15, 24103. [Google Scholar] [CrossRef]
- Htike, Z.Z.; Zaccardi, F.; Papamargaritis, D.; Webb, D.R.; Khunti, K.; Davies, M.J. Efficacy and safety of glucagon-like peptide-1 receptor agonists in type 2 diabetes: A systematic review and mixed-treatment comparison analysis. Diabetes Obes. Metab. 2017, 19, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Madsbad, S. Review of head-to-head comparisons of glucagon-like peptide-1 receptor agonists. Diabetes Obes. Metab. 2016, 18, 317–332. [Google Scholar] [CrossRef]
- Alfaris, N.; Waldrop, S.; Johnson, V.; Boaventura, B.; Kendrick, K.; Stanford, F.C. GLP-1 single, dual, and triple receptor agonists for treating type 2 diabetes and obesity: A narrative review. eClinicalMedicine 2024, 75, 102782. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Exenatide extended-release: A review of its use in type 2 diabetes mellitus. Drugs 2012, 72, 1679–1707. [Google Scholar] [CrossRef] [PubMed]
- McCormack, P.L. Exenatide twice daily: A review of its use in the management of patients with type 2 diabetes mellitus. Drugs 2014, 74, 325–351. [Google Scholar] [CrossRef]
- Pratley, R.E.; Aroda, V.R.; Lingvay, I.; Lüdemann, J.; Andreassen, C.; Navarria, A.; Viljoen, A.; SUSTAIN 7 Investigators. Semaglutide versus dulaglutide once weekly in patients with type 2 diabetes (SUSTAIN 7): A randomised, open-label, phase 3b trial. Lancet Diabetes Endocrinol. 2018, 6, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Patoulias, D.; Popovic, D.S.; Stoian, A.P.; Janez, A.; Sahebkar, A.; Rizzo, M. Effect of semaglutide versus other glucagon-like peptide-1 receptor agonists on cardio-metabolic risk factors in patients with type 2 diabetes: A systematic review and meta-analysis of head-to-head, phase 3, randomized controlled trials. J. Diabetes Complicat. 2023, 37, 108529. [Google Scholar] [CrossRef]
- Lingvay, I.; Bauer, R.; Baker-Knight, J.; Lawson, J.; Pratley, R. An Indirect Treatment Comparison of Semaglutide 2.0 mg vs Dulaglutide 3.0 mg and 4.5 mg Using Multilevel Network Meta-regression. J. Clin. Endocrinol. Metab. 2022, 107, 1461–1469. [Google Scholar] [CrossRef]
- Xie, Z.; Gan, B.; Cao, W.; Liu, J.; Chen, J. Semaglutide Versus Dulaglutide and Liraglutide in Chinese Patients with T2DM: A Multicenter Real-World Study. Obesity 2025, 34, 101–113. [Google Scholar] [CrossRef]
- Huthmacher, J.A.; Meier, J.J.; Nauck, M.A. Efficacy and Safety of Short- and Long-Acting Glucagon-Like Peptide 1 Receptor Agonists on a Background of Basal Insulin in Type 2 Diabetes: A Meta-analysis. Diabetes Care 2020, 43, 2303–2312. [Google Scholar] [CrossRef]
- Lee, W.C.; Dekoven, M.; Bouchard, J.; Massoudi, M.; Langer, J. Improved real-world glycaemic outcomes with liraglutide versus other incretin-based therapies in type 2 diabetes. Diabetes Obes. Metab. 2014, 16, 819–826. [Google Scholar] [CrossRef]
- Morieri, M.L.; Rigato, M.; Frison, V.; Simioni, N.; D’Ambrosio, M.; Tadiotto, F.; Paccagnella, A.; Lapolla, A.; Avogaro, A.; Fadini, G.P. Effectiveness of dulaglutide vs liraglutide and exenatide once-weekly. A real-world study and meta-analysis of observational studies. Metabolism 2020, 106, 154190. [Google Scholar] [CrossRef]
- Tanaka, K.; Okada, Y.; Tokutsu, A.; Tanaka, Y. Real-world effectiveness of liraglutide versus dulaglutide in Japanese patients with type 2 diabetes: A retrospective study. Sci. Rep. 2022, 12, 154. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, J.; Vázquez, L.; Del Prato, S.; Franco, D.R.; Weerakkody, G.; Dai, B.; Landó, L.F.; Bergman, B.K.; Rodríguez, A. Achieving Normoglycemia with Tirzepatide: Analysis of SURPASS 1-4 Trials. Diabetes Care 2023, 46, 1986–1992. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Lee, B.W.; Chawla, M.; Kim, J.; Huo, L.; Du, L.; Huang, Y.; Ji, L. Tirzepatide versus insulin glargine as second-line or third-line therapy in type 2 diabetes in the Asia-Pacific region: The SURPASS-AP-Combo trial. Nat. Med. 2023, 29, 1500–1510. [Google Scholar] [CrossRef]
- Rosenstock, J.; Frías, J.P.; Rodbard, H.W.; Tofé, S.; Sears, E.; Huh, R.; Fernández Landó, L.; Patel, H. Tirzepatide vs Insulin Lispro Added to Basal Insulin in Type 2 Diabetes: The SURPASS-6 Randomized Clinical Trial. J. Am. Med. Assoc. 2023, 330, 1631–1640. [Google Scholar] [CrossRef] [PubMed]
- Frías, J.P.; Davies, M.J.; Rosenstock, J.; Pérez Manghi, F.C.; Fernández Landó, L.; Bergman, B.K.; Liu, B.; Cui, X.; Brown, K.; SURPASS-2 Investigators. Tirzepatide versus Semaglutide Once Weekly in Patients with Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 503–515. [Google Scholar] [CrossRef]
- Mather, K.J.; Mari, A.; Heise, T.; DeVries, J.H.; Hua, M.; Urva, S.; Coskun, T.; Haupt, A.; Heine, R.J.; Pratt, E.; et al. Effects of Tirzepatide vs Semaglutide on β-Cell Function, Insulin Sensitivity, and Glucose Control During a Meal Test. J. Clin. Endocrinol. Metab. 2024, 109, 3046–3054. [Google Scholar] [CrossRef]
- Fujihara, K.; Matsubayashi, Y.; Kitazawa, M.; Sato, T.; Takeuchi, M.; Oura, T.; Sone, H. Achieving normoglycaemia with tirzepatide: Post hoc exploratory analysis of the SURPASS J-mono and J-combo studies. Diabetes Obes. Metab. 2024, 26, 5304–5311. [Google Scholar] [CrossRef]
- Rosenstock, J.; Frias, J.; Jastreboff, A.M.; Du, Y.; Lou, J.; Gurbuz, S.; Thomas, M.K.; Hartman, M.L.; Haupt, A.; Milicevic, Z.; et al. Retatrutide, a GIP, GLP-1 and glucagon receptor agonist, for people with type 2 diabetes: A randomised, double-blind, placebo and active-controlled, parallel-group, phase 2 trial conducted in the USA. Lancet 2023, 402, 529–544. [Google Scholar] [CrossRef]
- Katsi, V.; Koutsopoulos, G.; Fragoulis, C.; Dimitriadis, K.; Tsioufis, K. Retatrutide-A Game Changer in Obesity Pharmacotherapy. Biomolecules 2025, 15, 796. [Google Scholar] [CrossRef]
- Gonzalez-Rellan, M.J.; Drucker, D.J. New Molecules and Indications for GLP-1 Medicines. J. Am. Med. Assoc. 2025, 334, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Blüher, M.; Rosenstock, J.; Hoefler, J.; Manuel, R.; Hennige, A.M. Dose-response effects on HbA1c and bodyweight reduction of survodutide, a dual glucagon/GLP-1 receptor agonist, compared with placebo and open-label semaglutide in people with type 2 diabetes: A randomised clinical trial. Diabetologia 2024, 67, 470–482. [Google Scholar] [CrossRef]
- Xiao, Y.-J.; Yu, S.; Zhang, Y.-L.; Chen, J.; Liu, Y.-Q.; Liu, X.-L.; Sun, C.-F.; Deng, C.-L. Efficacy and safety of survodutide on glycemic control and weight loss in adults: A systematic review and meta-analysis. Diabetes Obes. Metab. 2025, 27, 7062–7074. [Google Scholar] [CrossRef] [PubMed]
- Moiz, A.; Filion, K.B.; Toutounchi, H.; Tsoukas, M.A.; Yu, O.H.Y.; Peters, T.M.; Eisenberg, M.J. Efficacy and Safety of Glucagon-Like Peptide-1 Receptor Agonists for Weight Loss Among Adults Without Diabetes: A Systematic Review of Randomized Controlled Trials. Ann. Intern. Med. 2025, 178, 199–217. [Google Scholar] [CrossRef]
- Elmaleh-Sachs, A.; Schwartz, J.L.; Bramante, C.T.; Nicklas, J.M.; Gudzune, K.A.; Jay, M. Obesity Management in Adults: A Review. J. Am. Med. Assoc. 2023, 330, 2000–2015. [Google Scholar] [CrossRef]
- Wadden, T.A.; Bailey, T.S.; Billings, L.K.; Davies, M.; Frias, J.P.; Koroleva, A.; Lingvay, I.; O’Neil, P.M.; Rubino, D.M.; Skovgaard, D.; et al. Effect of Subcutaneous Semaglutide vs Placebo as an Adjunct to Intensive Behavioral Therapy on Body Weight in Adults with Overweight or Obesity: The STEP 3 Randomized Clinical Trial. J. Am. Med. Assoc. 2021, 325, 1403–1413. [Google Scholar] [CrossRef]
- Bergmann, N.C.; Davies, M.J.; Lingvay, I.; Knop, F.K. Semaglutide for the treatment of overweight and obesity: A review. Diabetes Obes. Metab. 2023, 25, 18–35. [Google Scholar] [CrossRef]
- Garvey, W.T.; Batterham, R.L.; Bhatta, M.; Buscemi, S.; Christensen, L.N.; Frias, J.P.; Jódar, E.; Kandler, K.; Rigas, G.; Wadden, T.A.; et al. Two-year effects of semaglutide in adults with overweight or obesity: The STEP 5 trial. Nat. Med. 2022, 28, 2083–2091. [Google Scholar] [CrossRef]
- Gudzune, K.A.; Kushner, R.F. Medications for Obesity: A Review. J. Am. Med. Assoc. 2024, 332, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.J.; Deedwania, P.; Acharya, T.; Aguilar, D.; Bhatt, D.L.; Chyun, D.A.; Di Palo, K.E.; Golden, S.H.; Sperling, L.S.; American Heart Association Diabetes Committee of the Council on Lifestyle and Cardiometabolic Health; et al. Comprehensive Management of Cardiovascular Risk Factors for Adults with Type 2 Diabetes: A Scientific Statement From the American Heart Association. Circulation 2022, 145, e722–e759. [Google Scholar] [CrossRef]
- Grunvald, E.; Shah, R.; Hernaez, R.; Chandar, A.K.; Pickett-Blakely, O.; Teigen, L.M.; Harindhanavudhi, T.; Sultan, S.; Singh, S.; Davitkov, P.; et al. AGA Clinical Practice Guideline on Pharmacological Interventions for Adults with Obesity. Gastroenterology 2022, 163, 1198–1225. [Google Scholar] [CrossRef]
- Rubino, D.M.; Greenway, F.L.; Khalid, U.; O’Neil, P.M.; Rosenstock, J.; Sørrig, R.; Wadden, T.A.; Wizert, A.; Garvey, W.T.; STEP 8 Investigators. Effect of Weekly Subcutaneous Semaglutide vs Daily Liraglutide on Body Weight in Adults With Overweight or Obesity Without Diabetes: The STEP 8 Randomized Clinical Trial. J. Am. Med. Assoc. 2022, 327, 138–150. [Google Scholar] [CrossRef]
- Pi-Sunyer, X.; Astrup, A.; Fujioka, K.; Greenway, F.; Halpern, A.; Krempf, M.; Lau, D.C.W.; le Roux, C.W.; Violante Ortiz, R.; Jensen, C.B.; et al. A Randomized, Controlled Trial of 3.0 mg of Liraglutide in Weight Management. N. Engl. J. Med. 2015, 373, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; Bergenstal, R.; Bode, B.; Kushner, R.F.; Lewin, A.; Skjøth, T.V.; Andreasen, A.H.; Jensen, C.B.; DeFronzo, R.A.; NN8022-1922 Study Group. Efficacy of Liraglutide for Weight Loss Among Patients with Type 2 Diabetes: The SCALE Diabetes Randomized Clinical Trial. J. Am. Med. Assoc. 2015, 314, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Gao, Z.; Hao, Z.; Ma, H.; Duan, K.; Yang, C. Effect of long-acting versus short-acting glucagon-like peptide-1 receptor agonists on improving body weight and related metabolic parameters in type 2 diabetes: A head-to-head meta-analysis. Medicine 2023, 102, e35739. [Google Scholar] [CrossRef]
- Davies, M.J.; D’Alessio, D.A.; Fradkin, J.; Kernan, W.N.; Mathieu, C.; Mingrone, G.; Rossing, P.; Tsapas, A.; Wexler, D.J.; Buse, J.B. Management of Hyperglycemia in Type 2 Diabetes, 2018. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2018, 41, 2669–2701. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Agarwal, M.; Aggarwal, M.; Alexander, L.; Apovian, C.M.; Bindlish, S.; Bonnet, J.; Butsch, W.S.; Christensen, S.; Gianos, E.; et al. Nutritional priorities to support GLP-1 therapy for obesity: A joint Advisory from the American College of Lifestyle Medicine, the American Society for Nutrition, the Obesity Medicine Association, and The Obesity Society. Am. J. Clin. Nutr. 2025, 122, 344–367. [Google Scholar] [CrossRef]
- Xie, Z.; Zheng, G.; Liang, Z.; Li, M.; Deng, W.; Cao, W. Seven glucagon-like peptide-1 receptor agonists and polyagonists for weight loss in patients with obesity or overweight: An updated systematic review and network meta-analysis of randomized controlled trials. Metabolism 2024, 161, 156038. [Google Scholar] [CrossRef]
- Wong, H.J.; Sim, B.; Teo, Y.H.; Teo, Y.N.; Chan, M.Y.; Yeo, L.L.L.; Eng, P.C.; Tan, B.Y.Q.; Sattar, N.; Dalakoti, M.; et al. Efficacy of GLP-1 Receptor Agonists on Weight Loss, BMI, and Waist Circumference for Patients with Obesity or Overweight: A Systematic Review, Meta-analysis, and Meta-regression of 47 Randomized Controlled Trials. Diabetes Care 2025, 48, 292–300. [Google Scholar] [CrossRef]
- Rodriguez, P.J.; Goodwin Cartwright, B.M.; Gratzl, S.; Brar, R.; Baker, C.; Gluckman, T.J.; Stucky, N.L. Semaglutide vs Tirzepatide for Weight Loss in Adults with Overweight or Obesity. JAMA Intern. Med. 2024, 184, 1056–1064. [Google Scholar] [CrossRef]
- Heise, T.; DeVries, J.H.; Urva, S.; Li, J.; Pratt, E.J.; Thomas, M.K.; Mather, K.J.; Karanikas, C.A.; Dunn, J.; Haupt, A.; et al. Tirzepatide Reduces Appetite, Energy Intake, and Fat Mass in People with Type 2 Diabetes. Diabetes Care 2023, 46, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Aronne, L.J.; Sattar, N.; Horn, D.B.; Bays, H.E.; Wharton, S.; Lin, W.-Y.; Ahmad, N.N.; Zhang, S.; Liao, R.; Bunck, M.C.; et al. Continued Treatment with Tirzepatide for Maintenance of Weight Reduction in Adults with Obesity: The SURMOUNT-4 Randomized Clinical Trial. J. Am. Med. Assoc. 2024, 331, 38–48. [Google Scholar] [CrossRef]
- Horn, D.B.; Linetzky, B.; Davies, M.J.; Laffin, L.J.; Wang, H.; Murphy, M.A.; Zimner-Rapuch, S.; Lau, E.; Arad, A.D.; Lee, C.J. Cardiometabolic Parameter Change by Weight Regain on Tirzepatide Withdrawal in Adults with Obesity: A Post Hoc Analysis of the SURMOUNT-4 Trial. JAMA Intern. Med. 2025, 186, e256112. [Google Scholar] [CrossRef] [PubMed]
- Coskun, T.; Wu, Q.; Schloot, N.C.; Haupt, A.; Milicevic, Z.; Khouli, C.; Harris, C. Effects of retatrutide on body composition in people with type 2 diabetes: A substudy of a phase 2, double-blind, parallel-group, placebo-controlled, randomised trial. Lancet Diabetes Endocrinol. 2025, 13, 674–684. [Google Scholar] [CrossRef]
- le Roux, C.W.; Steen, O.; Lucas, K.J.; Startseva, E.; Unseld, A.; Hennige, A.M. Glucagon and GLP-1 receptor dual agonist survodutide for obesity: A randomised, double-blind, placebo-controlled, dose-finding phase 2 trial. Lancet Diabetes Endocrinol. 2024, 12, 162–173. [Google Scholar] [CrossRef]
- Sinha, B.; Ghosal, S. Efficacy and Safety of GLP-1 Receptor Agonists, Dual Agonists, and Retatrutide for Weight Loss in Adults with Overweight or Obesity: A Bayesian NMA. Obesity 2025, 33, 2046–2054. [Google Scholar] [CrossRef]
- Min, T.; Crockett, E.; Pavlou, A.; Bain, S.C. Cardiovascular outcome trials of GLP-1RAs and SGLT-2 inhibitors: A concise review from the clinicians’ perspective. Metab. Target Organ Damage 2023, 3, 6. [Google Scholar] [CrossRef]
- Rivera, F.B.; Cruz, L.L.A.; Magalong, J.V.; Ruyeras, J.M.M.J.; Aparece, J.P.; Bantayan, N.R.B.; Lara-Breitinger, K.; Gulati, M. Cardiovascular and renal outcomes of glucagon-like peptide 1 receptor agonists among patients with and without type 2 diabetes mellitus: A meta-analysis of randomized placebo-controlled trials. Am. J. Prev. Cardiol. 2024, 18, 100679. [Google Scholar] [CrossRef]
- Zhang, J.; Sanan, S.; Csanalosi, M.; Zheng, C.; Pfeiffer, A.F.H. Novel Dual and Triple Agonists Targeting GLP-1, GIP, Glucagon, and GDF15 for Type 2 Diabetes and Obesity Management. Endocrinology 2025, 166, bqaf130. [Google Scholar] [CrossRef]
- Hernandez, A.F.; Green, J.B.; Janmohamed, S.; D’Agostino, R.B.; Granger, C.B.; Jones, N.P.; Leiter, L.A.; Rosenberg, A.E.; Sigmon, K.N.; Somerville, M.C.; et al. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): A double-blind, randomised placebo-controlled trial. Lancet 2018, 392, 1519–1529. [Google Scholar] [CrossRef]
- Jacob, S.; Eckel, R.H.; Krentz, A.J. Improved cardiovascular outcomes with glucagon-like peptide-1 receptor agonists—What is the role of weight reduction? Cardiovasc. Endocrinol. Metab. 2025, 14, e00346. [Google Scholar] [CrossRef]
- Krüger, N.; Schneeweiss, S.; Desai, R.J.; Sreedhara, S.K.; Kehoe, A.R.; Fuse, K.; Hahn, G.; Schunkert, H.; Wang, S.V. Cardiovascular outcomes of semaglutide and tirzepatide for patients with type 2 diabetes in clinical practice. Nat. Med. 2025, 32, 342–352. [Google Scholar] [CrossRef]
- Dani, S.S.; Makwana, B.; Khadke, S.; Kumar, A.; Jhund, P.; Nasir, K.; Sattar, N.; Al-Kindi, S.; Fonarow, G.; Butler, J.; et al. An Observational Study of Cardiovascular Outcomes of Tirzepatide vs Glucagon-Like Peptide-1 Receptor Agonists. JACC Adv. 2025, 4, 101740. [Google Scholar] [CrossRef]
- Chuang, M.-H.; Chen, J.-Y.; Wang, H.-Y.; Jiang, Z.-H.; Wu, V.-C. Clinical Outcomes of Tirzepatide or GLP-1 Receptor Agonists in Individuals with Type 2 Diabetes. JAMA Netw. Open 2024, 7, e2427258. [Google Scholar] [CrossRef]
- Lincoff, A.M.; Brown-Frandsen, K.; Colhoun, H.M.; Deanfield, J.; Emerson, S.S.; Esbjerg, S.; Hardt-Lindberg, S.; Hovingh, G.K.; Kahn, S.E.; Kushner, R.F.; et al. Semaglutide and Cardiovascular Outcomes in Obesity without Diabetes. N. Engl. J. Med. 2023, 389, 2221–2232. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Zile, M.R.; Kramer, C.M.; Baum, S.J.; Litwin, S.E.; Menon, V.; Ge, J.; Weerakkody, G.J.; Ou, Y.; Bunck, M.C.; et al. Tirzepatide for Heart Failure with Preserved Ejection Fraction and Obesity. N. Engl. J. Med. 2025, 392, 427–437. [Google Scholar] [CrossRef]
- Kosiborod, M.N.; Abildstrøm, S.Z.; Borlaug, B.A.; Butler, J.; Rasmussen, S.; Davies, M.; Hovingh, G.K.; Kitzman, D.W.; Lindegaard, M.L.; Møller, D.V.; et al. Semaglutide in Patients with Heart Failure with Preserved Ejection Fraction and Obesity. N. Engl. J. Med. 2023, 389, 1069–1084. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.F.E.; Ørsted, D.D.; Brown-Frandsen, K.; Marso, S.P.; Poulter, N.R.; Rasmussen, S.; Tornøe, K.; Zinman, B.; Buse, J.B.; LEADER Steering Committee and Investigators. Liraglutide and Renal Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.F.E.; Hansen, T.; Idorn, T.; Leiter, L.A.; Marso, S.P.; Rossing, P.; Seufert, J.; Tadayon, S.; Vilsbøll, T. Effects of once-weekly subcutaneous semaglutide on kidney function and safety in patients with type 2 diabetes: A post-hoc analysis of the SUSTAIN 1–7 randomised controlled trials. Lancet Diabetes Endocrinol. 2020, 8, 880–893. [Google Scholar] [CrossRef]
- Holman, R.R.; Bethel, M.A.; Mentz, R.J.; Thompson, V.P.; Lokhnygina, Y.; Buse, J.B.; Chan, J.C.; Choi, J.; Gustavson, S.M.; Iqbal, N.; et al. Effects of Once-Weekly Exenatide on Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 1228–1239. [Google Scholar] [CrossRef]
- Mentz, R.J.; Thompson, V.P.; Aguilar, D.; Choi, J.; Gustavson, S.M.; Iqbal, N.; Kong, A.P.; Öhman, P.; Sattar, N.; Scott, R.S.; et al. Effects of Once-Weekly Exenatide on Clinical Outcomes in Patients with Preexisting Cardiovascular Disease. Circulation 2018, 138, 2576–2578. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.; Birkenfeld, A.L.; Donsmark, M.; Dungan, K.; Eliaschewitz, F.G.; Franco, D.R.; Jeppesen, O.K.; Lingvay, I.; Mosenzon, O.; Pedersen, S.D.; et al. Oral Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2019, 381, 841–851. [Google Scholar] [CrossRef]
- Ruff, C.T.; Baron, M.; Im, K.; O’Donoghue, M.L.; Fiedorek, F.T.; Sabatine, M.S. Subcutaneous infusion of exenatide and cardiovascular outcomes in type 2 diabetes: A non-inferiority randomized controlled trial. Nat. Med. 2022, 28, 89–95. [Google Scholar] [CrossRef]
- Reule, S.; Pickthorn, S.; Worwa, S.; Ishani, A.; Foley, R.; Reule, S.; Pickthorn, S.; Worwa, S.; Ishani, A.; Foley, R. Glucagon-like Peptide-1 Receptor Agonists and Survival in Advanced Chronic Kidney Disease and Type 2 Diabetes. Diabetology 2025, 6, 161. [Google Scholar] [CrossRef]
- Botros, F.T.; Gerstein, H.C.; Malik, R.; Nicolay, C.; Hoover, A.; Turfanda, I.; Colhoun, H.M.; Shaw, J.E. Dulaglutide and Kidney Function–Related Outcomes in Type 2 Diabetes: A REWIND Post Hoc Analysis. Diabetes Care 2023, 46, 1524–1530. [Google Scholar] [CrossRef]
- Perkovic, V.; Tuttle, K.R.; Rossing, P.; Mahaffey, K.W.; Mann, J.F.E.; Bakris, G.; Baeres, F.M.M.; Idorn, T.; Bosch-Traberg, H.; Lausvig, N.L.; et al. Effects of Semaglutide on Chronic Kidney Disease in Patients with Type 2 Diabetes. N. Engl. J. Med. 2024, 391, 109–121. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Sattar, N.; Pavo, I.; Haupt, A.; Duffin, K.L.; Yang, Z.; Wiese, R.J.; Tuttle, K.R.; Cherney, D.Z.I. Effects of tirzepatide versus insulin glargine on kidney outcomes in type 2 diabetes in the SURPASS-4 trial: Post-hoc analysis of an open-label, randomised, phase 3 trial. Lancet Diabetes Endocrinol. 2022, 10, 774–785. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Sattar, N.; Pavo, I.; Haupt, A.; Duffin, K.L.; Yang, Z.; Wiese, R.J.; Wilson, J.M.; Hemmingway, A.; Cherney, D.Z.I.; et al. Effects of Tirzepatide Versus Insulin Glargine on Cystatin C–Based Kidney Function: A SURPASS-4 Post Hoc Analysis. Diabetes Care 2023, 46, 1501–1506. [Google Scholar] [CrossRef]
- Packer, M.; Zile, M.R.; Kramer, C.M.; Murakami, M.; Ou, Y.; Borlaug, B.A.; SUMMIT Trial Study Group. Interplay of Chronic Kidney Disease and the Effects of Tirzepatide in Patients with Heart Failure, Preserved Ejection Fraction, and Obesity. J. Am. Coll. Cardiol. 2025, 85, 1721–1735. [Google Scholar] [CrossRef] [PubMed]
- Taal, M.W.; Selby, N.M. Glucagon-like Peptide-1 Receptor Agonists: New Evidence of Kidney and Cardiovascular Protection From the FLOW and SELECT Trials. Am. J. Kidney Dis. 2025, 85, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Kamalumpundi, V.; Codipilly, D.C. GLP1 and GIP Receptor Agonists: Effects on the Gastrointestinal Tract and Management Strategies for Primary Care Physicians. Mayo Clin. Proc. 2025, 100, 2228–2236. [Google Scholar] [CrossRef]
- Ismaiel, A.; Scarlata, G.G.M.; Boitos, I.; Leucuta, D.-C.; Popa, S.-L.; Al Srouji, N.; Abenavoli, L.; Dumitrascu, D.L. Gastrointestinal adverse events associated with GLP-1 RA in non-diabetic patients with overweight or obesity: A systematic review and network meta-analysis. Int. J. Obes. 2025, 49, 1946–1957. [Google Scholar] [CrossRef]
- Waldrop, G.; Zhong, J.; Peters, M.; Rajagopalan, S. Incretin-Based Therapy for Diabetes: What a Cardiologist Needs to Know. J. Am. Coll. Cardiol. 2016, 67, 1488–1496. [Google Scholar] [CrossRef]
- Liu, L.; Chen, J.; Wang, L.; Chen, C.; Chen, L. Association between different GLP-1 receptor agonists and gastrointestinal adverse reactions: A real-world disproportionality study based on FDA adverse event reporting system database. Front. Endocrinol. 2022, 13, 1043789. [Google Scholar] [CrossRef]
- Xie, X.; Yang, S.; Deng, S.; Liu, Y.; Xu, Z.; He, B. Comparative gastrointestinal adverse effects of GLP-1 receptor agonists and multi-target analogs in type 2 diabetes: A Bayesian network meta-analysis. Front. Pharmacol. 2025, 16, 1613610. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Z.; Ye, W.; Peng, P.; Wang, Y.; Wan, L.; Li, J.; Zhang, M.; Wang, Y.; Liu, R.; et al. Safety and effects of anti-obesity medications on weight loss, cardiometabolic, and psychological outcomes in people living with overweight or obesity: A systematic review and meta-analysis. eClinicalMedicine 2025, 79, 103020. [Google Scholar] [CrossRef] [PubMed]
- Kushner, R.F.; Almandoz, J.P.; Rubino, D.M. Managing Adverse Effects of Incretin-Based Medications for Obesity. J. Am. Med. Assoc. 2025, 334, 822–823. [Google Scholar] [CrossRef]
- Nadolsky, K.; Garvey, W.T.; Agarwal, M.; Bonnecaze, A.; Burguera, B.; Chaplin, M.D.; Griebeler, M.L.; Harris, S.R.; Schellinger, J.N.; Simonetti, J.; et al. American Association of Clinical Endocrinology Consensus Statement: Algorithm for the Evaluation and Treatment of Adults with Obesity/Adiposity-Based Chronic Disease—2025 Update. Endocr. Pract. 2025, 31, 1351–1394. [Google Scholar] [CrossRef] [PubMed]
- Eldor, R.; Avraham, N.; Rosenberg, O.; Shpigelman, M.; Golan-Cohen, A.; Cukierman-Yaffe, T.; Merzon, E.; Buch, A. Gradual Titration of Semaglutide Results in Better Treatment Adherence and Fewer Adverse Events: A Randomized Controlled Open-Label Pilot Study Examining a 16-Week Flexible Titration Regimen Versus Label-Recommended 8-Week Semaglutide Titration Regimen. Diabetes Care 2025, 48, 1607–1611. [Google Scholar] [CrossRef]
- Das, S.R.; Everett, B.M.; Birtcher, K.K.; Brown, J.M.; Januzzi, J.L.; Kalyani, R.R.; Kosiborod, M.; Magwire, M.; Morris, P.B.; Neumiller, J.J.; et al. 2020 Expert Consensus Decision Pathway on Novel Therapies for Cardiovascular Risk Reduction in Patients with Type 2 Diabetes: A Report of the American College of Cardiology Solution Set Oversight Committee. J. Am. Coll. Cardiol. 2020, 76, 1117–1145. [Google Scholar] [CrossRef]
- American Diabetes Association Professional Practice Committee. 13. Older Adults: Standards of Care in Diabetes—2025. Diabetes Care 2025, 48, S266–S282. [Google Scholar] [CrossRef]
- Fang, Y.E.; Paik, J.M.; Ortega-Montiel, J.; Tesfaye, H.; Wexler, D.J.; Patorno, E. Risk of Acute Pancreatitis and Biliary Events After Initiation of Incretin-Based Medications in Patients with Type 2 Diabetes. Diabetes Care 2025, 48, 2127–2137. [Google Scholar] [CrossRef]
- Nieto, L.M.; Martinez, J.; Narvaez, S.I.; Ko, D.; Kim, D.H.; Vega, K.J.; Chawla, S. Glucagon-Like Peptide-1 Receptor Agonists Use Does Not Increase the Risk for Acute Pancreatitis and Is Associated with Lower Complications in Patients with Type 2 Diabetes Who Develop Acute Pancreatitis: A Multicenter Analysis. Am. J. Gastroenterol. 2025, 121, 424–431. [Google Scholar] [CrossRef]
- Pratley, R.; Saeed, Z.I.; Casu, A. Incretin mimetics and acute pancreatitis: Enemy or innocent bystander? Curr. Opin. Gastroenterol. 2024, 40, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Jastreboff, A.M.; le Roux, C.W.; Stefanski, A.; Aronne, L.J.; Halpern, B.; Wharton, S.; Wilding, J.P.H.; Perreault, L.; Zhang, S.; Battula, R.; et al. Tirzepatide for Obesity Treatment and Diabetes Prevention. N. Engl. J. Med. 2025, 392, 958–971. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Xu, J.; Mu, X.; Shi, Y.; Fan, H.; Li, S. Safety issues of tirzepatide (pancreatitis and gallbladder or biliary disease) in type 2 diabetes and obesity: A systematic review and meta-analysis. Front. Endocrinol. 2023, 14, 1214334. [Google Scholar] [CrossRef]
- Nassar, M.; Nassar, O.; Abosheaishaa, H.; Misra, A. Decreased risk of recurrent acute pancreatitis with semaglutide and tirzepatide in people with type 2 diabetes or obesity with a history of acute pancreatitis: A propensity matched global federated TriNetX database-based retrospective cohort study. Diabetes Metab. Syndr. 2024, 18, 103116. [Google Scholar] [CrossRef]
- Blonde, L.; Umpierrez, G.E.; Reddy, S.S.; McGill, J.B.; Berga, S.L.; Bush, M.; Chandrasekaran, S.; DeFronzo, R.A.; Einhorn, D.; Galindo, R.J.; et al. American Association of Clinical Endocrinology Clinical Practice Guideline: Developing a Diabetes Mellitus Comprehensive Care Plan-2022 Update. Endocr. Pract. 2022, 28, 923–1049. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wang, J.; Ping, F.; Yang, N.; Huang, J.; Li, Y.; Xu, L.; Li, W.; Zhang, H. Association of Glucagon-Like Peptide-1 Receptor Agonist Use with Risk of Gallbladder and Biliary Diseases: A Systematic Review and Meta-analysis of Randomized Clinical Trials. JAMA Intern. Med. 2022, 182, 513–519. [Google Scholar] [CrossRef]
- Moiz, A.; Filion, K.B.; Tsoukas, M.A.; Yu, O.H.Y.; Peters, T.M.; Eisenberg, M.J. The expanding role of GLP-1 receptor agonists: A narrative review of current evidence and future directions. eClinicalMedicine 2025, 86, 103363. [Google Scholar] [CrossRef]
- Jalleh, R.J.; Marathe, C.S.; Rayner, C.K.; Jones, K.L.; Umapathysivam, M.M.; Wu, T.; Quast, D.R.; Plummer, M.P.; Nauck, M.A.; Horowitz, M. Physiology and Pharmacology of Effects of GLP-1-based Therapies on Gastric, Biliary and Intestinal Motility. Endocrinology 2024, 166, bqae155. [Google Scholar] [CrossRef] [PubMed]
- Natividade, G.R.; Spiazzi, B.F.; Baumgarten, M.W.; Bassotto, C.; Pereira, A.A.; Fraga, B.L.; Scalco, B.G.; Mattes, N.R.; Lavinsky, D.; Kramer, C.K.; et al. Ocular Adverse Events with Semaglutide: A Systematic Review and Meta-Analysis. JAMA Ophthalmol. 2025, 143, 759–768. [Google Scholar] [CrossRef]
- Ramsey, D.J.; Makwana, B.; Dani, S.S.; Patel, M.; Panchal, K.; Shah, J.; Khadke, S.; Kumar, A.; Patel, T.; Kosiborod, M.N.; et al. GLP-1 Receptor Agonists and Sight-Threatening Ophthalmic Complications in Patients with Type 2 Diabetes. JAMA Netw. Open 2025, 8, e2526321. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.I.; Kim, S.J.; Bailey, S.T.; Kovach, J.L.; Vemulakonda, G.A.; Ying, G.-S.; Flaxel, C.J.; American Academy of Ophthalmology Preferred Practice Pattern Retina/Vitreous Committee. Diabetic Retinopathy Preferred Practice Pattern®. Ophthalmology 2025, 132, P75–P162. [Google Scholar] [CrossRef] [PubMed]
- Katz, B.J.; Lee, M.S.; Lincoff, N.S.; Abel, A.S.; Chowdhary, S.; Ellis, B.D.; Najafi, A.; Nguyen, J.; Seay, M.D.; Warner, J.E.A. Ophthalmic Complications Associated with the Antidiabetic Drugs Semaglutide and Tirzepatide. JAMA Ophthalmol. 2025, 143, 215–220. [Google Scholar] [CrossRef]
- American Diabetes Association Professional Practice Committee. 12. Retinopathy, Neuropathy, and Foot Care: Standards of Care in Diabetes—2025. Diabetes Care 2025, 48, S252–S265. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Bedossa, P.; Fraessdorf, M.; Neff, G.W.; Lawitz, E.; Bugianesi, E.; Anstee, Q.M.; Hussain, S.A.; Newsome, P.N.; Ratziu, V.; et al. A Phase 2 Randomized Trial of Survodutide in MASH and Fibrosis. N. Engl. J. Med. 2024, 391, 311–319. [Google Scholar] [CrossRef]
- Rodriguez, P.J.; Zhang, V.; Gratzl, S.; Do, D.; Goodwin Cartwright, B.; Baker, C.; Gluckman, T.J.; Stucky, N.; Emanuel, E.J. Discontinuation and Reinitiation of Dual-Labeled GLP-1 Receptor Agonists Among US Adults with Overweight or Obesity. JAMA Netw. Open 2025, 8, e2457349. [Google Scholar] [CrossRef]
- Lim, C.-E.; Pasternak, B.; Eliasson, B.; Ueda, P. Treatment discontinuation among users of GLP-1 receptor agonists and SGLT2 inhibitors in a national population of individuals with type 2 diabetes. Diabetologia 2025, 68, 1680–1695. [Google Scholar] [CrossRef]
- Samuels, J.M.; Ye, F.; Irlmeier, R.; Silver, H.; Srivastava, G.; Spann, M. Real-world titration, persistence & weight loss of semaglutide and tirzepatide in an academic obesity clinic. Diabetes Obes. Metab. 2025, 27, 6200–6209. [Google Scholar] [CrossRef]
- Lassen, M.C.H.; Johansen, N.D.; Modin, D.; Catarig, A.-M.; Vistisen, B.K.; Amadid, H.; Zimmermann, E.; Gislason, G.; Biering-Sørensen, T. Adherence to glucagon-like peptide-1 receptor agonist treatment in type 2 diabetes mellitus: A nationwide registry study. Diabetes Obes. Metab. 2024, 26, 5239–5250. [Google Scholar] [CrossRef]
- Brown, E.; Heerspink, H.J.L.; Cuthbertson, D.J.; Wilding, J.P.H. SGLT2 inhibitors and GLP-1 receptor agonists: Established and emerging indications. Lancet 2021, 398, 262–276. [Google Scholar] [CrossRef]
- Kunutsor, S.K.; Seidu, S. Safety and Tolerability of Glucagon-Like Peptide-1 Receptor Agonists: A State-of-the-Art Narrative Review. Drugs 2025, 86, 11–36. [Google Scholar] [CrossRef]
- Chen, J.-C.; Fang, Y.-W.; Liu, Y.-F.; Chen, M.-T.; Tsai, M.-H. GLP-1 Receptor Agonist Therapy and Cardiorenal Outcomes in Patients ≥ 80 Years Old with Type 2 Diabetes. J. Am. Geriatr. Soc. 2025, 74, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Waqas, S.A.; Ali, D.; Afridi, M.K.; Siddiqui, H.F.; Nazir, A.; Greene, S.J.; Khan, M.S. Efficacy of GLP-1 receptor agonists among older adults: A meta-analysis of cardio-kidney outcome trials. Arch. Gerontol. Geriatr. 2025, 138, 105981. [Google Scholar] [CrossRef]
- de Boer, I.H.; Khunti, K.; Sadusky, T.; Tuttle, K.R.; Neumiller, J.J.; Rhee, C.M.; Rosas, S.E.; Rossing, P.; Bakris, G. Diabetes management in chronic kidney disease: A consensus report by the American Diabetes Association (ADA) and Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2022, 102, 974–989. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association Professional Practice Committee. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Care in Diabetes—2025. Diabetes Care 2025, 48, S181–S206. [Google Scholar] [CrossRef]
- LeRoith, D.; Biessels, G.J.; Braithwaite, S.S.; Casanueva, F.F.; Draznin, B.; Halter, J.B.; Hirsch, I.B.; McDonnell, M.E.; Molitch, M.E.; Murad, M.H.; et al. Treatment of Diabetes in Older Adults: An Endocrine Society* Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2019, 104, 1520–1574. [Google Scholar] [CrossRef] [PubMed]
- Kidney Disease: Improving Global Outcomes (KDIGO) Diabetes Work Group. KDIGO 2022 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease. Kidney Int. 2022, 102, S1–S127. [Google Scholar] [CrossRef]
- Navaneethan, S.D.; Bansal, N.; Cavanaugh, K.L.; Chang, A.; Crowley, S.; Delgado, C.; Estrella, M.M.; Ghossein, C.; Ikizler, T.A.; Koncicki, H.; et al. KDOQI US Commentary on the KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of CKD. Am. J. Kidney Dis. 2025, 85, 135–176. [Google Scholar] [CrossRef]
- Wyckoff, J.A.; Lapolla, A.; Asias-Dinh, B.D.; Barbour, L.A.; Brown, F.M.; Catalano, P.M.; Corcoy, R.; Di Renzo, G.C.; Drobycki, N.; Kautzky-Willer, A.; et al. Preexisting Diabetes and Pregnancy: An Endocrine Society and European Society of Endocrinology Joint Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2025, 110, 2405–2452. [Google Scholar] [CrossRef]
- Cesta, C.E.; Rotem, R.; Bateman, B.T.; Chodick, G.; Cohen, J.M.; Furu, K.; Gissler, M.; Huybrechts, K.F.; Kjerpeseth, L.J.; Leinonen, M.K.; et al. Safety of GLP-1 Receptor Agonists and Other Second-Line Antidiabetics in Early Pregnancy. JAMA Intern. Med. 2024, 184, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Maya, J.; Pant, D.; Fu, Y.; James, K.; Batlle, C.; Hsu, S.; Soria-Contreras, D.C.; Shook, L.L.; Mow, C.; Hivert, M.-F.; et al. Gestational Weight Gain and Pregnancy Outcomes After GLP-1 Receptor Agonist Discontinuation. J. Am. Med. Assoc. 2025, 334, e2520951. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, A.-S.; Blair, R.; Peters, A.L. Management of Preexisting Diabetes in Pregnancy: A Review. J. Am. Med. Assoc. 2019, 321, 1811–1819. [Google Scholar] [CrossRef]
- Małecki, M.T.; Batterham, R.L.; Sattar, N.; Levine, J.A.; Rodríguez, Á.; Bergman, B.K.; Wang, H.; Ghimpeteanu, G.; Lee, C.J. Predictors of ≥15% Weight Reduction and Associated Changes in Cardiometabolic Risk Factors with Tirzepatide in Adults with Type 2 Diabetes in SURPASS 1-4. Diabetes Care 2023, 46, 2292–2299. [Google Scholar] [CrossRef]
- Yao, H.; Zhang, A.; Li, D.; Wu, Y.; Wang, C.-Z.; Wan, J.-Y.; Yuan, C.-S. Comparative effectiveness of GLP-1 receptor agonists on glycaemic control, body weight, and lipid profile for type 2 diabetes: Systematic review and network meta-analysis. BMJ 2024, 384, e076410. [Google Scholar] [CrossRef]
- Caruso, I.; Di Gioia, L.; Di Molfetta, S.; Cignarelli, A.; Palmer, S.C.; Natale, P.; Strippoli, G.F.M.; Perrini, S.; Natalicchio, A.; Laviola, L.; et al. Glucometabolic outcomes of GLP-1 receptor agonist-based therapies in patients with type 2 diabetes: A systematic review and network meta-analysis. eClinicalMedicine 2023, 64, 102181. [Google Scholar] [CrossRef] [PubMed]
- Vozza, A.; Triggiani, D.; Fanelli, M.; Lisco, G.; Coletto, D.; Custodero, C.; Volpe, S.; Racaniello, D.; Colaianni, V.; Lavarra, V.; et al. Predictive factors of body weight loss in patients with type 2 diabetes treated with GLP-1 receptor agonists: A 52-week prospective real-life study. Front. Endocrinol. 2025, 16, 1674308. [Google Scholar] [CrossRef]
- Giorgino, F.; Lingvay, I.; Van Gaal, L.F.; Sharma, P.; Rodríguez, Á.; Kiljański, J.; Torcello-Gómez, A.; Levine, J.A. Early Fasting Serum Glucose or Weight Reduction with Tirzepatide and Metabolic Outcomes in People with Type 2 Diabetes: A Post Hoc Analysis of the SURPASS Trials. Diabetes Care 2025, 48, 790–798. [Google Scholar] [CrossRef]
- Bihan, H.; Ng, W.L.; Magliano, D.J.; Shaw, J.E. Predictors of efficacy of GLP-1 agonists and DPP-4 inhibitors: A systematic review. Diabetes Res. Clin. Pract. 2016, 121, 27–34. [Google Scholar] [CrossRef]
- Deacon, C.F.; Mannucci, E.; Ahrén, B. Glycaemic efficacy of glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors as add-on therapy to metformin in subjects with type 2 diabetes-a review and meta analysis. Diabetes Obes. Metab. 2012, 14, 762–767. [Google Scholar] [CrossRef]
- Ahmad, E.; Lim, S.; Lamptey, R.; Webb, D.R.; Davies, M.J. Type 2 diabetes. Lancet 2022, 400, 1803–1820. [Google Scholar] [CrossRef]
- Sharma, A.; Mariam, A.; Zacherle, E.; Milinovich, A.; Bauman, J.; Sugano, D.S.; Noone, J.; Rajpura, J.R.; Zimmerman, R.S.; Burguera, B.; et al. Elucidating the role of weight loss and glycaemic control in patients with type 2 diabetes. Diabetes Obes. Metab. 2024, 26, 5347–5357. [Google Scholar] [CrossRef]
- Gilbert, O.; Gulati, M.; Gluckman, T.J.; Kittleson, M.M.; Rikhi, R.; Saseen, J.J.; Tchang, B.G. 2025 Concise Clinical Guidance: An ACC Expert Consensus Statement on Medical Weight Management for Optimization of Cardiovascular Health: A Report of the American College of Cardiology Solution Set Oversight Committee. J. Am. Coll. Cardiol. 2025, 86, 536–555. [Google Scholar] [CrossRef]
- Karagiannis, T.; Malandris, K.; Avgerinos, I.; Stamati, A.; Kakotrichi, P.; Liakos, A.; Vasilakou, D.; Kakaletsis, N.; Tsapas, A.; Bekiari, E. Subcutaneously administered tirzepatide vs semaglutide for adults with type 2 diabetes: A systematic review and network meta-analysis of randomised controlled trials. Diabetologia 2024, 67, 1206–1222. [Google Scholar] [CrossRef] [PubMed]
- Yanovski, S.Z.; Yanovski, J.A. Approach to Obesity Treatment in Primary Care: A Review. JAMA Intern. Med. 2024, 184, 818–829. [Google Scholar] [CrossRef]
- Lingvay, I.; Sumithran, P.; Cohen, R.V.; le Roux, C.W. Obesity management as a primary treatment goal for type 2 diabetes: Time to reframe the conversation. Lancet 2022, 399, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Frias, J.P.; Nauck, M.A.; Van, J.; Kutner, M.E.; Cui, X.; Benson, C.; Urva, S.; Gimeno, R.E.; Milicevic, Z.; Robins, D.; et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: A randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet 2018, 392, 2180–2193. [Google Scholar] [CrossRef]
- Karagiannis, T.; Avgerinos, I.; Liakos, A.; Del Prato, S.; Matthews, D.R.; Tsapas, A.; Bekiari, E. Management of type 2 diabetes with the dual GIP/GLP-1 receptor agonist tirzepatide: A systematic review and meta-analysis. Diabetologia 2022, 65, 1251–1261. [Google Scholar] [CrossRef]
- Center for Drug Evaluation and Research. Approved Drug Products with Therapeutic Equivalence Evaluations|Orange Book. FDA. December 2025. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/approved-drug-products-therapeutic-equivalence-evaluations-orange-book (accessed on 23 December 2025).
- American Diabetes Association Professional Practice Committee. 8. Obesity and Weight Management for the Prevention and Treatment of Type 2 Diabetes: Standards of Care in Diabetes—2025. Diabetes Care 2025, 48, S167–S180. [Google Scholar] [CrossRef] [PubMed]
- Updike, W.H.; Pane, O.; Franks, R.; Saber, F.; Abdeen, F.; Balazy, D.D.; Carris, N.W. Is it Time to Expand Glucagon-like Peptide-1 Receptor Agonist Use for Weight Loss in Patients Without Diabetes? Drugs 2021, 81, 881–893. [Google Scholar] [CrossRef]
- Rathmann, W.; Bongaerts, B. Pharmacogenetics of novel glucose-lowering drugs. Diabetologia 2021, 64, 1201–1212. [Google Scholar] [CrossRef]
- Tonin, G.; Goričar, K.; Blagus, T.; Janež, A.; Dolžan, V.; Klen, J. Genetic variability in sodium-glucose cotransporter 2 and glucagon-like peptide 1 receptor effect on glycemic and pressure control in type 2 diabetes patients treated with SGLT2 inhibitors and GLP-1RA in the everyday clinical practice. Front. Endocrinol. 2025, 16, 1547920. [Google Scholar] [CrossRef]
- Guan, Z.; Du, Y.; Li, R.; Zhang, S.; Xu, Y.; Zhang, X.; Zhang, F.; Yin, Y.; Wu, K.; Li, X.; et al. Association between glucagon-like peptide-1 receptor gene polymorphism and treatment response to GLP1R agonists in Chinese patients with type 2 diabetes: A prospective cohort study. Eur. J. Clin. Pharmacol. 2022, 78, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Mashayekhi, M.; Safa, B.I.; Nian, H.; Devin, J.K.; Gamboa, J.L.; Yu, C.; Chen, R.; Beckman, J.A.; Koethe, J.R.; Silver, H.J.; et al. RISING STARS: Effects of a GLP-1 receptor polymorphism on responses to liraglutide. J. Endocrinol. 2025, 267, e250174. [Google Scholar] [CrossRef]
- Bonnefond, A.; Florez, J.C.; Loos, R.J.F.; Froguel, P. Dissection of type 2 diabetes: A genetic perspective. Lancet Diabetes Endocrinol. 2025, 13, 149–164. [Google Scholar] [CrossRef]
- Jacobi, S.F.; Khajavi, N.; Kleinau, G.; Teumer, A.; Scheerer, P.; Homuth, G.; Völzke, H.; Wiegand, S.; Kühnen, P.; Krude, H.; et al. Evaluation of a rare glucose-dependent insulinotropic polypeptide receptor variant in a patient with diabetes. Diabetes Obes. Metab. 2019, 21, 1168–1176. [Google Scholar] [CrossRef] [PubMed]
- Rees, T.A.; Buttle, B.J.; Tasma, Z.; Yang, S.-H.; Harris, P.W.R.; Walker, C.S. Tirzepatide, GIP(1-42) and GIP(1-30) display unique signaling profiles at two common GIP receptor variants, E354 and Q354. Front. Pharmacol. 2024, 15, 1463313. [Google Scholar] [CrossRef] [PubMed]
- Kyriakidou, A.; Koufakis, T.; Goulis, D.G.; Vasilopoulos, Y.; Zebekakis, P.; Kotsa, K. Pharmacogenetics of the Glucagon-like Peptide-1 Receptor Agonist Liraglutide: A Step Towards Personalized Type 2 Diabetes Management. Curr. Pharm. Des. 2021, 27, 1025–1034. [Google Scholar] [CrossRef]
- Klen, J.; Dolžan, V. Glucagon-like Peptide-1 Receptor Agonists in the Management of Type 2 Diabetes Mellitus and Obesity: The Impact of Pharmacological Properties and Genetic Factors. Int. J. Mol. Sci. 2022, 23, 3451. [Google Scholar] [CrossRef] [PubMed]
- Bracchiglione, J.; Meza, N.; Franco, J.V.; Escobar Liquitay, C.M.; Novik, A.V.; Ocara Vargas, M.; Lazcano, G.; Poloni, D.; Rinaldi Langlotz, F.; Roqué-Figuls, M.; et al. Semaglutide for adults living with obesity. Cochrane Database Syst. Rev. 2025, 10, CD015092. [Google Scholar] [CrossRef] [PubMed]
- Maccora, C.; Ciuoli, C.; Goracci, A.; Benenati, N.; Formichi, C.; Pilli, T.; Verdino, V.; Mnutr, O.N.; Bufano, A.; Tirone, A.; et al. One Month Weight Loss Predicts the Efficacy of Liraglutide in Obese Patients: Data from a Single Center. Endocr. Pract. 2020, 26, 235–240. [Google Scholar] [CrossRef]
- Mehrtash, F.; Dushay, J.; Manson, J.E. Integrating Diet and Physical Activity When Prescribing GLP-1s-Lifestyle Factors Remain Crucial. JAMA Intern. Med. 2025, 185, 1151–1152. [Google Scholar] [CrossRef]
- Wang, J.; Lin, C.; Cai, X.; Wang, Y.; Wei, T.; Wang, Y.; Tie, C.; Yang, Y.; Lv, F.; Yang, W.; et al. Glucagon-like peptide-1 receptor agonist treatment associated weight fluctuation and influencing factors in patients with overweight or obesity. Diabetes Obes. Metab. 2025, 27, 5042–5051. [Google Scholar] [CrossRef]
- Squire, P.; Naude, J.; Zentner, A.; Bittman, J.; Khan, N. Factors associated with weight loss response to GLP-1 analogues for obesity treatment: A retrospective cohort analysis. BMJ Open 2025, 15, e089477. [Google Scholar] [CrossRef] [PubMed]
- Jia, I.-I.-T.T.; Bloomfield, G.C.; Chen, M.Y.; Cunningham, M.H.; Azagury, D.E.; Alimi, Y.R.; Prindeze, N.J. Analysis of the long-term impact of glucagon-like peptide-1 (GLP-1) receptor agonists for control of obesity and obesity-related comorbidities: A meta-analysis. Surg. Endosc. 2025, 39, 8580–8589. [Google Scholar] [CrossRef]
- Wilcox, T.; De Block, C.; Schwartzbard, A.Z.; Newman, J.D. Diabetic Agents, From Metformin to SGLT2 Inhibitors and GLP1 Receptor Agonists: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 1956–1974. [Google Scholar] [CrossRef] [PubMed]
- Clinical Management of Obesity. Available online: https://cdn.csdurant.com/media/books.pcibooks.com/ebooks/obesity/index.html?page=1 (accessed on 23 December 2025).
- Kelly, C.A.; Sipos, J.A. Approach to the Patient with Thyroid Nodules: Considering GLP-1 Receptor Agonists. J. Clin. Endocrinol. Metab. 2025, 110, e2080–e2087. [Google Scholar] [CrossRef]
- Honigberg, M.C.; Chang, L.-S.; McGuire, D.K.; Plutzky, J.; Aroda, V.R.; Vaduganathan, M. Use of Glucagon-Like Peptide-1 Receptor Agonists in Patients with Type 2 Diabetes and Cardiovascular Disease: A Review. JAMA Cardiol. 2020, 5, 1182–1190. [Google Scholar] [CrossRef]
- Vangoitsenhoven, R.; Mathieu, C.; Van der Schueren, B. GLP1 and cancer: Friend or foe? Endocr. Relat. Cancer 2012, 19, F77–F88. [Google Scholar] [CrossRef]
- Glasgow, K.; Jiminez, V.; Garcia, N.; Gillis, A. Incidental detection of multiple endocrine neoplasia and medullary thyroid carcinoma before starting GLP-1 agonist: A case report. Heliyon 2024, 10, e33420. [Google Scholar] [CrossRef]
- Kupnicka, P.; Król, M.; Żychowska, J.; Łagowski, R.; Prajwos, E.; Surówka, A.; Chlubek, D. GLP-1 Receptor Agonists: A Promising Therapy for Modern Lifestyle Diseases with Unforeseen Challenges. Pharmaceuticals 2024, 17, 1470. [Google Scholar] [CrossRef]
- Mehta, A.E.; Lomeli, L.D.; Pantalone, K.M. Glucagon-like peptide-1 receptor agonists and pancreatitis: A reconcilable divorce. Clevel. Clin. J. Med. 2025, 92, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Staller, K.; Parkman, H.P.; Greer, K.B.; Leiman, D.A.; Zhou, M.J.; Singh, S.; Camilleri, M.; Altayar, O.; AGA Clinical Guidelines Committee. AGA Clinical Practice Guideline on Management of Gastroparesis. Gastroenterology 2025, 169, 828–861. [Google Scholar] [CrossRef] [PubMed]
- Jalleh, R.J.; Plummer, M.P.; Marathe, C.S.; Umapathysivam, M.M.; Quast, D.R.; Rayner, C.K.; Jones, K.L.; Wu, T.; Horowitz, M.; Nauck, M.A. Clinical Consequences of Delayed Gastric Emptying with GLP-1 Receptor Agonists and Tirzepatide. J. Clin. Endocrinol. Metab. 2024, 110, 1–15. [Google Scholar] [CrossRef]
- Chiang, C.-H.; Jaroenlapnopparat, A.; Colak, S.C.; Yu, C.-C.; Xanthavanij, N.; Wang, T.-H.; See, X.Y.; Lo, S.-W.; Ko, A.; Chang, Y.-C.; et al. Glucagon-Like Peptide-1 Receptor Agonists and Gastrointestinal Adverse Events: A Systematic Review and Meta-Analysis. Gastroenterology 2025, 169, 1268–1281. [Google Scholar] [CrossRef]
- Zayed, M.F.; Khyat, N.O.; Alsibyani, R.W.; Alshami, R.Z.; Alsubahi, R.A.; Alamoudi, M.K. An Insight into Pharmaceutical Design and Pharmacokinetic Characteristics of GLP-1 RAs. Curr. Pharm. Des. 2026, 32, 1095–1106. [Google Scholar] [CrossRef]
- Gentilella, R.; Pechtner, V.; Corcos, A.; Consoli, A. Glucagon-like peptide-1 receptor agonists in type 2 diabetes treatment: Are they all the same? Diabetes Metab. Res. Rev. 2019, 35, e3070. [Google Scholar] [CrossRef]
- Di Dalmazi, G.; Coluzzi, S.; Baldassarre, M.P.A.; Ghit, A.; Graziano, G.; Rossi, M.C.; Ciappini, B.; Milo, M.; Carrieri, F.; Nicolucci, A.; et al. Effectiveness and Tolerability of Once-Weekly GLP-1 Receptor Agonists in Clinical Practice: A Focus on Switching Between Once-Weekly Molecules in Type 2 Diabetes. Front. Endocrinol. 2022, 13, 892702. [Google Scholar] [CrossRef]
- Samson, S.L.; Garber, A.J. A Plethora of GLP-1 Agonists: Decisions About What to Use and When. Curr. Diabetes Rep. 2016, 16, 120. [Google Scholar] [CrossRef] [PubMed]
- Derington, C.G.; Sarwal, A.; Wei, G.; Hartsell, S.E.; Throolin, M.; Singh, R.; Nevers, M.R.; Zhang, C.; Katkam, N.; Takyi, A.; et al. Liraglutide vs Semaglutide vs Dulaglutide in Veterans with Type 2 Diabetes. JAMA Netw. Open 2025, 8, e2537297. [Google Scholar] [CrossRef]
- Overgaard, R.V.; Lindberg, S.Ø.; Thielke, D. Impact on HbA1c and body weight of switching from other GLP-1 receptor agonists to semaglutide: A model-based approach. Diabetes Obes. Metab. 2019, 21, 43–51. [Google Scholar] [CrossRef]
- Ciudin, A.; Johansson, E.; Zimner-Rapuch, S.; Dimitriadis, G.K.; Bertrand, M.; Curteis, T.; Clark, L.J.; Fan, L.; Sapin, H.; Bergmann, J.-F. Indirect comparative efficacy and safety of tirzepatide 10 and 15 mg versus semaglutide 2.4 mg for the management of obesity and overweight in patients with type 2 diabetes. Diabetes Obes. Metab. 2025, 27, 4709–4719. [Google Scholar] [CrossRef]
- Ding, Y.; Shi, Y.; Guan, R.; Yan, S.; Liu, H.; Wang, Z.; Li, J.; Wang, T.; Cai, W.; Ma, G. Evaluation and comparison of efficacy and safety of tirzepatide and semaglutide in patients with type 2 diabetes mellitus: A Bayesian network meta-analysis. Pharmacol. Res. 2024, 199, 107031. [Google Scholar] [CrossRef] [PubMed]
- Mondoh, A.; Crotty, M.; le Roux, C.W. Tirzepatide vs. semaglutide: Clinical decision-making in the GLP-1 landscape. Expert Opin. Pharmacother. 2025, 26, 1841–1854. [Google Scholar] [CrossRef] [PubMed]
- Osumili, B.; Fan, L.; Paik, J.S.; Pantalone, K.M.; Ranta, K.; Sapin, H.; Tofé, S. Tirzepatide 5, 10 and 15 mg versus injectable semaglutide 0.5 mg for the treatment of type 2 diabetes: An adjusted indirect treatment comparison. Diabetes Res. Clin. Pract. 2024, 212, 111717. [Google Scholar] [CrossRef]
- Lyseng-Williamson, K.A. Glucagon-Like Peptide-1 Receptor Analogues in Type 2 Diabetes: Their Use and Differential Features. Clin. Drug Investig. 2019, 39, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.C.; Lim, J.; Loo, L.S.; Jung, H.; Konig, M.; Tham, L.S. Practical Applications of a Nausea and Vomiting Model in the Clinical Development of Additional Doses of Dulaglutide. J. Clin. Pharmacol. 2024, 64, 215–226. [Google Scholar] [CrossRef]
- Kalyani, R.R.; Neumiller, J.J.; Maruthur, N.M.; Wexler, D.J. Diagnosis and Treatment of Type 2 Diabetes in Adults: A Review. J. Am. Med. Assoc. 2025, 334, 984–1002. [Google Scholar] [CrossRef]
- Qaseem, A.; Obley, A.J.; Shamliyan, T.; Hicks, L.A.; Harrod, C.S.; Crandall, C.J.; Clinical Guidelines Committee of the American College of Physicians; Balk, E.M.; Cooney, T.G.; Cross, J.T.; et al. Newer Pharmacologic Treatments in Adults with Type 2 Diabetes: A Clinical Guideline From the American College of Physicians. Ann. Intern. Med. 2024, 177, 658–666. [Google Scholar] [CrossRef]
- Neves, J.S.; Borges-Canha, M.; Vasques-Nóvoa, F.; Green, J.B.; Leiter, L.A.; Granger, C.B.; Carvalho, D.; Leite-Moreira, A.; Hernandez, A.F.; Del Prato, S.; et al. GLP-1 Receptor Agonist Therapy With and Without SGLT2 Inhibitors in Patients with Type 2 Diabetes. J. Am. Coll. Cardiol. 2023, 82, 517–525. [Google Scholar] [CrossRef]
- Neuen, B.L.; Fletcher, R.A.; Heath, L.; Perkovic, A.; Vaduganathan, M.; Badve, S.V.; Tuttle, K.R.; Pratley, R.; Gerstein, H.C.; Perkovic, V.; et al. Cardiovascular, Kidney, and Safety Outcomes with GLP-1 Receptor Agonists Alone and in Combination with SGLT2 Inhibitors in Type 2 Diabetes: A Systematic Review and Meta-Analysis. Circulation 2024, 150, 1781–1790. [Google Scholar] [CrossRef]
- Apperloo, E.M.; Neuen, B.L.; Fletcher, R.A.; Jongs, N.; Anker, S.D.; Bhatt, D.L.; Butler, J.; Cherney, D.Z.I.; Herrington, W.G.; Inzucchi, S.E.; et al. Efficacy and safety of SGLT2 inhibitors with and without glucagon-like peptide 1 receptor agonists: A SMART-C collaborative meta-analysis of randomised controlled trials. Lancet Diabetes Endocrinol. 2024, 12, 545–557. [Google Scholar] [CrossRef]
- Colombijn, J.M.T.; de Leijer, J.F.; Visseren, F.L.J.; Verhaar, M.C.; van Raalte, D.H.; Sattar, N.; Vernooij, R.W.M.; van Sloten, T.T. Effectiveness and safety of combining SGLT2 inhibitors and GLP-1 receptor agonists in individuals with type 2 diabetes: A systematic review and meta-analysis of cohort studies. Diabetologia 2026, 69, 36–49. [Google Scholar] [CrossRef]
- Chai, S.; Niu, Y.; Liu, F.; Wu, S.; Yang, Z.; Sun, F. Comparison of GLP-1 Receptor Agonists, SGLT-2 Inhibitors, and DPP-4 Inhibitors as an Add-On Drug to Insulin Combined with Oral Hypoglycemic Drugs: Umbrella Review. J. Diabetes Res. 2024, 2024, 8145388. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.; Retnakaran, R.; Zinman, B.; Kramer, C.K. Efficacy of glucagon-like peptide-1 receptor agonists compared to dipeptidyl peptidase-4 inhibitors for the management of type 2 diabetes: A meta-analysis of randomized clinical trials. Diabetes Obes. Metab. 2018, 20, 68–76. [Google Scholar] [CrossRef]
- González-Luis, A.; Llinares-Arvelo, V.; Martínez-Alberto, C.E.; Hernández-Carballo, C.; Mora-Fernández, C.; Navarro-González, J.F.; Donate-Correa, J. Glucagon-like peptide-1 receptor agonists and muscle health: Potential role in sarcopenia prevention and treatment. Eur. J. Endocrinol. 2025, 193, R31–R44. [Google Scholar] [CrossRef] [PubMed]
- Maihemuti, A.; Cui, C.; Wang, Q.; Amuti, R.; Chau, W.W.; Chai, S.; Zhang, N.; Wong, R.M.Y.; Ko, H.; Kwok, T.C.-Y.; et al. Glucagon Like Peptide-1 Receptor Agonists for Sarcopenia and Muscle Wasting Disorders: A Systematic Review of Efficacy and Mechanisms. Aging Dis. 2025, 18, 2. [Google Scholar] [CrossRef]
- Celletti, F.; Farrar, J.; De Regil, L. World Health Organization Guideline on the Use and Indications of Glucagon-Like Peptide-1 Therapies for the Treatment of Obesity in Adults. J. Am. Med. Assoc. 2025, 335, 434. [Google Scholar] [CrossRef]
- Hwang, J.H.; Laiteerapong, N.; Huang, E.S.; Kim, D.D. Lifetime Health Effects and Cost-Effectiveness of Tirzepatide and Semaglutide in US Adults. JAMA Health Forum 2025, 6, e245586. [Google Scholar] [CrossRef]
- Betensky, D.J.; Smith, K.C.; Katz, J.N.; Yang, C.; Hunter, D.J.; Collins, J.E.; Feldman, C.H.; Messier, S.P.; Kim, J.S.; Selzer, F.; et al. The Cost-Effectiveness of Semaglutide and Tirzepatide for Patients with Knee Osteoarthritis and Obesity. Ann. Intern. Med. 2025, 178, 1549–1560. [Google Scholar] [CrossRef]
- Liu, L.; Cui, J.; Neidecker, M.V.; Nahata, M.C. Tirzepatide vs semaglutide and liraglutide for weight loss in patients with overweight or obesity without diabetes: A short-term cost-effectiveness analysis in the United States. J. Manag. Care Spec. Pharm. 2025, 31, 441–450. [Google Scholar] [CrossRef]
- Valentine, W.J.; Hoog, M.; Mody, R.; Belger, M.; Pollock, R. Long-term cost-effectiveness analysis of tirzepatide versus semaglutide 1.0 mg for the management of type 2 diabetes in the United States. Diabetes Obes. Metab. 2023, 25, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; McAdam Marx, C. Short-term cost-effectiveness analysis of tirzepatide for the treatment of type 2 diabetes in the United States. J. Manag. Care Spec. Pharm. 2023, 29, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, J.T.; Landsteiner, A.; Drake, T.; Sultan, S.; Langsetmo, L.; Kaka, A.; Anthony, M.; Billington, C.J.; Kalinowski, C.; Ullman, K.; et al. Cost-Effectiveness of Newer Pharmacologic Treatments in Adults with Type 2 Diabetes: A Systematic Review of Cost-Effectiveness Studies for the American College of Physicians. Ann. Intern. Med. 2024, 177, 633–642. [Google Scholar] [CrossRef]
- Laddu, D.; Neeland, I.J.; Carnethon, M.; Stanford, F.C.; Mongraw-Chaffin, M.; Barone Gibbs, B.; Ndumele, C.E.; Longenecker, C.T.; Chung, M.L.; Rao, G.; et al. Implementation of Obesity Science Into Clinical Practice: A Scientific Statement From the American Heart Association. Circulation 2024, 150, e7–e19. [Google Scholar] [CrossRef] [PubMed]
- Podolsky, M.I.; Raquib, R.; Shafer, P.R.; Hempstead, K.; Ellis, R.P.; Stokes, A.C. Factors Associated with Semaglutide Initiation Among Adults with Obesity. JAMA Netw. Open 2025, 8, e2455222. [Google Scholar] [CrossRef]
- Wright, D.R.; Guo, J.; Hernandez, I. A Prescription for Achieving Equitable Access to Antiobesity Medications. JAMA Health Forum 2023, 4, e230493. [Google Scholar] [CrossRef]
- Klebanoff, M.J.; Li, P.; Long, J.A.; Doshi, J.A. Medicare Part D Coverage and Costs for Glucagon-Like Peptide-1 Receptor Agonists. J. Am. Med. Assoc. 2025, 334, 1848–1850. [Google Scholar] [CrossRef]
- Liu, X.; Lu, C.A.; Shih, Y.-C.T.; Jiang, C. Coverage and Prior Authorization Policies for Semaglutide and Tirzepatide in Medicare Part D Plans. JAMA Netw. Open 2025, 8, e2529842. [Google Scholar] [CrossRef]
- Li, P.; Varghese, J.S.; Shah, M.K.; Galindo, R.J.; Pasquel, F.J.; Ali, M.K.; Shao, H. Prescribing Trends of Glucagon-Like Peptide 1 Receptor Agonists for Type 2 Diabetes or Obesity. JAMA Netw. Open 2025, 8, e2540890. [Google Scholar] [CrossRef]
- Radwan, R.M.; Lee, Y.A.; Kotecha, P.; Wright, D.R.; Hernandez, I.; Ramon, R.; Donahoo, W.T.; Chen, Y.; Allen, J.M.; Bian, J.; et al. Regional trends and disparities in newer GLP1 receptor agonist initiation among real-world adult patients eligible for obesity treatment. Diabetes Obes. Metab. 2025, 27, 3113–3123. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ross, J.S.; Jastreboff, A.M.; Meeker, D.; Freedman, H.; Krumholz, H.M.; Lu, Y. Uptake of and Disparities in Semaglutide and Tirzepatide Prescribing for Obesity in the US. J. Am. Med. Assoc. 2025, 333, 2203–2206. [Google Scholar] [CrossRef] [PubMed]
- Lamprea-Montealegre, J.A.; Madden, E.; Tummalapalli, S.L.; Peralta, C.; Neilands, T.B.; Garcia, P.K.; Muiru, A.; Karliner, L.; Shlipak, M.G.; Estrella, M.M. Association of Race and Ethnicity with Prescription of SGLT2 Inhibitors and GLP1 Receptor Agonists Among Patients with Type 2 Diabetes in the Veterans Health Administration System. J. Am. Med. Assoc. 2022, 328, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, T.; Bekiari, E.; Tsapas, A. Socioeconomic aspects of incretin-based therapy. Diabetologia 2023, 66, 1859–1868. [Google Scholar] [CrossRef]
- Gasoyan, H.; Pfoh, E.R.; Schulte, R.; Sullivan, E.; Le, P.; Rothberg, M.B. Association of patient characteristics and insurance type with anti-obesity medications prescribing and fills. Diabetes Obes. Metab. 2024, 26, 1687–1696. [Google Scholar] [CrossRef]
- Eberly, L.A.; Yang, L.; Essien, U.R.; Eneanya, N.D.; Julien, H.M.; Luo, J.; Nathan, A.S.; Khatana, S.A.M.; Dayoub, E.J.; Fanaroff, A.C.; et al. Racial, Ethnic, and Socioeconomic Inequities in Glucagon-Like Peptide-1 Receptor Agonist Use Among Patients with Diabetes in the US. JAMA Health Forum 2021, 2, e214182. [Google Scholar] [CrossRef]
- Hernandez, I.; Wright, D.R.; Guo, J.; Shrank, W.H. Medicare Part D Coverage of Anti-obesity Medications: A Call for Forward-Looking Policy Reform. J. Gen. Intern. Med. 2024, 39, 306–308. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.H.; Laiteerapong, N.; Huang, E.S.; Mozaffarian, D.; Fendrick, A.M.; Kim, D.D. Fiscal Impact of Expanded Medicare Coverage for GLP-1 Receptor Agonists to Treat Obesity. JAMA Health Forum 2025, 6, e250905. [Google Scholar] [CrossRef]
- Hoog, M.M.; Vallarino, C.; Maldonado, J.M.; Grabner, M.; Teng, C.-C.; Terrell, K.; Richard, E.L. Real-World Effectiveness of Tirzepatide versus Semaglutide on HbA1c and Weight in Patients with Type 2 Diabetes. Diabetes Ther. 2025, 16, 2237–2256. [Google Scholar] [CrossRef]
- Thomsen, R.W.; Mailhac, A.; Løhde, J.B.; Pottegård, A. Real-world evidence on the utilization, clinical and comparative effectiveness, and adverse effects of newer GLP-1RA-based weight-loss therapies. Diabetes Obes. Metab. 2025, 27, 66–88. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Tittel, S.R.; Schloot, N.C.; Heitmann, E.; Otto, T.; Lebrec, J.; Pavel, M.; Lanzinger, S. Clinical characteristics, treatment patterns, and persistence in individuals with type 2 diabetes initiating a glucagon-like peptide-1 receptor agonist: A retrospective analysis of the Diabetes Prospective Follow-Up Registry. Diabetes Obes. Metab. 2023, 25, 1813–1822. [Google Scholar] [CrossRef]
- Spinelli, K.J.; Oakes, A.H.; Chiu, S.-T.; Imboden, M.T.; Miller, A.; Jain, S.; Gluckman, T.J. Health Disparity Clusters of Off Label Prescriptions for Glucagon-Like Peptide 1 Receptor Agonists. Am. J. Med. Open 2025, 13, 100100. [Google Scholar] [CrossRef]
- Parkman, H.P.; Rim, D.S.; Anolik, J.R.; Dadparvar, S.; Maurer, A.H. Glucagonlike Peptide-1 Receptor Agonists: The Good, the Bad, and the Ugly—Benefits for Glucose Control and Weight Loss with Side Effects of Delaying Gastric Emptying. J. Nucl. Med. Technol. 2024, 52, 3–7. [Google Scholar] [CrossRef]
- Sodhi, M.; Rezaeianzadeh, R.; Kezouh, A.; Etminan, M. Risk of Gastrointestinal Adverse Events Associated with Glucagon-Like Peptide-1 Receptor Agonists for Weight Loss. J. Am. Med. Assoc. 2023, 330, 1795–1797. [Google Scholar] [CrossRef]
- Zhang, D.; Gencerliler, N.; Mukhopadhyay, A.; Blecker, S.; Grams, M.E.; Wright, D.R.; Wang, V.H.-C.; Rajan, A.; Butt, E.; Shin, J.-I.; et al. Association of Patient Cost Sharing with Adherence to GLP-1RA and Adverse Health Outcomes. Diabetes Care 2025, 48, 1329–1336. [Google Scholar] [CrossRef]
- Farmer, A.J.; Prevost, A.T.; Hardeman, W.; Craven, A.; Sutton, S.; Griffin, S.J.; Kinmonth, A.-L.; Support and Advice for Medication Trial Group. Protocol for SAMS (Support and Advice for Medication Study): A randomised controlled trial of an intervention to support patients with type 2 diabetes with adherence to medication. BMC Fam. Pract. 2008, 9, 20. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gershkowitz, B.D.; Hillert, C.J.; Crotty, B.H. Digital Coaching Strategies to Facilitate Behavioral Change in Type 2 Diabetes: A Systematic Review. J. Clin. Endocrinol. Metab. 2021, 106, e1513–e1520. [Google Scholar] [CrossRef] [PubMed]
- Aroda, V.R.; Rosenstock, J.; Terauchi, Y.; Altuntas, Y.; Lalic, N.M.; Morales Villegas, E.C.; Jeppesen, O.K.; Christiansen, E.; Hertz, C.L.; Haluzík, M.; et al. PIONEER 1: Randomized Clinical Trial of the Efficacy and Safety of Oral Semaglutide Monotherapy in Comparison with Placebo in Patients with Type 2 Diabetes. Diabetes Care 2019, 42, 1724–1732. [Google Scholar] [CrossRef]
- Rosenstock, J.; Allison, D.; Birkenfeld, A.L.; Blicher, T.M.; Deenadayalan, S.; Jacobsen, J.B.; Serusclat, P.; Violante, R.; Watada, H.; Davies, M.; et al. Effect of Additional Oral Semaglutide vs Sitagliptin on Glycated Hemoglobin in Adults with Type 2 Diabetes Uncontrolled with Metformin Alone or with Sulfonylurea: The PIONEER 3 Randomized Clinical Trial. J. Am. Med. Assoc. 2019, 321, 1466–1480. [Google Scholar] [CrossRef]
- Wharton, S.; Lingvay, I.; Bogdanski, P.; Duque do Vale, R.; Jacob, S.; Karlsson, T.; Shaji, C.; Rubino, D.; Garvey, W.T.; OASIS 4 Study Group. Oral Semaglutide at a Dose of 25 mg in Adults with Overweight or Obesity. N. Engl. J. Med. 2025, 393, 1077–1087. [Google Scholar] [CrossRef]
- Bækdal, T.A.; Breitschaft, A.; Donsmark, M.; Maarbjerg, S.J.; Søndergaard, F.L.; Borregaard, J. Effect of Various Dosing Conditions on the Pharmacokinetics of Oral Semaglutide, a Human Glucagon-Like Peptide-1 Analogue in a Tablet Formulation. Diabetes Ther. 2021, 12, 1915–1927. [Google Scholar] [CrossRef]
- van Hout, M.; Forte, P.; Jensen, T.B.; Boschini, C.; Bækdal, T.A. Effect of Various Dosing Schedules on the Pharmacokinetics of Oral Semaglutide: A Randomised Trial in Healthy Subjects. Clin. Pharmacokinet. 2023, 62, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Wharton, S.; Aronne, L.J.; Stefanski, A.; Alfaris, N.F.; Ciudin, A.; Yokote, K.; Halpern, B.; Shukla, A.P.; Zhou, C.; Macpherson, L.; et al. Orforglipron, an Oral Small-Molecule GLP-1 Receptor Agonist for Obesity Treatment. N. Engl. J. Med. 2025, 393, 1796–1806. [Google Scholar] [CrossRef]
- Lilly’s Oral GLP-1, Orforglipron, Superior to Oral Semaglutide in Head-to-Head Trial|Eli Lilly and Company. Available online: https://investor.lilly.com/news-releases/news-release-details/lillys-oral-glp-1-orforglipron-superior-oral-semaglutide-head (accessed on 27 December 2025).
- Pratt, E.; Ma, X.; Liu, R.; Robins, D.; Haupt, A.; Coskun, T.; Sloop, K.W.; Benson, C. Orforglipron (LY3502970), a novel, oral non-peptide glucagon-like peptide-1 receptor agonist: A Phase 1a, blinded, placebo-controlled, randomized, single- and multiple-ascending-dose study in healthy participants. Diabetes Obes. Metab. 2023, 25, 2634–2641. [Google Scholar] [CrossRef] [PubMed]
- Escobar, J.; Monday, O.; Vemoori, Y.; Yadav, I.; Maslamani, A.N.J.; Al Kutabi, S.; Saeed, L.; Khan, A. Safety and Efficacy of Efpeglenatide in Patients with Type 2 Diabetes: A Meta-Analysis of Randomized Controlled Trials. Cureus 2023, 15, e45927. [Google Scholar] [CrossRef]
- Del Prato, S.; Kang, J.; Trautmann, M.E.; Stewart, J.; Sorli, C.H.; Derwahl, M.; Soto, A.; Yoon, K.-H. Efficacy and safety of once-monthly efpeglenatide in patients with type 2 diabetes: Results of a phase 2 placebo-controlled, 16-week randomized dose-finding study. Diabetes Obes. Metab. 2020, 22, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Metsera. A Phase 2b, Multi-Center, Randomized, Double-Blind, Placebo-Controlled Study to Investigate the Efficacy, Safety and Tolerability of Multiple Dose Regimens of Once-Weekly Switching to Once-Monthly MET097 in Participants with Obesity or Overweight (VESPER-3). Clinicaltrials.gov, Clinical Trial Registration NCT06973720. May 2025. Available online: https://clinicaltrials.gov/study/NCT06973720 (accessed on 27 December 2025).
- Sarma, S.; Palcu, P. Weight loss between glucagon-like peptide-1 receptor agonists and bariatric surgery in adults with obesity: A systematic review and meta-analysis. Obesity 2022, 30, 2111–2121. [Google Scholar] [CrossRef]
- Eli Lilly and Company. A Randomized, Double-Blind, Phase 3 Study to Investigate the Efficacy and Safety of LY3437943 Once Weekly Compared to Placebo in Participants with Severe Obesity and Established Cardiovascular Disease. Clinicaltrials.gov, Clinical Trial Registration NCT05882045. July 2025. Available online: https://clinicaltrials.gov/study/NCT05882045 (accessed on 27 December 2025).
- Bossart, M.; Wagner, M.; Elvert, R.; Evers, A.; Hübschle, T.; Kloeckener, T.; Lorenz, K.; Moessinger, C.; Eriksson, O.; Velikyan, I.; et al. Effects on weight loss and glycemic control with SAR441255, a potent unimolecular peptide GLP-1/GIP/GCG receptor triagonist. Cell Metab. 2022, 34, 59–74.e10. [Google Scholar] [CrossRef]
- Hanmi Pharmaceutical Company Limited. A Phase 2, Randomized, Double-Blind, Placebo-Controlled, Parallel Group Study to Evaluate Efficacy, Safety and Tolerability of HM15211(Efocipegtrutide) Treatment for 12 Months in Subjects with Biopsy Confirmed NASH. Clinicaltrials.gov, Clinical Trial Registration NCT04505436. November 2025. Available online: https://clinicaltrials.gov/study/NCT04505436 (accessed on 27 December 2025).
- Hope, D.C.D.; Tan, T.M.-M. Glucagon and energy expenditure; Revisiting amino acid metabolism and implications for weight loss therapy. Peptides 2023, 162, 170962. [Google Scholar] [CrossRef]
- Nair, K.S.; Halliday, D.; Matthews, D.E.; Welle, S.L. Hyperglucagonemia during insulin deficiency accelerates protein catabolism. Am. J. Physiol. 1987, 253, E208–E213. [Google Scholar] [CrossRef]
- Enebo, L.B.; Berthelsen, K.K.; Kankam, M.; Lund, M.T.; Rubino, D.M.; Satylganova, A.; Lau, D.C.W. Safety, tolerability, pharmacokinetics, and pharmacodynamics of concomitant administration of multiple doses of cagrilintide with semaglutide 2·4 mg for weight management: A randomised, controlled, phase 1b trial. Lancet 2021, 397, 1736–1748. [Google Scholar] [CrossRef] [PubMed]
- Kawao, N.; Nishikawa, A.; Matsumura, D.; Yamada, A.; Ohira, T.; Mizukami, Y.; Kaji, H. Roles of Insulin-Like Growth Factor-1 in Muscle Wasting and Osteopenia in Mice with Hyponatremia. Calcif. Tissue Int. 2025, 116, 61. [Google Scholar] [CrossRef]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.-S.; Harrison, S.A.; NN9931-4296 Investigators. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef]
- Malhotra, A.; Bednarik, J.; Chakladar, S.; Dunn, J.P.; Weaver, T.; Grunstein, R.; Fietze, I.; Redline, S.; Azarbarzin, A.; Sands, S.A.; et al. Tirzepatide for the treatment of obstructive sleep apnea: Rationale, design, and sample baseline characteristics of the SURMOUNT-OSA phase 3 trial. Contemp. Clin. Trials 2024, 141, 107516. [Google Scholar] [CrossRef]
- Edison, P.; Femminella, G.D.; Ritchie, C.; Nowell, J.; Holmes, C.; Walker, Z.; Ridha, B.; Raza, S.; Livingston, N.R.; Frangou, E.; et al. Liraglutide in mild to moderate Alzheimer’s disease: A phase 2b clinical trial. Nat. Med. 2025, 32, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Novo Nordisk A/S. A Randomised Double-blind Placebo-controlled Clinical Trial Investigating the Effect and Safety of Oral Semaglutide in Subjects with Early Alzheimer’s Disease (EVOKE Plus). Clinicaltrials.gov, Clinical Trial Registration NCT04777409. November 2025. Available online: https://clinicaltrials.gov/study/NCT04777409 (accessed on 27 December 2025).
- Aranäs, C.; Edvardsson, C.E.; Shevchouk, O.T.; Zhang, Q.; Witley, S.; Blid Sköldheden, S.; Zentveld, L.; Vallöf, D.; Tufvesson-Alm, M.; Jerlhag, E. Semaglutide reduces alcohol intake and relapse-like drinking in male and female rats. eBioMedicine 2023, 93, 104642. [Google Scholar] [CrossRef]
- Scheen, A.J. Glucagon-like peptide-1 receptor agonists and alcohol use disorders: An emerging unexpected beneficial effect. Diabetes Obes. Metab. 2025, 27, 4083–4091. [Google Scholar] [CrossRef] [PubMed]
- Do, D.; Lee, T.; Peasah, S.K.; Good, C.B.; Inneh, A.; Patel, U. GLP-1 Receptor Agonist Discontinuation Among Patients with Obesity and/or Type 2 Diabetes. JAMA Netw. Open 2024, 7, e2413172. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.