Abstract
A convenient synthetic approach to previously unknown NH-iminophosphonates bearing 2-, 3-, and 4-pyridyldifluoromethyl groups at the imine carbon atom was developed. The synthetic potential of these novel building blocks was demonstrated by their conversion into highly functionalized acyclic and heterocyclic aminophosphonates and phosphonic acids combining in their structure biorelevant aminophosphonic fragment, difluoromethyl group, and pyridyl, piperidyl, thiazolidin-4-one, or thiazidinan-4-one heterocyclic moieties in a single molecular platform.
Keywords:
iminophosphonates; aminophosphonates; NH-imines; nitriles; difluoromethyl; pyridine; piperidine; E-Z isomerism 1. Introduction
α-Aminophosphonates are phosphorus analogs of amino acids in which the planar carboxylic group is replaced with a tetrahedral bioisosteric phosphonic unit. They exhibit a wide range of biological activity and play a significant role in the development of antibiotics, antiviral, antihypertensive, antitumor agents and other bioactive substances [1,2,3,4,5]. Of particular interest are fluorinated aminophosphonates. Modification of organic molecule with fluorine has become almost a standard tool in modern drug design. It is generally accepted that introduction of a fluorine containing group may improve the pharmacodynamic and the pharmacokinetic profiles of the compound by concomitant alteration of its electronic, lipohilic, and steric characteristics as well as its metabolic stability [6,7,8,9].
α-Iminophosphonates, i.e., the compounds bearing (RO)2P(O)C=N-fragment, are valuable precursors of α-aminophosphonates. Unprotected NH-iminophosphonates are especially promising in this respect. They show enhanced reactivity and allow straightforward preparation of aminophosphonates with a free NH2 group avoiding tedious N-deprotection step. Recently we have prepared the first representatives of NH-iminophosphonates and demonstrated their potential for the introduction of pharmacophore α-fluoroalkylated aminophosphonic unit into various structures.
NH-Iminophosphonates incorporating heterocyclic residue were unknown so far. In the present work we report the synthesis of NH-iminophosphonates bearing α-, β-, or γ-pyridyldifluoromethyl group at the imine carbon atom and their use for the preparation of aminophosphonic acids derivatives bearing difluoromethyl group and heterocyclic residue. Note, that compounds with 2-pyridyldifluoromethyl fragment were used for the development of effective thrombin and trypsin inhibitors [10] and highly selective antifungal agents [11]. The presence of a pyridine ring offers additional synthetic possibilities connected with the hydrogenation of heterocyclic ring and preparation of compounds containing aminophosphonic unit, difluoromethyl group and piperidine fragment in the same molecule. It is worth of noting that according to [12] piperidine and pyridine are most frequent nitrogen heterocycles in U.S. FDA approved drugs.
2. Results and Discussion
Analysis of the literature data shows that hydrophosphoryl compounds can react with nitriles by different schemes. Thus, reaction of dialkylphosphites with alkyl, cycloalkyl, aryl, or pyridyl nitriles in the presence of acids [13,14] or under free radical conditions [15] leads to aminobisphosphonates I, as the main products, formed in low to moderate yields (Scheme 1). At the same time, under basic conditions benzonitrile reacts with dialkyl or diphenyl phosphites to form phosphorylamino phosphonates II [16]. Recently we have shown that highly electrophilic polyfluoroalkylated nitriles in the reactions with dialkyl or diphenyl phosphite afford first representatives of NH-iminophosphonates III [17,18]. At the same time, mixed perfluoro(chloro) acetonitriles react with (RO)2POH (R = Et, Ph) by two competitive routes: addition to the C≡N bond affording the respective N-unprotected iminophosphonates III, or reductive dehalogenation leading to chloro(fluoro) acetonitriles IV and the respective halogenophosphates V [19].
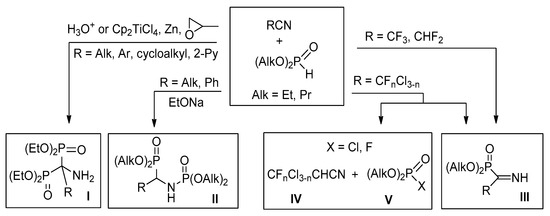
Scheme 1.
Reactions of nitriles with hydrophosphoryl compounds.
Thus, the results of reactions of hydrophosphoryl compounds with nitriles depend on reactants structure and conditions.
We have found that all three isomeric pyridyldifluoroacetonitriles 1a–c react with diethyl phosphite under mild conditions (20 mol.% Et3N, r.t.) to afford respective NH-iminophosphonates 2a–c (Scheme 2). It should be noted that 2-pyridyl nitrile 1a reacts much more slowly than the 3- and 4-isomers 1b,c: under the same conditions the reaction is completed within 120 and 12 h, respectively. Similar differences have previously been noted for pKa values of the isomeric pyridineacetic acids [20] which may be associated with the specific effect of the α-nitrogen atom.
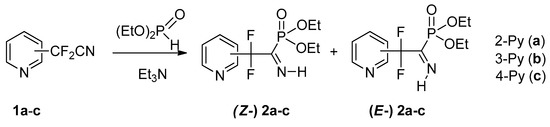
Scheme 2.
Synthesis of pyridyldifluoromethylated NH-iminophosphonates.
Formation of the P-C=N-H fragment is confirmed by 13C NMR spectra of compounds 2, in which the imine C-atom signal (δC171–173 ppm) with a large direct C-P coupling constant (1JCP 153–154 Hz for Z-izomers 2a–c; 210 Hz for E-isomer 2a) was identified, and 1H, 31P NMR spectra showing characteristic spin-spin interaction of the N-H proton and phosphorus atom (3JP-C-N-H 40–62 Hz).
Iminophosphonates 2a–c are formed as an equilibrium mixture of Z/E isomers and, similarly to other fluoroalkylated NH-iminophosphonates, a more sterically hindered Z configuration is preferred. We have shown previously that substituent at the C=N bond of iminophosphonates markedly affect E/Z isomeric ratio: C-trifluoromethylated iminophosphonates exist preferentially in Z configuration, whereas for their C-arylated analogs E-configuration prevails [21,22]. Table 1 shows the values of equilibrium Z/E ratios and phosphorus chemical shifts for pyridyldifluoromethylphosphonates 2a–c and some other related iminophosphonates. It is seen that 31P NMR data and Z/E ratios for iminophosphonates 2b,c are very close and markedly differ from those for isomer 2a. It is very interesting to note from analysis of Table 1 that imines 2b,c bearing more strong electron withdrawing 3- and 4-pyridyl substituent (σp 0.25 and 0.44, respectively [23] are in this respect more similar to trifluoromethylated analog 3, whereas isomer 2a with less electron withdrawing 2-pyridyl group (σp 0.17) is more like benzimidoyl phosphonates 4, 5.

Table 1.
Z/E isomeric ratio and selected 13C, 31P NMR data of the compounds 2 and some related iminophosphonates.
Identification of Z,E-isomers 2a–c is based mainly on the significant difference in coupling constants of the N-H proton and phosphorus atom (3JP-C-N-H ~40 and ~61–62 Hz, respectively) and high-field shift of phosphorus signals of Z-isomers in 31P NMR spectra, previously noted for other α-iminophosphonates [17,18,21,22].
Iminophosphonates 2 contain a polarized C=N bond activated by the presence of electron-withdrawing group at the imine carbon atom. Thus, imines 2a–c readily add methanol across the C=N bond to afford adduct 6a–c (Scheme 3). The reactivity decreases in the order 2a > 2b,c: after 8 h the conversion is 100%, 44%, and 46%, respectively. The complete conversion of 2b,c proceeds within 2 days.
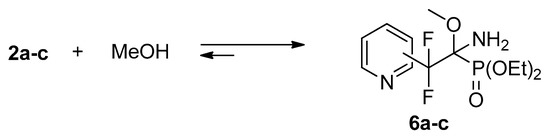
Scheme 3.
Reversible reaction of iminophosphonates 2 with methanol.
Similar to trifluoromethylated analogs [17,18,21], compounds 6a–c in CDCl3 solution partially (~15–20%) dissociate to the starting compounds. This property of NH-imine adducts was successfully explored for the improvement of the enantiomeric excess in catalytic asymmetric reduction [24].
It is very interesting to note, that addition of methanol to iminophosphonates 2 is accompanied by the change in the Z/E ratio of the starting compounds and Z-E equilibrium is established very quickly. Thus, immediately after dissolution of imines 2a–c in methanol, Z/E ratios of imines 2a–c, according to 31P, 19F NMR data, are 1:1, 2.5:1, and 2:1, respectively. Evaporation of methanol and dissolution in deuterochloroform leads to recovery of Z/E isomeric ratios, presented in Table 1, i.e., 2:1, 6:1, and 7:1, respectively. Similar increase of E-isomer content on going from aprotic to protic solvent was shown recently for the sodium salts of N-methyliminophosphonates, bearing aryl or hetaryl substituents at the imine carbon atom [22].
Synthesis of biorelevant pirydyldifluoroemethylated aminophosphonates and aminophosphonic acids. We have found that selective reduction of the C=N bond in iminophosphonates 2a–c with borane-dimethylsulfide (BMS) proceeds under mild conditions to give aminophosphonates 7a–c incorporating pyridyldifluoromethyl residue (Scheme 4). Free aminophosphonates 7a–c are not very stable (the stability decreases in the order 7c > 7b > 7a) and should be stored at the reduced temperature in refrigerator.
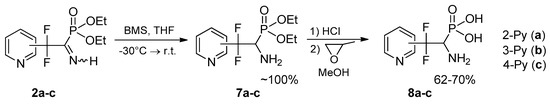
Scheme 4.
Selective reduction of the C=N bond in the iminophosphonates 2a–c.
Preparation of heterocycle-containing aminophosphonic acids from their esters are often complicated by side processes. It was found that hydrolysis of aminophosphonates 7a–c with conc. HCl proceeds cleanly and affords the first representatives of fluoroalkylated aminophosphonic acids 8a–c, incorporating heterocyclic moiety in the β-position.
The possibility of reducing the pyridine ring was studied using compound 7c as the most stable pyridyl substituted aminophosphonate. It was found that catalytic hydrogenation of 7c leads to complete reduction of pyridine ring to afford 4-piperidyl substituted aminophosphonate 9 (Scheme 5).
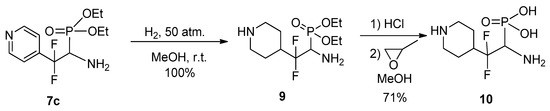
Scheme 5.
Catalytic hydrogenation of the aminophosphonate 7c.
The latter should be stored at a reduced temperature, since under ordinary conditions it gradually decomposes, most likely undergoing O-N transfer of the ethyl group. More stable free aminophosphonic acid 10 was obtained by hydrolysis of 9 followed by treatment with propylene oxide.
Cyclocondensation with mercaptocarboxylic acids. Highly polarized azomethyne bond in the imines 2 can be readily functionalized by the reactions with the bifunctional compounds. Thus, cyclocondensation of iminophosphonates 2a–c with thioglycolic or 3-mercaptopropionic acid leads to highly functionalized thiazolidin-4-ones 11a–c or thiazidinan-4-ones 12a–c, respectively (Scheme 6). Obviously, the reaction proceeds via primary nucleophilic addition to the C=N bond followed by intramolecular ring closure in the intermediate A. The ease of cyclization of the latter is due to the spatial accessibility of the unsubstituted N-nucleophilic center, and the experimentally found noticeably faster formation of thiazolidinones 11 as compared to thiazinanones 12 is explained by the proximity of the reaction centers upon creation of the 5-membered cycle. We were able to detect by 31P NMR adduct A (Scheme 6, n = 2) resulting from addition of 3-mercaptopropionic acid (δP 16 ppm).
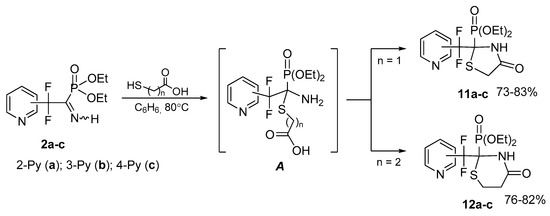
Scheme 6.
Cyclocondensation with mercaptocarboxylic acids.
3. Conclusions
In summary, the triethylamine catalyzed addition of diethyl phosphite to isomeric pyridyldifluoroacetonitriles leads to α-iminopyridyldifluoroethyl phosphonates, existing as equilibrium mixture of E-Z isomers. The synthetic potential of these novel electrophilic building blocks was demonstrated by their reduction to biorelevant compounds combining in their structure aminophosphonic residue, difluoromethyl group and isomeric pyridine or piperidine moiety. Cyclocondensation with mercaptocarboxylic acids allow preparation of hybrid molecules bearing two bioactive heterocyclic moieties (pyridine and thiazolidine or thiazinane) and aminophosphonic fragment in a single molecular platform. These reactions proceed under mild neutral conditions and lead directly to N-unprotected aminophosphonic derivatives.
4. Materials and Methods
1H, 19F, and 13C NMR spectra were recorded using Bruker Avance NMR spectrometers operating at 302, 400 and 499.8 1H frequencies; 75.8, 125.7 and 150.8 MHz for 13C experiments; 188, 376.5 and 470.3 MHz for 19F; 81 and 202.3 MHz for 31P. Chemical shifts are reported relative to internal TMS (1H) or CFCl3 (19F) and external 85%-H3PO4 (31P) standards. Melting points are uncorrected. Solvents were dried before use according to standard methods. Elemental analysis was carried out in the analytical laboratory of Institute of Organic Chemistry, NAS of Ukraine. See the Supplementary File for the 1H, 19F, and 13C NMR spectra of compounds synthesized.
4.1. Synthesis of Diethyl (2,2-Difluoro-1-imino-2-(pyridinyl)ethyl)phosphonates 2a–c: General Procedure
To equimolar mixture of pyridinylacetonitrile 1 (0.45 g, 2.9 mmol) and diethyl phosphite (0.4 g, 2.9 mmol) was added triethylamine (0.58 mmol, 0.059 g). After 12 h for 2b,c and 120 h for 2a triethylamine was removed under vacuum.
4.1.1. Diethyl (2,2-Difluoro-1-imino-2-(pyridin-2-yl)ethyl)phosphonate 2a
Yield 0.85 g (100%); light brown oil. Z/E = 2:1. Z-isomer: 1H NMR (499.8 MHz, CDCl3) δ (ppm): 1.33 (t, 3JHH = 7.2 Hz, 6H, CH3), 4.16–4.22 (m, 4H, CH2), 7.40 (t, 3JHH = 7.9 Hz, 1H, Py), 7.74 (d, 3JHH = 7.9 Hz, 1H, Py), 7.84–7.88 (m, 1H, Py), 8.66 (d, 3JHH = 4.9 Hz, 1H, Py), 12.27 (d, 3JHP = 39.9 Hz, 1H, NH).
13C NMR (75.8 MHz, CDCl3) δ (ppm): 16.0 (d, 3JCP = 6 Hz, CH3), 63.8 (d, 2JCP = 6 Hz, CH2), 116.1 (td, 1JCF = 246 Hz, 2JCP = 33 Hz, CF2), 120.9 (t, 3JCF = 4 Hz, CPy), 125.1 (s, CPy), 136.9 (s, CPy), 149.3 (s, CPy), 152.8 (t, 2JCF = 28.5 Hz, CCF2), 173.2 (dt, 1JCP = 153 Hz, 2JCF = 33 Hz, C=N).
19F NMR (470.3 MHz, CDCl3) δ (ppm): −100.2.
31P NMR (202.3 MHz, CDCl3) δ (ppm): 5.9 (m, 3JPH = 39.9 Hz).
E-isomer: 1H NMR (499.8 MHz, CDCl3) δ (ppm): 1.26 (t, 3JHH = 7.1 Hz, 6H, CH3), 4.10–4.15 (m, 4H, CH2), 7.40 (t, 3JHH = 7.7 Hz, 1H, Py), 7.80–7.84 (m, 2H, Py), 8.6 (d, 3JHH = 4.9 Hz, 1H, Py), 11.97 (dt, 3JHP = 61.6 Hz, 4JHF = 4.0 Hz, 1H, NH).
13C NMR (75.8 MHz, CDCl3) δ (ppm): 16.2 (d, 3JCP = 6 Hz, CH3), 63.7 (d, 2JCP = 6 Hz, CH2), 113.9 (td, 1JCF = 249 Hz, 2JCP = 42 Hz, CF2), 120.8 (t, 3JCF = 4 Hz, CPy), 125.4 (s, CPy), 137.3 (s, CPy), 149.1 (s, CPy), 151.5 (t, 2JCF = 29 Hz, CCF2), 170.8 (dt, 1JCP = 210 Hz, 2JCF = 30 Hz, C=N).
19F NMR (470.3 MHz, CDCl3) δ (ppm): −103.5.
31P NMR (202.3 MHz, CDCl3) δ (ppm): 6.6 (m, 3JPH = 61.6 Hz).
Anal. Calc. for C11H15F2N2O3P: C, 45.21; H, 5.17; N, 9.59. Found: C, 45.38; H, 5.29; N, 9.67.
4.1.2. Diethyl (2,2-Difluoro-1-imino-2-(pyridin-3-yl)ethyl)phosphonate 2b
Yield 0.85 g (100%); yellow oil. Z/E = 6:1. Z-isomer: 1H NMR (302 MHz, CDCl3) δ (ppm): 1.27 (t, 3JHH = 7.1 Hz, 6H, CH3), 3.93–4.20 (m, 4H, CH2), 7.29 (dd, 3JHH = 8.1 Hz, 3JHH = 5.1 Hz, Py), 7.81 (d, 3JHH = 8.1 Hz, 1H, Py), 8.63 (d, 3JHH = 5.1 Hz, 1H, Py), 8.74 (s, 1H, Py), 12.17 (d, 3JHP = 39.6 Hz, 1H, NH).
13C NMR (75.8 MHz, CDCl3) δ (ppm): 16.1 (d, 3JCP = 6 Hz, CH3), 63.9 (d, 2JCP = 6 Hz, CH2), 117.3 (td, 1JCF = 246 Hz, 2JCP = 35 Hz, CF2), 122.9 (s, CPy), 129.9 (t, 2JCF = 27 Hz, CCF2), 133.8 (t, 3JCF = 6 Hz, CPy), 147.3 (t, 3JCF = 6.5 Hz, CPy), 151.6 (s, CPy), 173.0 (dt, 1JCP = 154 Hz, 2JCF = 35 Hz, C=N).
19F NMR (470.3 MHz, CDCl3) δ (ppm): −97.5.
31P NMR (81 MHz, CDCl3) δ (ppm): 1.2 (m, 3JPH = 39.6 Hz).
E-isomer: 1H NMR (302 MHz, CDCl3) δ (ppm): 1.15 (t, 3JHH = 6.9 Hz, 6H, CH3), 3.93–4.20 (m, 4H, CH2), 7.30–7.35 (m, 1H, Py), 7.85 (d, 3JHH = 6.7 Hz, 1H, Py), 8.66 (d, 3JHH = 7.2 Hz, 1H, Py), 8.78 (s, 1H, Py), 11.85 (d, 3JHP = 61.2 Hz, 1H, NH).
19F NMR (470.3 MHz, CDCl3) δ (ppm): −100.5.
31P NMR (81 MHz, CDCl3) δ (ppm): 2.9 (m, 3JPH = 61.2 Hz).
Anal. Calc. for C11H15F2N2O3P: C, 45.21; H, 5.17; N, 9.59. Found: C, 45.46; H, 5.04; N, 9.43.
4.1.3. Diethyl (2,2-Difluoro-1-imino-2-(pyridin-4-yl)ethyl)phosphonate 2c
Yield 0.85 g (100%); light orange oil. Z/E = 7:1. Z-isomer: 1H NMR (400 MHz, CDCl3) δ (ppm): 1.34 (t, 3JHH = 7.1 Hz, 6H, CH3), 4.14–4.27 (m, 4H, CH2), 7.50 (d, 3JHH = 6 Hz, 2H, Py), 8.74 (d, 3JHH = 6 Hz, 2H, Py), 12.21 (d, 3JHP = 39.5 Hz, 1H, NH).
13C NMR (75.8 MHz, CDCl3) δ (ppm): 16.3 (d, 3JCP = 6 Hz, CH3), 64.1 (d, 2JCP = 6 Hz, CH2), 116.9 (td, 1JCF = 246 Hz, 2JCP = 34 Hz, CF2), 120.4 (t, 3JCF = 6 Hz, 2CPy), 142.2 (td, 2JCF = 27 Hz, 3JCP = 6 Hz, CCF2), 150.2 (s, 2CPy), 172.9 (dt, 1JCP = 154 Hz, 2JCF = 35 Hz, C=N).
19F NMR (470.3 MHz, CDCl3) δ (ppm): −99.4.
31P NMR (202.3 MHz, CDCl3) δ (ppm): 0.1 (m, 3JPH = 39.5 Hz).
E-isomer: 1H NMR (400 MHz, CDCl3) δ (ppm): 1.26 (t, 3JHH = 7.1 Hz, 6H, CH3), 4.05–4.13 (m, 4H, CH2), 7.45 (d, 3JHH = 5 Hz, 2H, Py), 8.76 (d, 3JHH = 5 Hz, 2H, Py), 11.81 (d, 3JHP = 60.9 Hz, 1H, NH).
19F NMR (470.3 MHz, CDCl3) δ (ppm): −102.4.
31P NMR (202.3 MHz, CDCl3) δ (ppm): 1.8 (m, 3JPH = 60.9 Hz).
Anal. Calc. for C11H15F2N2O3P: C, 45.21; H, 5.17; N, 9.59. Found: C, 45.03; H, 5.23; N, 9.41.
4.2. Reactions of Iminophosphonates 2a–c with Methanol
Iminophosphonates 2a–c (0.02 g, 0.07 mmol) were dissolved in methanol (0.6 mL) at room temperature. According to 31P, 19F NMR the reaction was completed after 8 h (2a) or 2 days (2b,c). The spectral data are similar to those of methanol adduct with trifluoromethyl analog [17].
Spectral data for adduct 6a: 1H NMR (400 MHz, CDCl3) δ (ppm): 1.30–1.34 (m, 6H, CH3), 3.21 (s, 3H, OCH3), 4.20–4.30 (m, 4H, CH2), 7.36 (dd, 3JHH = 8.1 Hz, 3JHH = 3.9 Hz, 1H, Py), 7.68 (d, 3JHH = 8.1 Hz, 1H, Py), 7.78 (t, 3JHH = 8.1 Hz, 1H, Py), 8.60 (d, 3JHH = 3.9 Hz, 1H, Py). 19F NMR (470.3 MHz, MeOH) δ (ppm): −103.0 (d, 2JFAFB = 255 Hz), −110.5 (d, 2JFBFA= 255 Hz). 31PNMR (202.3 MHz, MeOH) δ (ppm): 15.1.
Spectral data for adduct 6b: 1H NMR (400 MHz, CDCl3) δ (ppm): 1.09 (t, 3JHH = 7.5 Hz, 3H, CH3), 1.27 (t, 3JHH = 7.6 Hz, 3H, CH3), 2.40 (d, 3JHP = 20 Hz, 2H, NH2), 3.37 (s, 3H, OCH3), 3.94–4.08 (m, 2H, OCH2), 4.13–4.18 (m, 2H, OCH2), 7.30 (dd, 3JHH = 7.3 Hz, 3JHH = 4.2 Hz, 1H, Py), 7.87 (d, 3JHH = 7.3 Hz, 1H, Py), 8.63 (d, 3JHH = 4.2 Hz, 1H, Py), 8.77 (s, 1H, Py). 19F NMR (470.3 MHz, MeOH) δ (ppm): −103.9 (d, 2JFAFB = 257 Hz), −106.6 (dd, 2JFBFA = 257 Hz, 3JFP = 7 Hz). 31PNMR (202.3 MHz, MeOH) δ (ppm): 14.8 (m, 3JPF = 7 Hz).
Spectral data for adduct 6c: 1H NMR (400 MHz, CDCl3) δ (ppm): 1.13 (t, 3JHH = 6.9 Hz, 3H, CH3), 1.30 (t, 3JHH = 6.6 Hz, 3H, CH3), 2.40 (d, 3JHP = 22.5 Hz, 2H, NH2), 3.38 (s, 3H, OCH3), 3.97–4.12 (m, 2H, OCH2), 4.13–4.21 (m, 2H, OCH2), 7.49 (d, 3JHH = 5.3 Hz, 2H, Py), 8.66 (d, 3JHH = 5.3 Hz, 2H, Py). 19F NMR (470.3 MHz, MeOH) δ (ppm): −106.4 (d, 2JFAFB = 254 Hz), −108.4 (dd, 2JFBFA= 254 Hz, 3JFP = 7 Hz). 31PNMR (202.3 MHz, MeOH) δ (ppm): 13.1 (m, 3JPF = 7 Hz).
4.3. Synthesis of Diethyl (1-Amino-2,2-difluoro-2-(pyridinyl)ethyl)phosphonates 7a–c: General Procedure
To a solution of respective iminophosphonate 2 (0.65 g, 2.22 mmol) in THF (8 mL) borane-dimethyl sulfide complex (0.25 g, 3.34 mmol) was added dropwise at −30 °C in argon atmosphere. The reaction mixture was kept at this temperature for 0.5 h and allowed to warm to r.t. After that the reaction mixture was quenched with methanol (2 mL) and stirred for 15 min. Then solvents were evaporated under reduced pressure and residue was dried in vacuo.
4.3.1. Diethyl (1-Amino-2,2-difluoro-2-(pyridin-2-yl)ethyl)phosphonate 7a
Yield 0.65 g (100%); yellow crystals; mp 52–53 °C. 1H NMR (499.8 MHz, CDCl3) δ (ppm): 1.22 (t, 3JHH = 7.1 Hz, 3H, CH3), 1.25 (t, 3JHH = 7.1 Hz, 3H, CH3), 1.85 (br s, 2H, NH2), 4.18–4.00 (m, 4H, CH2), 4.23 (m, 1H, CH), 7.37 (dd, 3JHH = 7.8, 3JHH = 4.8 Hz, 1H, Py), 7.7 (d, 3JHH = 7.8 Hz, 1H, Py), 7.81 (t, 3JHH = 7.8 Hz, 1H, Py), 8.64 (d, 3JHH = 4.8 Hz, 1H, Py). 13C NMR (125.7 MHz, CDCl3) δ (ppm):16.3 (d, 3JCP = 5.9 Hz, CH3), 16.4 (d, 3JCP = 6.1 Hz, CH3), 53.7 (dt, 1JCP = 154.9 Hz, 2JCF = 28 Hz, CH), 62.7 (d, 2JCP = 6.9Hz, CH2), 63.0 (d, 2JCP = 6.7 Hz, CH2), 120.2 (td, 1JCF = 248 Hz, 2JCP = 5 Hz, CF2), 121.0 (t, 3JCF = 4.8 Hz, CPy), 124.9 (s, CPy), 137.0 (s, CPy), 149.2 (s, CPy), 153.6 (t, 2JCF = 29 Hz, CCF2). 19F NMR (470.3 MHz, CDCl3) δ (ppm): -103.8 (ddd, 2JFAFB = 256.5 Hz, 3JFH = 15.4 Hz, 3JFP = 15.4 Hz), −106.3 (dd, 2JFBFA = 256.5 Hz, 3J = 15.1 Hz). 31P NMR (202.3 MHz, CDCl3) δ (ppm): 19.8. Anal. Calc. for C11H17F2N2O3P: C, 44.90; H, 5.82; N, 9.52. Found: C, 44.67; H, 5.73; N, 9.41.
4.3.2. Diethyl (1-Amino-2,2-difluoro-2-(pyridin-3-yl)ethyl)phosphonate 7b
Yield 0.65 g (100%); white-yellow oil. 1H NMR (302 MHz, CDCl3) δ (ppm): 1.13–1.43 (m, 6H, CH3), 1.85 (br s, 2H, NH2), 3.62 (m, 1H, CH), 4.00–4.25 (m, 4H, CH2), 7.54–7.64 (m, 1H, Py), 8.15 (d, 3JHH = 8.1 Hz, 1H, Py), 8.68 (d, 3JHH = 5.8 Hz, 1H, Py), 8.82 (s, 1H, Py). 13C NMR (125.7 MHz, DMSO-d6) δ (ppm): 16.0 (d, 3JCP = 5 Hz, CH3), 16.2 (d, 3JCP = 5 Hz, CH3), 53.8 (dt, 1JCP = 150 Hz, 2JCF = 29 Hz, CH), 62.0 (d, 2JCP = 6 Hz, CH2), 62.5 (d, 2JCP = 6 Hz, CH2), 119.8 (td, 1JCF = 248 Hz, 2JCP = 6 Hz, CF2), 125.8 (s, CPy), 133.6 (td, 2JCF = 28 Hz, 3JCP = 4 Hz, CCF2), 138.0 (t, 3JCF = 6 Hz, CPy), 144.8 (t, 3JCF = 6 Hz, CPy), 148.4 (s, CPy). 19F NMR (470.3 MHz, CDCl3) δ (ppm): −100.0 (ddd, 2JFAFB = 259.1 Hz, 3JFH = 11.8 Hz, 3JFP = 11.8 Hz), −100.8 (ddd, 2JFBFA = 259.1 Hz, 3JFH = 12.4Hz, 3JFP = 12.4Hz). 31P NMR (202.3 MHz, CDCl3) δ (ppm): 18.8. Anal. Calc. for C11H17F2N2O3P: C, 44.90; H, 5.82; N, 9.52. Found: C, 44.71; H, 5.74; N, 9.39.
4.3.3. Diethyl (1-Amino-2,2-difluoro-2-(pyridin-4-yl)ethyl)phosphonate 7c
Yield 0.65 g (100%); white-yellow powder; mp 105–107 °C. 1H NMR (302 MHz, CDCl3) δ (ppm): 1.12–1.48 (m, 6H, CH3), 1.81 (br s, 2H, NH2), 3.60 (m, 1H, CH), 4.00–4.23 (m, 4H, CH2), 7.70 (d, 3JHH = 6 Hz, 2H, Py), 8.70 (d, 3JHH = 6 Hz, 2H, Py). 13C NMR (150.8 MHz, CDCl3) δ (ppm): 16.3 (d, 3JCP = 5.9 Hz, CH3), 16.4 (d, 3JCP = 5.9 Hz, CH3), 53.4 (dt, 1JCP = 151.3 Hz, 2JCF = 29.2 Hz, CH), 63.2 (d, 2JCP = 6.8 Hz, CH2), 63.5 (d, 2JCP = 6.8 Hz, CH2), 119.0 (td, 1JCF = 248 Hz, 2JCP = 5.7 Hz, CF2), 123.0 (t, 3JCF = 5.9 Hz, CPy), 146.5 (t, 2JCF = 28.2 Hz, CCF2), 147.5 (s, CPy). 19F NMR (470.3 MHz, CDCl3) δ (ppm): −102.4 (ddd, 2JFAFB = 249.4 Hz, 3JFH = 11.4 Hz, 3JFP = 11.4 Hz), −102.7 (ddd, 2JFBFA = 249.4 Hz, 3JFH = 11.8 Hz, 3JFP = 11.8 Hz). 31P NMR (202.3 MHz, CDCl3) δ (ppm): 17.95 (m, 2JPH = 18.2, 3JPF = 8.6 Hz). Anal. Calc. for C11H17F2N2O3P: C, 44.90; H, 5.82; N, 9.52. Found: C, 44.78; H, 5.68; N, 9.43.
4.4. Synthesis of (1-Amino-2,2-difluoro-2-(pyridinyl)ethyl)phosphonic Acids 8a–c: General Procedure
To respective aminophosphonate 7 (0.3 g, 1.02 mmol) was added concentrated HCl (2 mL), the reaction mixture was heated at 110 °C for 2.5 h. After that HCl/H2O was evaporated, the residue was dissolved in MeOH (2 mL), and treated with propylene oxide (0.35 g, 6.12 mmol). After 24 h the precipitate formed was separated by filtration, washed with MeOH (2 mL) and dried in vacuo.
4.4.1. (1-Amino-2,2-difluoro-2-(pyridin-2-yl)ethyl)phosphonic Acid 8a
Yield 0.17 g (70%); white powder; mp 235–237 °C (decomp.). 1H NMR (499.8 MHz, D2O) δ (ppm): 4.13–4.30 (m, 1H, CH), 7.58–7.64 (m, 1H, Py), 7.86 (d, 3JHH = 8.2 Hz, 1H, Py), 8.04 (t, 3JHH = 8.2 Hz, 1H, Py), 8.64 (d, 3JHH = 4.9 Hz, 1H, Py). 13C NMR (75.8 MHz, D2O) δ (ppm): 53.8 (dt, 1JCP = 126.5 Hz, 2JCF = 27.8 Hz, CH), 119.1 (td, 1JCF = 247 Hz, 2JCP = 6.0 Hz, CF2), 121.4 (s, CPy), 126.3 (s, CPy), 138.7 (s, CPy), 149.3 (s, CPy), 151.3 (t, 2JCF = 27.5 Hz, CCF2). 19F NMR (470.3 MHz, D2O) δ (ppm): −93.6 (d, 2JFAFB = 257 Hz), −110.0 (dd, 2JFBFA = 257 Hz, 3J = 21.5 Hz). 31P NMR (202.3 MHz, D2O) δ (ppm): 3.9. Anal. Calc. for C7H9F2N2O3P: C, 35.31; H, 3.81; N, 11.76. Found: C, 35.13; H, 3.66; N, 11.55.
4.4.2. (1-Amino-2,2-difluoro-2-(pyridin-3-yl)ethyl)phosphonic Acid 8b
Yield 0.15 g (62%); white powder; mp 210–125 °C (decomp.). 1H NMR (499.8 MHz, D2O) δ (ppm): 3.62 (m, 1H, CH), 7.66 (dd, 3JHH = 8.2 Hz, 3JHH = 5.1 Hz, 1H, Py), 8.18 (d, 3JHH = 8.2 Hz, 1H, Py), 8.72 (d, 3JHH = 5.1 Hz, 1H, Py), 8.83 (s, 1H, Py). 13C NMR (75.8 MHz, D2O) δ (ppm): 54.8 (dt, 1JCP = 132.7 Hz, 2JCF = 24.1 Hz, CH), 119.3 (t, 1JCF = 248 Hz, CF2), 124.5 (s, CPy), 130.2 (td, 2JCF = 26 Hz, 3JCP = 4.6 Hz, CCF2), 136.3 (t, 3JCF = 5.7 Hz, CPy), 145.6 (t, 3JCF = 7.9 Hz, CPy), 150.4 (s, CPy). 19F NMR (470.3 MHz, D2O) δ (ppm): −86.1 (dd, 2JFAFB = 253 Hz, 3J = 24.7 Hz), −110.1 (dd, 2JFBFA = 253 Hz, 3J = 24.5 Hz). 31P NMR (202.3 MHz, D2O) δ (ppm): 2.8 (d, 2JPH = 24.3 Hz). Anal. Calc. for C7H9F2N2O3P: C, 35.31; H, 3.81; N, 11.76. Found: C, 35.04; H, 3.72; N, 11.59.
4.4.3. (1-Amino-2,2-difluoro-2-(pyridin-4-yl)ethyl)phosphonic Acid 8c
Yield 0.16 g (67%); white powder; mp 218–220 °C (decomp.). 1H NMR (499.8 MHz, D2O) δ (ppm): 4.08 (m, 1H, CH), 7.73 (d, 3JHH = 5.3 Hz, 2H, Py), 8.72 (d, 3JHH = 5.3 Hz, 2H, Py). 13C NMR (75.8 MHz, D2O) δ (ppm): 54.3 (ddd, 1JCP = 129.6 Hz, 2JCFA = 27.6 Hz, 2JCFB = 20.1 Hz, CH), 119.0 (td, 1JCF = 247.3 Hz, 2JCP = 2.8 Hz, CF2), 121.4 (dd, 3JCFA = 7.3 Hz, 3JCFB = 4.8 Hz, CPy), 143.3 (t, 2JCF = 26.2 Hz, CCF2), 148.8 (s, CPy). 19F NMR (470.3 MHz, D2O) δ (ppm): −89.8 (d, 2JFAFB = 250 Hz), −110.2 (dd, 2JFBFA = 250 Hz, 3J = 23.3 Hz). 31P NMR (202.3 MHz, D2O) δ (ppm): 3.3. Anal. Calc. for C7H9F2N2O3P: C, 35.31; H, 3.81; N, 11.76. Found: C, 35.12; H, 3.77; N, 11.63.
4.5. Diethyl (1-Amino-2,2-difluoro-2-(piperidin-4-yl)ethyl)phosphonate 9
To a solution of aminophosphonate 7c (0.3 g, 1.02 mmol) in MeOH (10 mL) was added Rh/Al2O3 (0.1 g, 10%), the mixture was hydrogenated under 50 atm. of H2 for 10 h. After completion of the reaction, the catalyst was filtered off, the solvent was evaporated to give 0.31 g of 9 (yield 100%) as colorless oil. 1H NMR (302 MHz, DMSO-d6) δ (ppm): 1.09–1.29 (m, 6H, CH3), 1.30–1.93 (m, 4H, CH2), 2.69–2.85 (m, 2H, CH2), 2.87–3.07 (m, 1H), 3.15–3.30 (m, 2H, CH2), 3.47 (m, 1H, CHP), 3.93–4.17 (m, 4H, OCH2), 8.39 (s, 1H, NH). 13C NMR (75.8 MHz, DMSO-d6) δ (ppm): 16.1 (d, 3JCP = 5.2 Hz, CH3), 16.2 (d, 3JCP = 5.2 Hz, CH3), 21.4 (t, 3JCF = 4.1 Hz, CF2CHCH2), 22.0 (t, 3JCF = 4.7 Hz, CF2CHCH2), 37.3 (td, 2JCF = 23.7 Hz, 3JCP = 4.2 Hz, CHCF2), 42.8 (s, 2CH2), 50.7 (dt, 1JCP = 147.3 Hz, 2JCF = 28.4 Hz, CHP), 61.8 (d, 2JCP = 6.8 Hz, OCH2), 62.1 (d, 2JCP = 6.7 Hz, OCH2), 124.0 (td, 1JCF = 248 Hz, 2JCP = 30 Hz, CF2).19F NMR (470.3 MHz, DMSO-d6) δ (ppm): −111.2 (m), −111.5 (m). 31P NMR (202.3 MHz, DMSO-d6) δ (ppm): 21.3. Anal. Calc. for C11H23F2N2O3P: C, 44.00; H, 7.72; N, 9.33. Found: C, 44.21; H, 7.65; N, 9.28.
4.6. (1-Amino-2,2-difluoro-2-(piperidin-4-yl)ethyl)phosphonic Acid 10
To aminophosphonate 9 (0.29 g, 0.97 mmol) was added concentrated HCl (1 mL), the reaction mixture was heated at 110 °C for 2.5 h. After completion of reaction HCl/H2O was evaporated, the residue was dissolved in MeOH (1 mL), and treated with propylene oxide (0.23 g, 4 mmol). After 24 h the precipitate formed was separated by filtration, washed with MeOH (2 mL) and dried to give 0.17 g of 10 (yield 71%) as white powder; mp 250 °C (decomp.). 1H NMR (499.8 MHz, D2O) δ (ppm): 1.65–1.91 (m, 4H), 2.19 (t, 2JHH = 14.9 Hz, 2H), 3.07 (t, 2JHH = 14.1 Hz, 2H), 3.48–3.58 (m, 2H), 3.70–3.78 (m, 1H, CH). 13C NMR (150.8 MHz, D2O) δ (ppm): 27.4 (s, CH2), 28.3 (s, CH2), 37.4 (t, 2JCF = 22.4 Hz, CHCF2), 42.8 (s, CH2), 43.1 (s, CH2), 52.0 (d, 1JCP = 125.6 Hz, CH), 122.5 (t, 1JCF = 248 Hz, CF2).19F NMR (470.3 MHz, D2O) δ (ppm): −107.3 (ddd, 2JFAFB = 246 Hz, 3JFH = 26.5 Hz, 3JFP = 26.5 Hz), −111.6 (dd, 2JFBFA = 246 Hz, 3J = 26.5 Hz). 31P NMR (202.3 MHz, D2O) δ (ppm): 3.5. Anal. Calc. for C7H15F2N2O3P: C, 34.43; H, 6.19; N, 11.47. Found: C, 34.17; H, 6.07; N, 11.63.
4.7. Synthesis of Diethyl (2-(Difluoro(pyridinyl)methyl)-4-oxothiazolidin-2-yl)phosphonates 11a–c: General Procedure
To solution of respective iminophosphonate 2 (0.12 g, 0.41 mmol) in benzene (1 mL) was added mercaptoacetic acid (0.04 g, 0.41 mmol). The reaction mixture was refluxed for 2 h, the solvent was evaporated in vacuum and the residue was triturated with hexane.
4.7.1. Diethyl (2-(Difluoro(pyridin-2-yl)methyl)-4-oxothiazolidin-2-yl)phosphonate 11a
Yield 0.11 g (73%); yellow oil. 1H NMR (302 MHz, CDCl3) δ (ppm): 1.11–1.30 (m, 3JHH = 7.1 Hz, 6H, CH3), 3.44 (dd, 2JHAHB = 15.1 Hz, 4JHAP = 5.8 Hz, 1H, SCHAHB), 3.6 (d, 2JHBHA = 15.1 Hz, 1H, SCHAHB), 3.93–4.22 (m, 4H, OCH2), 7.45 (dd, 3JHH = 7.9 Hz, 3JHH = 4.9 Hz, 1H, Py), 7.74 (d, 3JHH = 7.9 Hz, 1H, Py), 7.86 (t, 3JHH = 7.9 Hz, 1H, Py), 8.20 (s, 1H, NH), 8.65 (d, 3JHH = 4.9 Hz, 1H, Py). 13C NMR (150.8 MHz, CDCl3) δ (ppm): 16.3 (d, 3JCP = 5.9 Hz, CH3), 16.4 (d, 3JCP = 5.6 Hz, CH3), 32.1 (s, SCH2), 64.8 (d, 2JCP = 7.7 Hz, OCH2), 65.2 (d, 2JCP = 7.6 Hz, OCH2), 68.5 (dt, 1JCP = 170.7 Hz, 2JCF = 32.6 Hz, CP), 116.0 (td, 1JCF = 254 Hz, 2JCP = 21 Hz, CF2), 121.7 (t, 3JCF = 4.5Hz, CPy),125.6 (s, CPy), 137.5 (s, CPy), 149.0 (s, CPy), 152.2 (t, 2JCF = 30Hz, CCF2), 174.1 (d, 3JCP = 5.2 Hz, C(O)). 19F NMR (470.3 MHz, CDCl3) δ (ppm): −98.5 (dd, 2JFAFB = 260 Hz, 3JFP = 12 Hz), −108.4 (d,2JFBFA = 260 Hz). 31P NMR (202.3 MHz, CDCl3) δ (ppm):13.5. Anal. Calc. for C13H17F2N2O4PS: C, 42.62; H, 4.68; N, 7.65; S, 8.75. Found: C, 42.44; H, 4.53; N, 7.51; S, 8.62.
4.7.2. Diethyl (2-(Difluoro(pyridin-3-yl)methyl)-4-oxothiazolidin-2-yl)phosphonate 11b
Yield 0.125 g (83%); white powder; mp 162–164 °C. 1H NMR (302 MHz, CDCl3) δ (ppm): 1.30 (t, 3JHH = 6.8 Hz, 6H, CH3), 3.05 (dd, 2JHAHB = 15.3 Hz, 4JHAP = 5.9 Hz, 1H, SCHAHB), 3.39 (d, 2JHBHA = 15.3 Hz, 1H, SCHAHB), 4.09–4.45 (m, 4H, OCH2), 7.38 (dd, 3JHH = 8.0 Hz, 3JHH = 4.9 Hz, 1H, Py), 7.99 (d, 3JHH = 8.0 Hz, 1H, Py), 8.73 (d, 3JHH = 4.9 Hz, 1H, Py), 8.73 (s, 1H, NH), 8.91 (s, 1H, Py). 13C NMR (150.8 MHz, CDCl3) δ (ppm): 16.3 (d, 3JCP = 6 Hz, CH3), 16.4 (d, 3JCP = 6 Hz, CH3), 32.1 (s, SCH2), 65.0 (d, 2JCP = 8 Hz, OCH2), 65.1 (d, 2JCP = 8 Hz, OCH2), 68.0 (dt, 1JCP = 167 Hz, 2JCF = 32 Hz, CP), 120.7 (td, 1JCF = 255 Hz, 2JCP = 12 Hz, CF2), 122.7 (s, CPy), 128.2 (td, 2JCF = 27 Hz, 3JCP = 4 Hz, CCF2), 136.0 (t, 3JCF = 7 Hz, CPy), 148.6 (t, 3JCF = 5 Hz, CPy), 151.4 (s, CPy), 174.8 (d, 3JCP = 7 Hz, C(O)). 19F NMR (188 MHz, CDCl3) δ (ppm): −96.7 (d, 2JFAFB = 255.5 Hz), −101.9 (d, 2JFBFA = 255.5 Hz). 31P NMR (202.3 MHz, CDCl3) δ (ppm):13.4. Anal. Calc. for C13H17F2N2O4PS: C, 42.62; H, 4.68; N, 7.65; S, 8.75. Found: C, 42.47; H, 4.73; N, 7.41; S, 8.58.
4.7.3. Diethyl (2-(Difluoro(pyridin-4-yl)methyl)-4-oxothiazolidin-2-yl)phosphonate 11c
Yield 0.116 g (77%); white powder; mp 80–82 °C. 1H NMR (499.8 MHz, CDCl3) δ (ppm): 1.31 (t, 3JHH = 7 Hz, 6H, CH3), 3.1 (dd, 2JHAHB = 15.2 Hz, 4JHAP = 5.6 Hz, 1H, SCHAHB), 3.4 (d, 2JHBHA = 15.2 Hz, 1H, SCHAHB), 4.12–4.33 (m, 4H, OCH2), 7.58 (d, 3JHH = 5.3 Hz, 2H, Py), 8.26 (s, 1H, NH), 8.73 (d, 3JHH = 5.3 Hz, 2H, Py).13C NMR (150.8 MHz, CDCl3) δ (ppm): 16.3 (d, 3JCP = 5.7 Hz, CH3), 16.4 (d, 3JCP = 5.7 Hz, CH3), 32.1 (s, SCH2), 65.0 (d, 2JCP = 7.3 Hz, OCH2), 65.2 (d, 2JCP = 7.3 Hz, OCH2), 67.6 (dt, 1JCP = 167.1 Hz, 2JCF = 34.2 Hz, CP), 120.2 (td, 1JCF = 253 Hz, 2JCP = 11.6 Hz, CF2), 122.4 (t, 3JCF = 5.6 Hz, 2CPy), 140.7 (td, 2JCF = 27.6 Hz, 3JCP = 2.8 Hz, CCF2), 149.3 (s, 2CPy), 174.8 (d, 3JCP = 6.5 Hz, CO). 19F NMR (470.3 MHz, CDCl3) δ (ppm): −98.9 (d, 2JFAFB = 252 Hz), −104.0 (d,2JFBFA = 252 Hz). 31P NMR (202.3 MHz, CDCl3) δ (ppm): 13.2.Anal. Calc. for C13H17F2N2O4PS: C, 42.62; H, 4.68; N, 7.65; S, 8.75. Found: C, 42.39; H, 4.56; N, 7.48; S, 8.64.
4.8. Synthesis of Diethyl (2-(Difluoro(pyridinyl)methyl)-4-oxo-1,3-thiazinan-2-yl)phosphonates 12a–c: General Procedure
To a solution of respective iminophosphonate 2 (0.13 g, 0.44 mmol) in benzene (1 mL) was added 3-mercaptopropionic acid (0.05 g, 0.44 mmol). The reaction mixture was refluxed for 8 h, the solvent was evaporated in vacuum and the residue was triturated with Et2O.
4.8.1. Diethyl (2-(Difluoro(pyridine-2-yl)methyl)-4-oxo-1,3-thiazinan-2-yl)phosphonate 12a
Yield 0.13 g (76%); yellow oil. 1H NMR (302 MHz, CDCl3) δ (ppm): 1.13 (t, 3JHH = 7.1 Hz, 3H, CH3), 1.25 (t, 3JHH = 7.1 Hz, 3H, CH3), 2.73–2.84 (m, 2H, CH2), 2.91–3.01 (m, 1H, CH), 3.22–3.35 (m, 1H, CH), 3.96–4.28 (m, 4H, 2OCH2), 7.46 (dd, 3JHH = 7.9 Hz, 3JHH = 5.1 Hz, 1H), 7.70 (d, 3JHH = 7.9 Hz, 1H), 7.86 (t, 3JHH = 7.9 Hz, 1H), 8.09 (s, 1H, NH), 8.70 (d, 3JHH = 5.1 Hz, 1H). 13C NMR (125.7 MHz, CDCl3) δ (ppm):16.2 (d, 3JCP = 6 Hz, CH3), 16.3 (d, 3JCP = 5 Hz, CH3), 23.5 (s, SCH2), 33.3 (s, CH2CO), 64.7 (d, 2JCP = 7.7 Hz, OCH2), 65.3 (d, 2JCP = 7.6 Hz, OCH2), 66.6 (dm, 1JCP = 168.7 Hz, CP), 116.0 (td, 1JCF = 256.2 Hz, 2JCP = 15 Hz, CF2), 122.1 (s, CPy),125.5 (s, CPy), 137.2 (s, CPy), 148.8 (s, CPy), 152.2 (td, 2JCF = 30 Hz, 3JCP = 2.5 Hz, CCF2), 170.9 (d, 3JCP = 3.1 Hz, CO). 19F NMR (470.3 MHz, CDCl3) δ (ppm): −99 (dd, 2JFAFB = 255 Hz, 3JFP = 9 Hz), −106.4 (d,2JFBFA = 255 Hz). 31P NMR (202.3 MHz, CDCl3) δ (ppm): 13.2 (m, 3J = 9 Hz). Anal. Calc. for C14H19F2N2O4PS: C, 44.21; H, 5.04; N, 7.37; S, 8.42. Found: C, 44.39; H, 4.91; N, 7.43; S, 8.34.
4.8.2. Diethyl (2-(Difluoro(pyridine-3-yl)methyl)-4-oxo-1,3-thiazinan-2-yl)phosphonate 12b
Yield 0.14 g (82%); white powder; mp 158–160 °C.1H NMR (400 MHz, CDCl3) δ (ppm): 1.31 (t, 3JHH = 7.2 Hz, 6H, CH3), 2.32–2.44 (m, 2H, CH2), 2.53–2.68 (m, 1H, CH2), 2.80–2.89 (m, 1H, CH2), 4.10–4.35 (m, 4H, OCH2), 6.72 (s, 1H, NH), 7.37 (dd, 3JHH = 8.1 Hz, 3JHH = 4.9 Hz, 1H, Py), 7.92 (d, 3JHH = 8.1Hz, 1H, Py), 8.72 (d, 3JHH = 4.9 Hz, 1H, Py), 8.87 (s, 1H, Py). 13C NMR (150.8 MHz, CDCl3) δ (ppm): 16.40 (d, 3JCP = 5.7 Hz, CH3), 16.43 (d, 3JCP = 5.6 Hz, CH3), 23.4 (s, SCH2), 33.4 (s, CH2CO), 65.0 (d, 2JCP = 7.5 Hz, OCH2), 65.2 (d, 2JCP = 7.5 Hz, OCH2), 65.5 (dt, 1JCP = 161.2 Hz, 2JCF = 29.5 Hz, CP), 120.5 (td, 1JCF = 255.7 Hz, 2JCP = 7.4 Hz, CF2), 122.5 (s, CPy), 128.3 (td, 2JCF = 26.7 Hz, 3JCP = 3.6 Hz, CCF2), 135.9 (t, 3JCF = 5.9 Hz, CPy), 148.9 (t, 3JCF = 6.4 Hz, CPy), 151.5 (s, CPy), 170.4 (d, 3JCP = 3.1Hz, C(O)). 19F NMR (470.3 MHz, CDCl3) δ (ppm): -98.8 (d, 2JFAFB = 253.6 Hz), −102.2 (d,2JFBFA = 253.6 Hz). 31P NMR (202.3 MHz, CDCl3) δ (ppm): 13.5. Anal. Calc. for C14H19F2N2O4PS: C, 44.21; H, 5.04; N, 7.37; S, 8.42. Found: C, 44.08; H, 4.97; N, 7.48; S, 8.31.
4.8.3. Diethyl (2-(Difluoro(pyridine-4-yl)methyl)-4-oxo-1,3-thiazinan-2-yl)phosphonate 12c
Yield 0.13 g (76%); white powder; mp 110 °C. 1H NMR (400 MHz, CDCl3) δ (ppm): 1.27–1.38 (m, 3JHH = 7.4 Hz, 6H, CH3), 2.41–2.53 (m, 2H, CH2), 2.56–2.67 (m, 1H, CH), 2.84–2.92 (m, 1H, CH), 4.19–4.31 (m, 4H, OCH2), 6.39 (s, 1H, NH), 7.65 (d, 3JHH = 5.1 Hz, 2H, Py), 8.76 (d, 3JHH = 5.1Hz, 2H, Py). 13C NMR (150.8 MHz, CDCl3) δ (ppm): 16.4 (br d, 3JCP = 5.7 Hz, CH3), 23.4 (s, SCH2), 33.4 (s, CH2CO), 65.2 (d, 2JCP = 7.5 Hz, OCH2), 65.2 (dt, 1JCP = 161.4 Hz, 2JCF = 30.8 Hz, CP), 65.3 (d, 2JCP = 7.5 Hz, OCH2), 119.9 (td, 1JCF = 257 Hz, 2JCP = 8 Hz, CF2), 122.4 (t, 3JCF = 5.7 Hz, 2CPy), 140.8 (td, 2JCF = 28.1 Hz, 3JCP = 4.8 Hz, CCF2), 149.3 (s, 2CPy), 170.6 (d, 3JCP = 2.7 Hz, CO). 19F NMR (470.3 MHz, CDCl3) δ (ppm): −100.4 (d, 2JFAFB = 251 Hz), −103.5 (d,2JFBFA = 251 Hz). 31P NMR (202.3 MHz, CDCl3) δ (ppm): 12.9. Anal. Calc. for C14H19F2N2O4PS: C, 44.21; H, 5.04; N, 7.37; S, 8.42. Found: C, 44.48; H, 4.88; N, 7.28; S, 8.28.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/org2020007/s1, 1H, 19F, and 13C NMR spectra of compounds synthesized.
Author Contributions
Conceptualization, P.P.O. and Y.V.R.; methodology, P.P.O., Y.V.R., O.M.S., L.V.B. and A.V.B.; validation, P.P.O., Y.V.R., O.M.S. and L.V.B.; investigation, O.M.S., L.V.B. and A.V.B.; data curation, P.P.O. and Y.V.R.; writing—original draft preparation, P.P.O. and Y.V.R.; writing—review and editing, P.P.O., Y.V.R. and O.M.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article and supplementary material.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kukhar, V.P.; Hudson, H.R. Aminophosphonic and Aminophosphinic Acids. Chemistry and Biological Activity; John Wiley & Sons: New York, NY, USA, 2000. [Google Scholar]
- Kafarski, P.; Lejczak, B. Biological activity of aminophosphonic acids. Phosphorus Sulfur Silicon Relat. Elem. 1991, 63, 193–215. [Google Scholar] [CrossRef]
- Mucha, A.; Kafarski, P.; Berlicki, L. Remarkable potential of the α-aminophosphonate/phosphinate structural motif in medicinal chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef]
- Orsini, F.; Sello, G.; Sisti, M. Aminophosphonic acids and derivatives. Synthesis and biological applications. Curr. Med. Chem. 2010, 17, 264–289. [Google Scholar] [CrossRef] [PubMed]
- Marrs, E.C.L.; Varadi, L.; Bedernjak, A.F.; Day, K.M.; Gray, M.; Jones, A.L.; Cummings, S.P.; Anderson, R.J.; Perry, J.D. Phosphonopeptides revisited, in an era of increasing antimicrobial resistance. Molecules 2020, 25, 1445. [Google Scholar] [CrossRef] [PubMed]
- Smart, B.E. Fluorine substituent effects (on bioactivity). J. Fluor. Chem. 2001, 109, 3–11. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Wanat, W.; Dziuk, B.; Kafarsky, P. New crystal structures of fluorinated α-aminophosphonic acid analogues of phenylglycine. Struct. Chem. 2020, 31, 1197–1209. [Google Scholar] [CrossRef]
- Cytlak, T.; Kazmierczak, M.; Skibinska, M.; Koroniak, H. Latest achievements in the preparation of fluorinated aminophosphonates and aminophosphonic acids. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 602–620. [Google Scholar] [CrossRef]
- Burgey, C.S.; Robinson, K.A.; Lyle, T.A.; Sanderson, P.E.J.; Lewis, S.D.; Lucas, B.J.; Krueger, J.A.; Singh, R.; Miller-Stein, C.; White, R.B.; et al. Metabolism-directed optimization of 3-aminopyrazinone acetamide thrombin inhibitors. Development of an orally bioavailable series containing P1 and P3 pyridines. J. Med. Chem. 2003, 46, 461–473. [Google Scholar] [CrossRef]
- Qian, A.; Zheng, Y.; Wang, R.; Weie, J.; Cui, Y.; Cao, X.; Yang, Y. Design, synthesis, and structure-activity relationship studies of novel tetrazole antifungal agents with potent activity, broad antifungal spectrum and high selectivity. Bioorg. Med. Chem. Lett. 2018, 28, 344–350. [Google Scholar] [CrossRef]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef]
- Nifant’ev, E.Y. Chemistry of Hydrophosphoryl Compounds; Nauka: Moscow, Russia, 1983; pp. 84–87. (In Russian) [Google Scholar]
- Orlovskii, V.V.; Vovsi, B.A. Interaction of dialkyl phosphites with nitriles. Zhurnal Obshch. Khimii 1976, 46, 297–300. (In Russian) [Google Scholar]
- Midrier, C.; Lantsoght, M.; Volle, J.-N.; Pirat, J.-L.; Virieux, D.; Stevens, C.V. Hydrophosphonylation of alkenes or nitriles by double radical transfer mediated by titanocene/propylene oxide. Tetrahedron Lett. 2011, 52, 6693–6696. [Google Scholar] [CrossRef]
- Zimin, M.G.; Dvoinishnikova, T.A.; Konovalova, I.V.; Pudovik, A.N. Reaction of sodium salts of dialkylphosphorous acids with carboxylic acid nitriles. Russ. Chem. Bull. 1978, 27, 436–437. [Google Scholar] [CrossRef]
- Rassukana, Y.V.; Kolotylo, M.V.; Synytsya, O.A.; Pirozhenko, V.V.; Onys’ko, P.P. α-Iminotrifluoroethylphosphonates: The first representatives of NH imidoyl phosphonates. Synthesis 2007, 17, 2627–2630. [Google Scholar] [CrossRef]
- Rassukana, Y.V.; Yelenich, I.P.; Synytsya, A.D.; Onys’ko, P.P. Fluorinated NH-iminophosphonates and iminocarboxylates: Novel synthons for the preparation of biorelevant α-aminophosphonates and carboxylates. Tetrahedron 2014, 70, 2928–2937. [Google Scholar] [CrossRef]
- Rassukana, Y.V.; Yelenich, I.P.; Bezdudny, A.V.; Mironov, V.F.; Onys’ko, P.P. Chemoselectivity of reactions of haloacetonitriles with hydrogen phosphonates: Dramatic effect of the nature of halogen atom. Tetrahedron Lett. 2014, 55, 4771–4773. [Google Scholar] [CrossRef]
- Blanch, J.J. Determination of the Hammett substituent constants for the 2-, 3-, and 4-pyridyl and -pyridinium groups. J. Chem. Soc. B 1966, 937–939. [Google Scholar] [CrossRef]
- Khomutnik, Y.Y.; Onys’ko, P.P.; Rassukanaya, Y.V.; Pirozhenko, V.V.; Synytsya, A.D. N-aryltrifluoroacetimidoylphosphonates. Russ. J. Gen. Chem. 2013, 83, 445–452. [Google Scholar] [CrossRef]
- Onys’ko, P.P.; Chudakova, T.I.; Pirozhenko, V.V.; Rozhenko, A.B. α-Ketophosphonates in the synthesis of α-iminophosphonates. Curr. Green Chem. 2020, 7, 226–238. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Rassukana, Y.V.; Onys’ko, P.P.; Kolotylo, M.V.; Sinitsa, A.D.; Lyzwa, P.; Mikolajczyk, M. A new strategy for asymmetric synthesis of aminophosphonic acid derivatives: The first enantioselective catalytic reduction of C-phosphorylated N-H imines. Tetrahedron Lett. 2009, 50, 288–290. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).