
Z,E-Isomerism in a Series of Substituted Iminophosphonates: Quantum Chemical Research

 , and
, and
Abstract
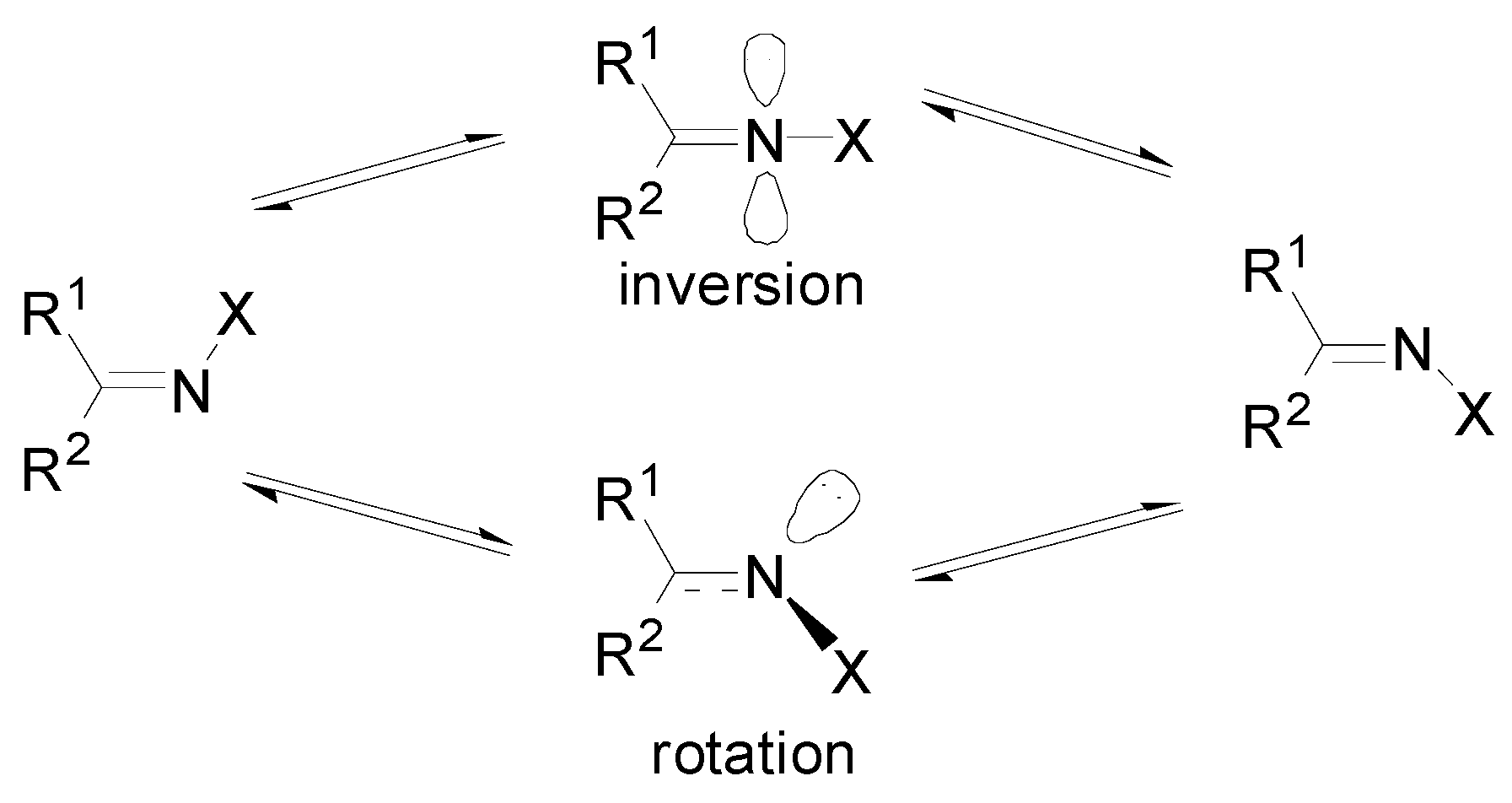
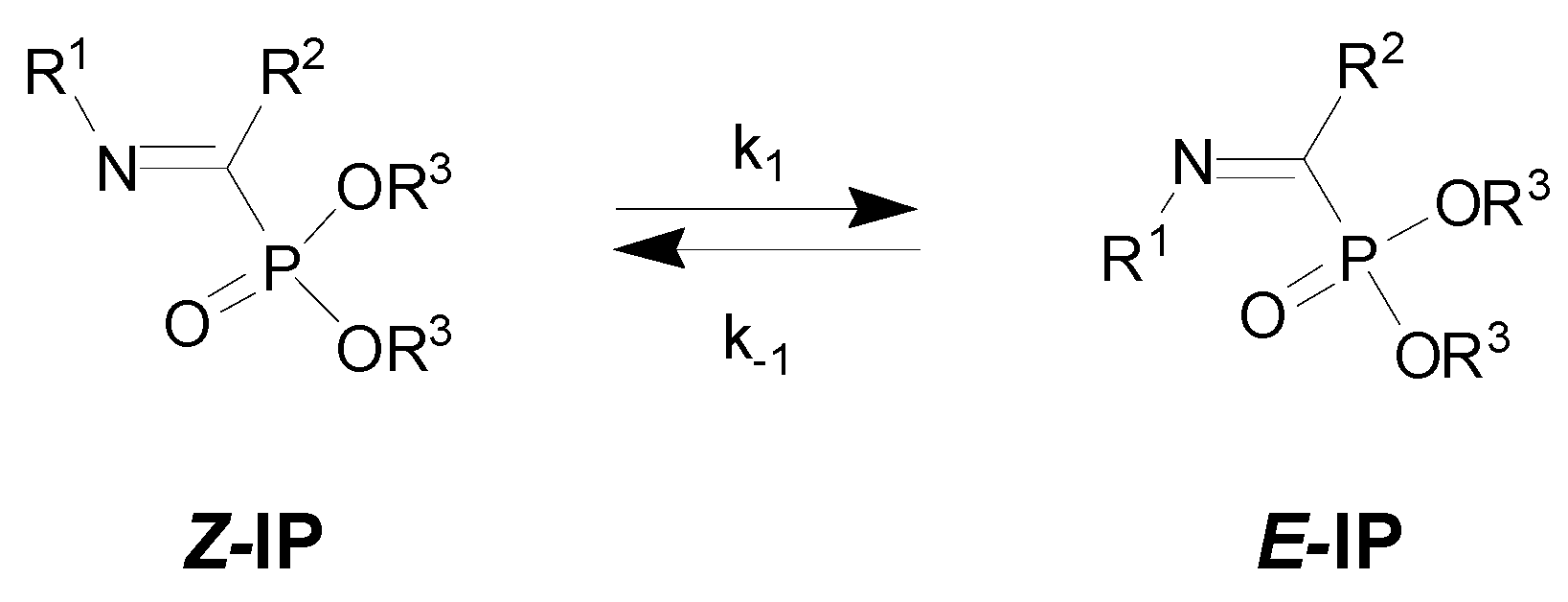
1. Introduction
2. Methods of Calculations
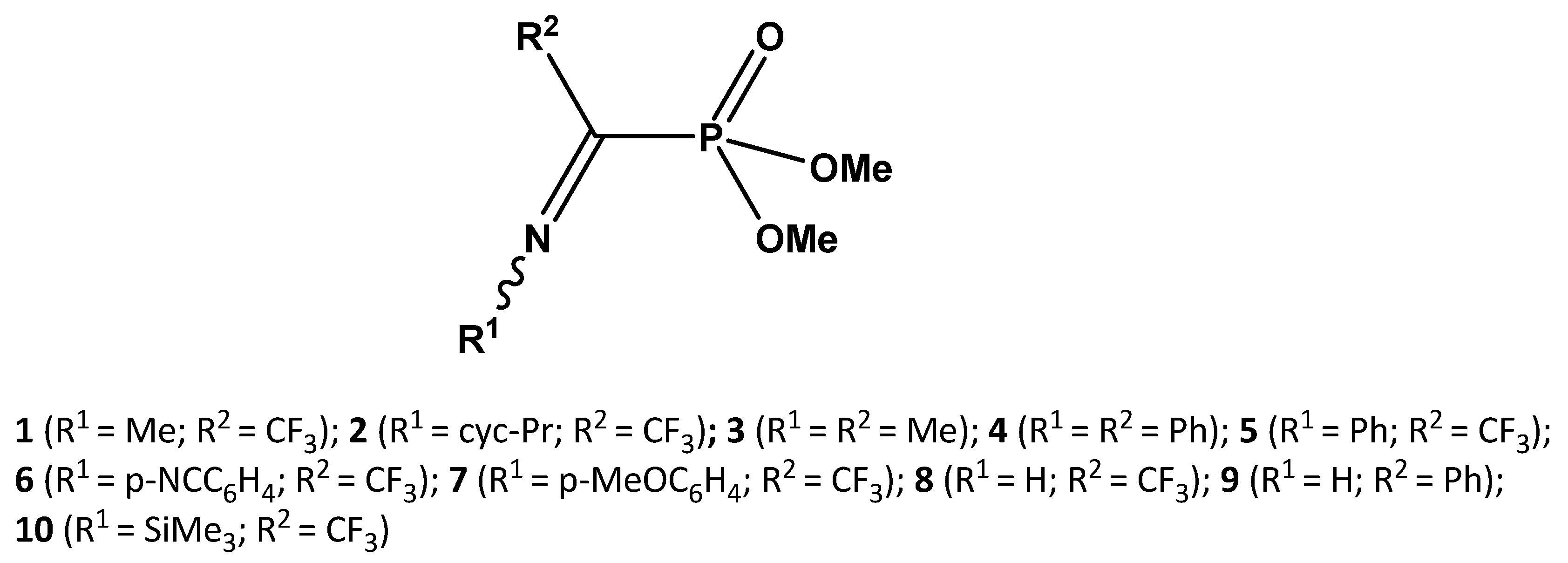
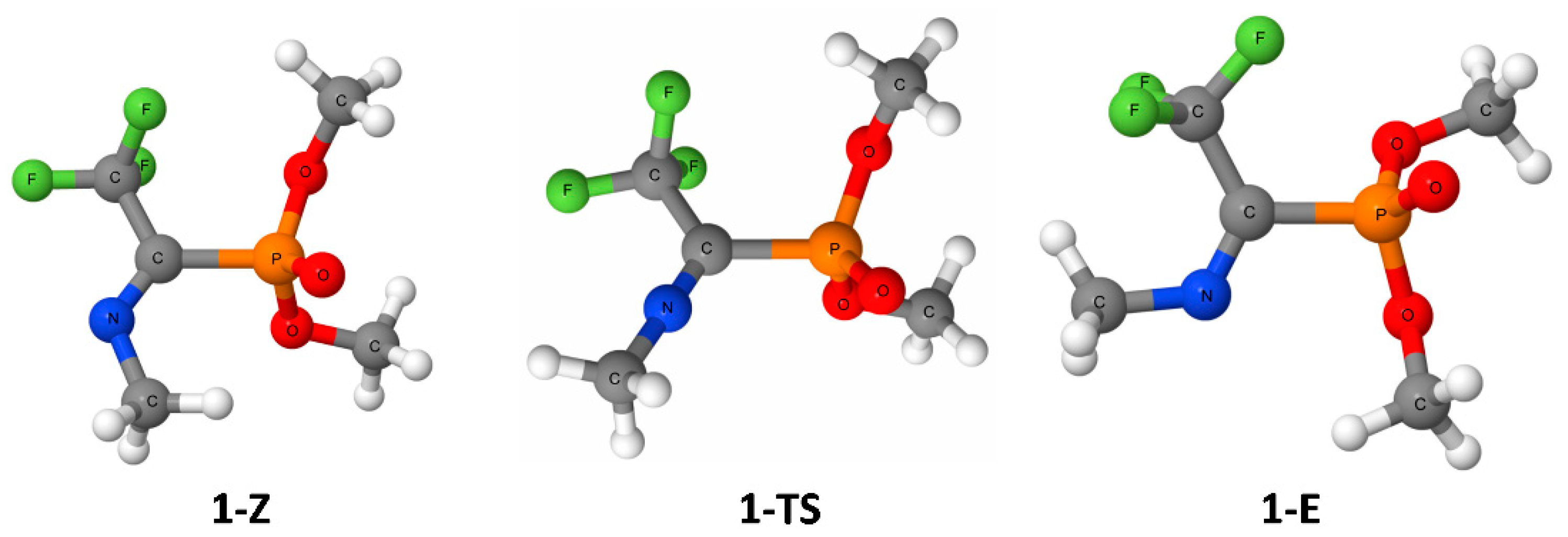
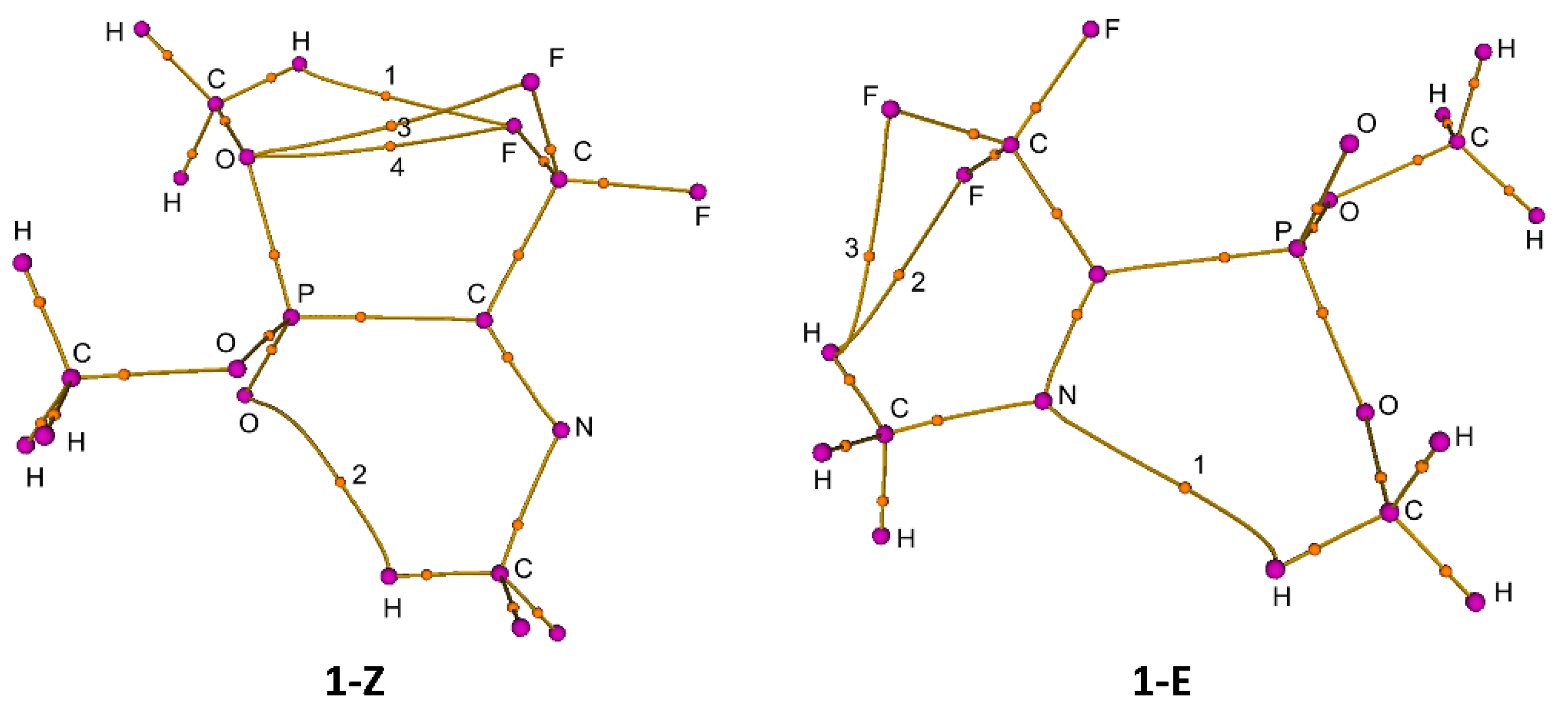
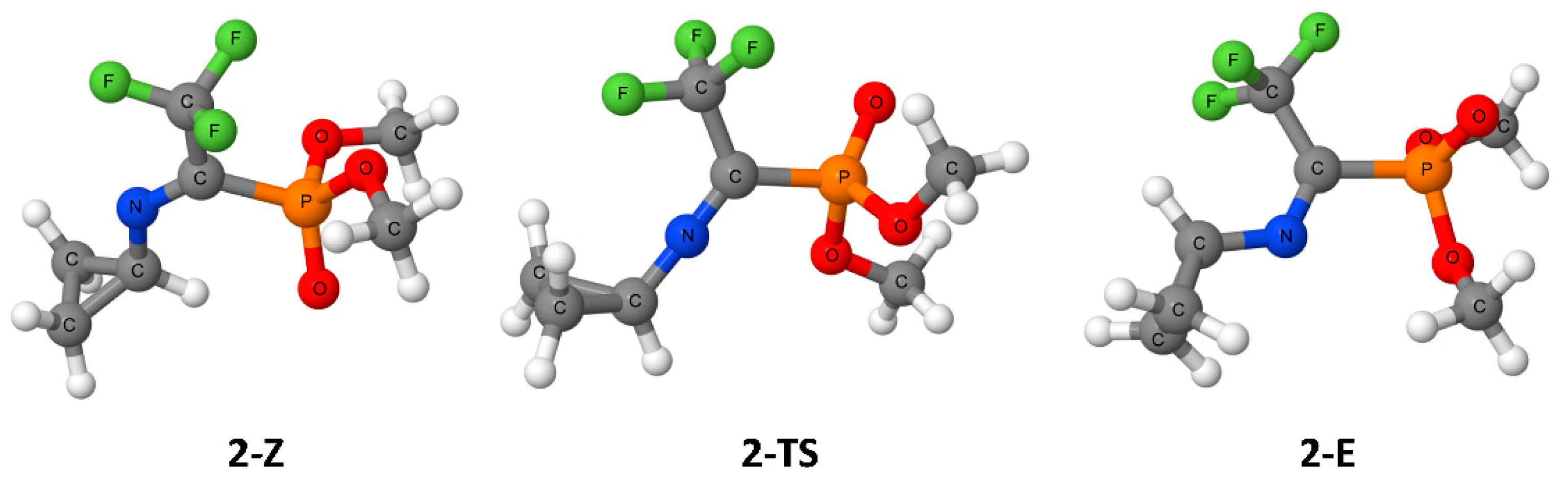
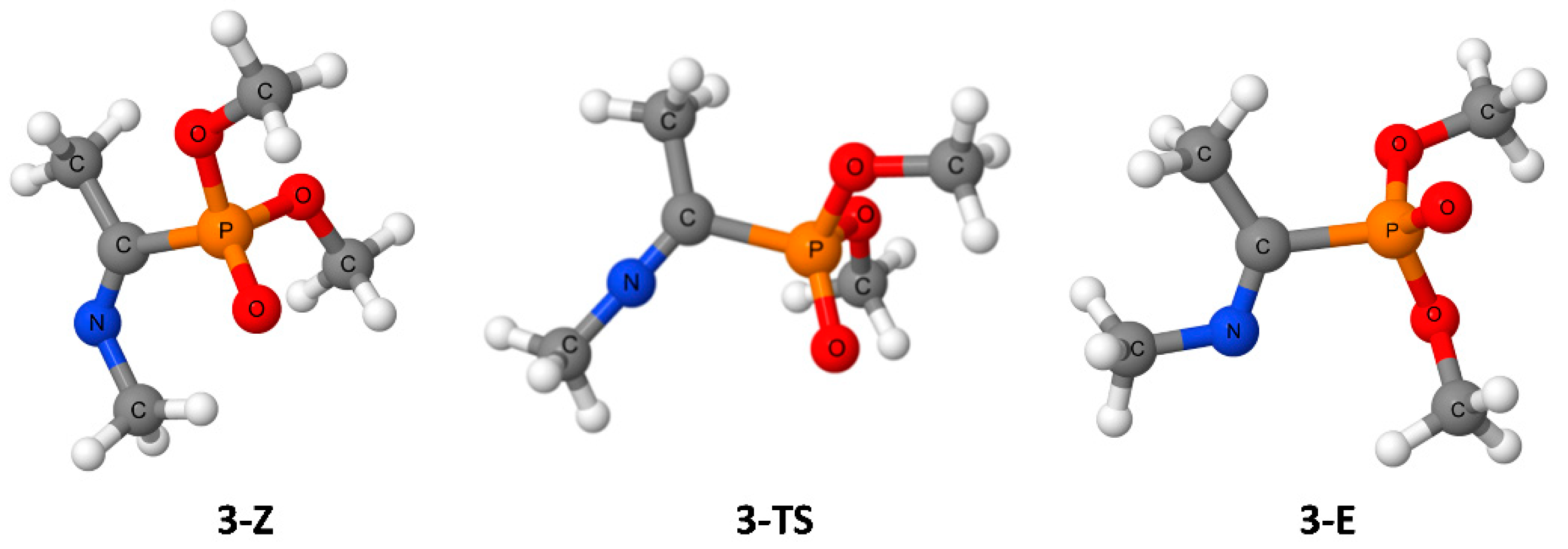
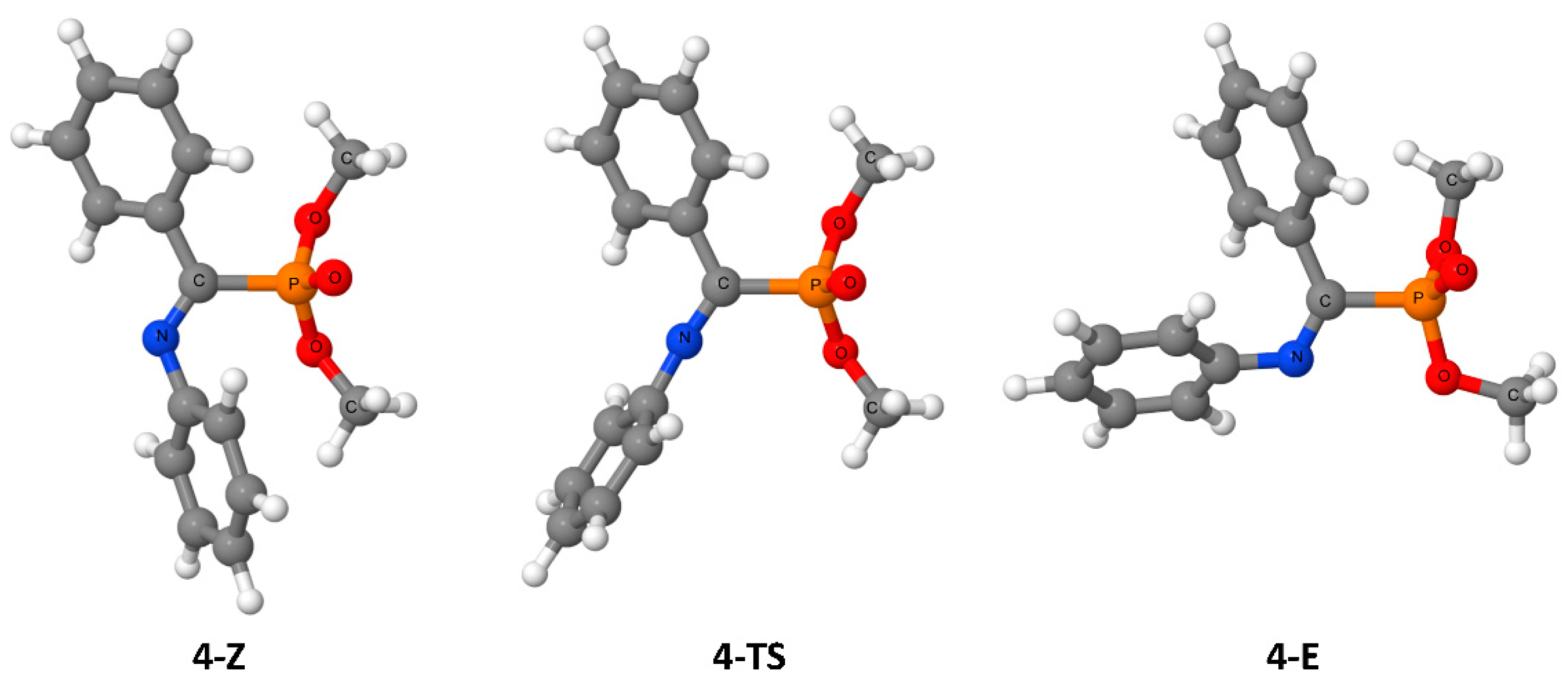
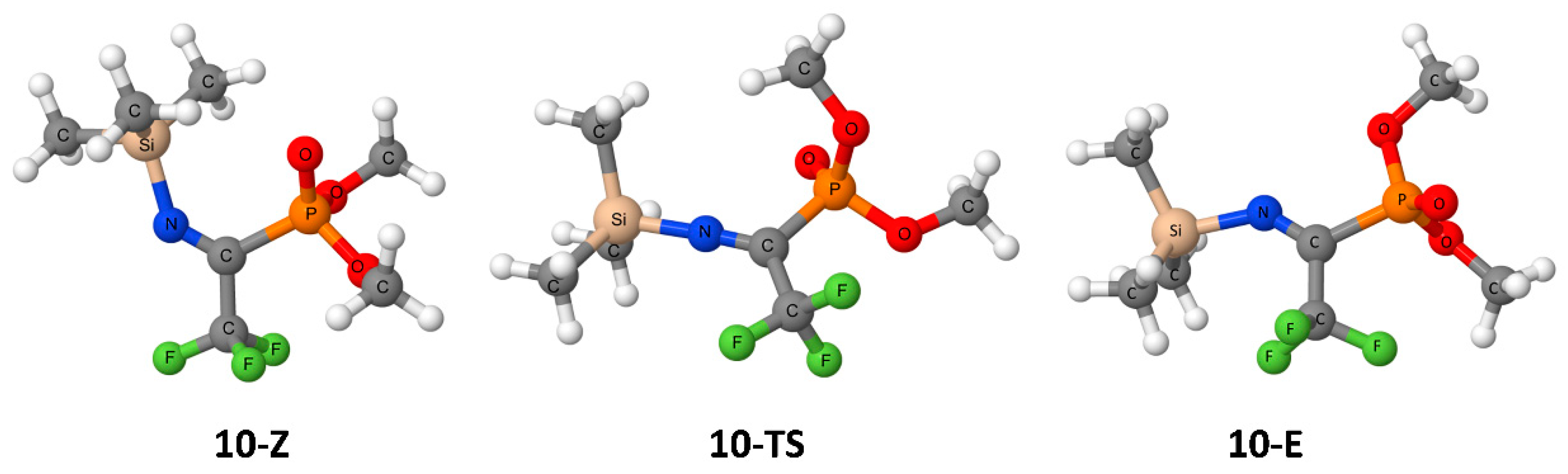
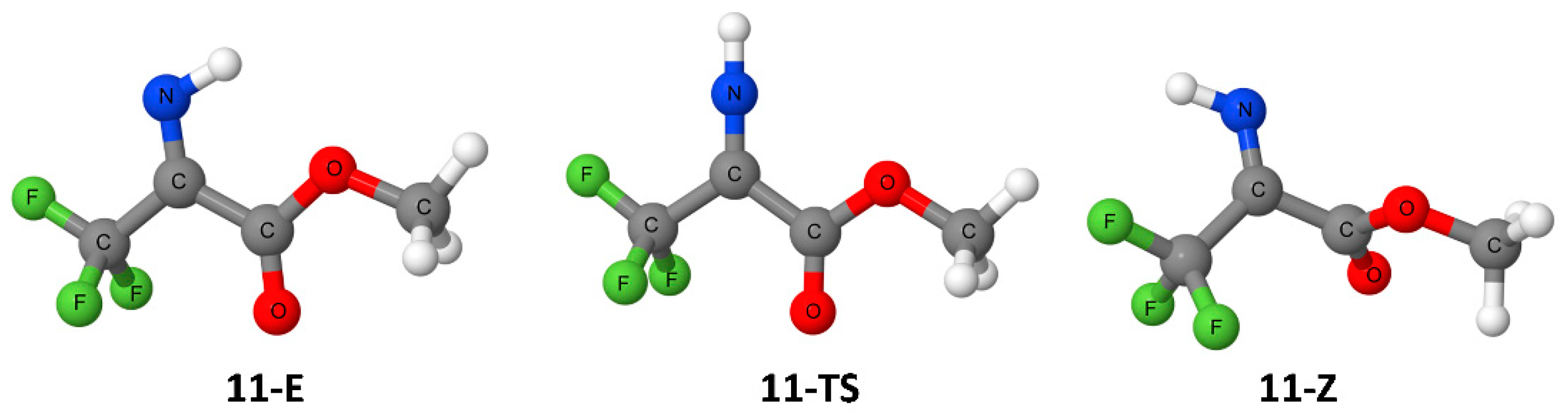
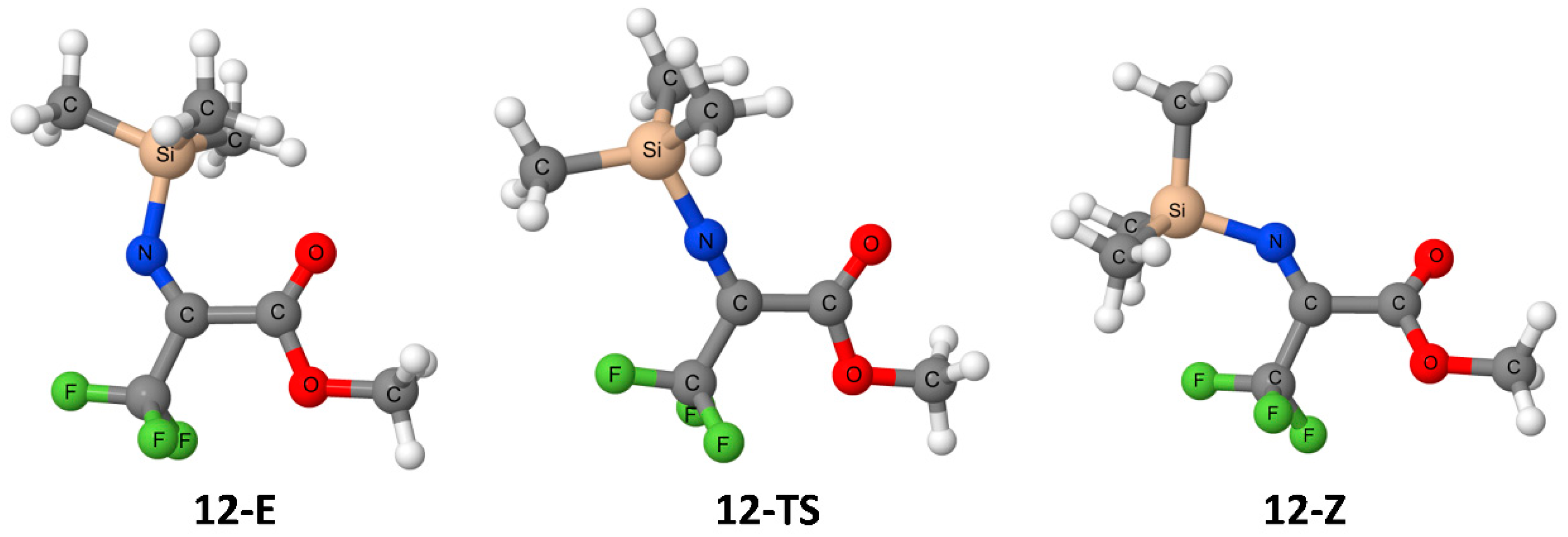
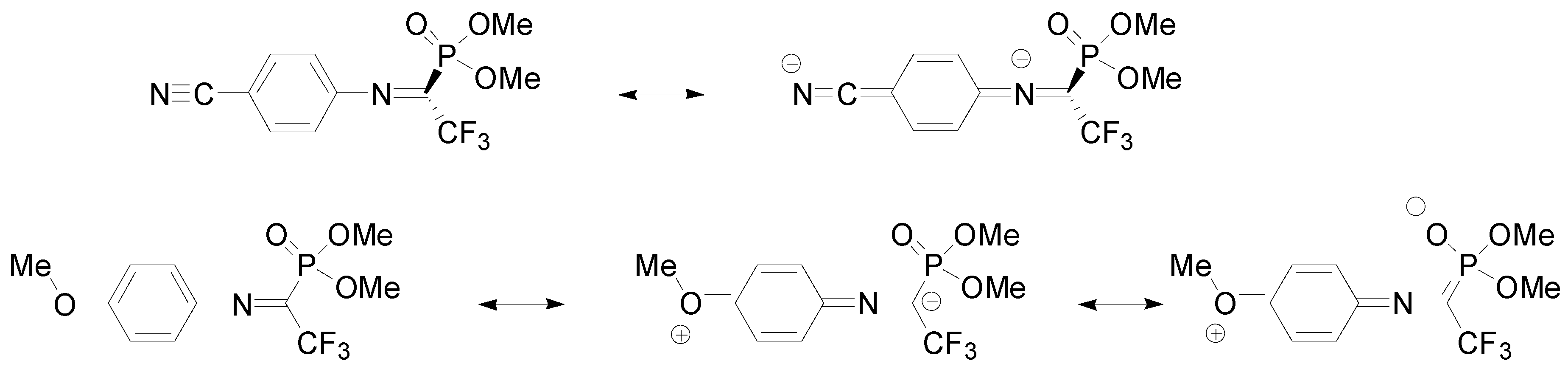
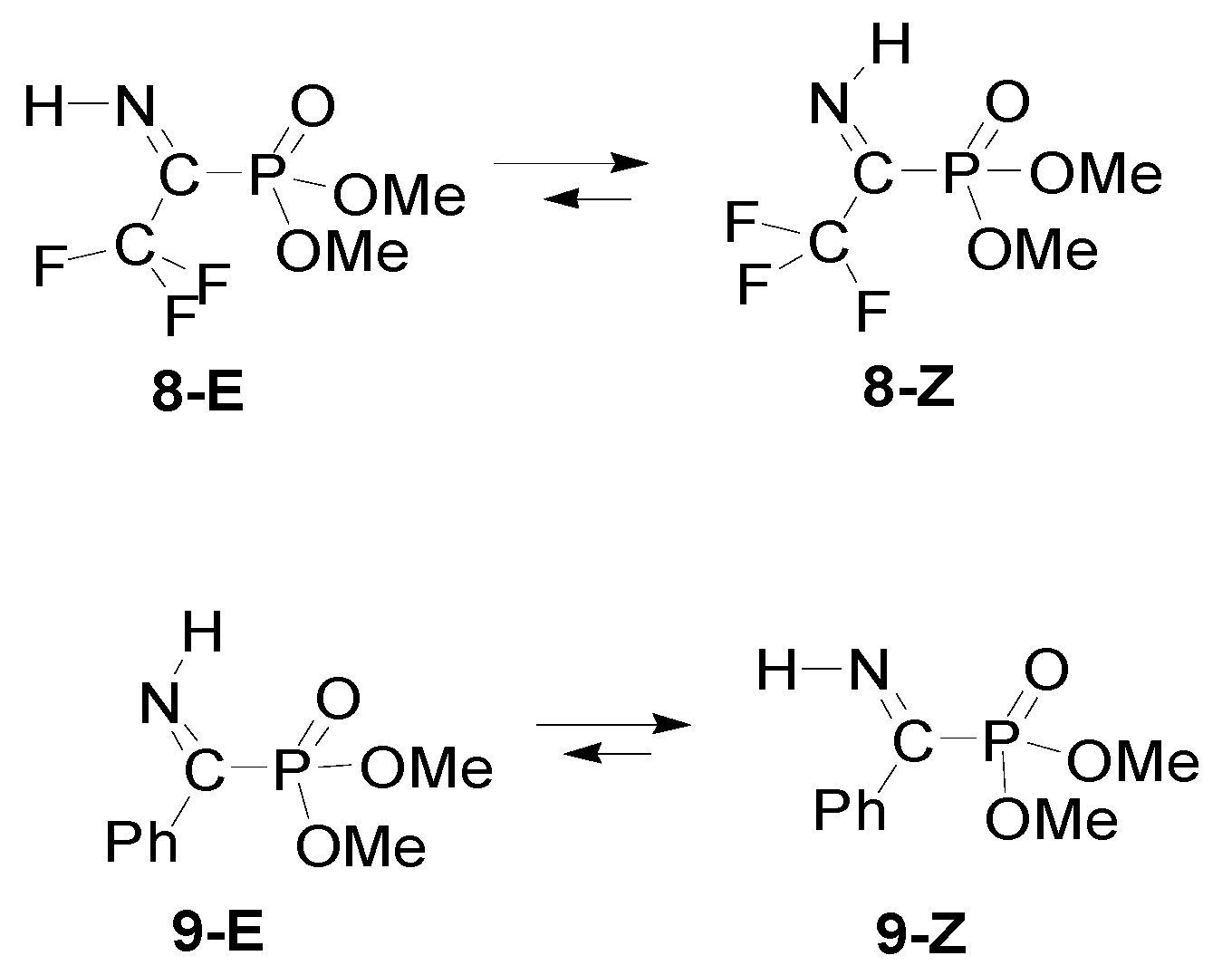
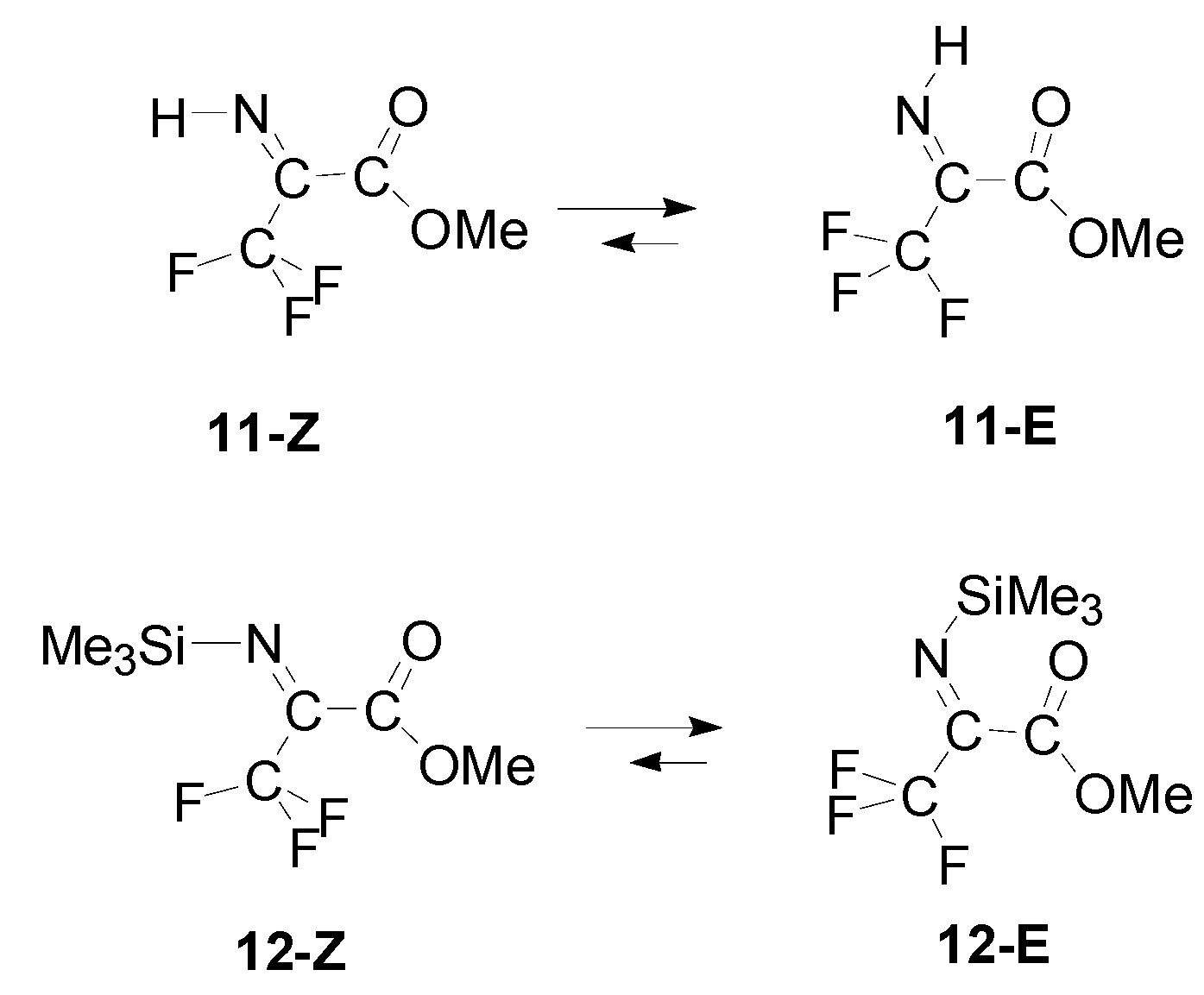
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kafarski, P.; Lejczak, B. Biological activity of aminophosphonic acids. Phosphorus Sulfur Silicon Relat. Elem. 1991, 63, 193–215. [Google Scholar] [CrossRef]
- Turcheniuk, K.V.; Kukhar, V.P.; Röschenthaler, G.-V.; Aceña, J.L.; Soloshonok, V.A.; Sorochinsky, A.E. Recent advances in the synthesis of fluorinated aminophosphonates and aminophosphonic acids. RSC Adv. 2013, 3, 6693–6716. [Google Scholar] [CrossRef]
- Su, Q.; Li, Y.; Wang, B.; Liu, M.; Wang, H.; Wang, W.; Liu, F. Combining the Advantages of Alkene and Azo E–Z Photoisomerizations: Mechanistic Insights into Ketoimine Photoswitches. J. Phys. Chem. A 2017, 121, 2588–2596. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Smart, B.E. Fluorine substituent effects (on bioactivity). J. Fluor. Chem. 2001, 109, 3–11. [Google Scholar] [CrossRef]
- Knorr, R.; Ruhdorfer, J.; Mehlstäubl, J.; Böhrer, P.; Stephenson, D.S. (E, Z)1-Equilibria, 17 Demonstration of the Nitrogen Inversion Mechanism of Imines in a Schiff Base Model. Eur. J. Inorg. Chem. 1993, 126, 747–754. [Google Scholar] [CrossRef]
- Pirozhenko, V.V.; Rozhenko, A.B.; Avdeenko, A.P.; Konovalova, S.A.; Santalova, A.A. Z,E-Isomerization mechanism forN-arylthio-1,4-benzoquinonimines: DNMR and DFT investigations. Magn. Reson. Chem. 2008, 46, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Gálvez, J.; Guirado, A. A theoretical study of topomerization of imine systems: Inversion, rotation or mixed mechanisms? J. Comput. Chem. 2009, 31, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Khomutnik, Y.Y.; Onys’Ko, P.P.; Rassukanaya, Y.V.; Pirozhenko, V.V.; Sinitsa, A.D. N-aryltrifluoroacetimidoylphosphonates. Russ. J. Gen. Chem. 2013, 83, 445–452. [Google Scholar] [CrossRef]
- Roberts, J.D.; Hall, G.E.; Middleton, W.J. Nuclear magnetic resonance spectroscopy. Kinetics of isomerization of p-substituted hexafluoroacetone N-phenylimines. J. Am. Chem. Soc. 1971, 93, 4778–4781. [Google Scholar] [CrossRef]
- Onys’Ko, P.P.; Klyukovskii, D.V.; Bezdudnyi, A.V.; Pirozhenko, V.V.; Pustovit, Y.M.; Synytsya, A.D. N-(R-Cyclopropyl)trifluoroacetimidoyl Phosphonates. Phosphorus. Sulfur. Silicon Relat. Elem. 2014, 189, 1094–1101. [Google Scholar] [CrossRef]
- Onys’Ko, P.; Rassukana, Y.; Kolotylo, M.; Sinitsa, O.; Pirozhenko, V. α-Iminotrifluoroethylphosphonates: The First Representatives of N-H Imidoyl Phosphonates. Synthesis 2007, 2007, 2627–2630. [Google Scholar] [CrossRef]
- Wahbi, A.; Slimani, H.; Touil, S. Multinuclear NMR structural study of novel γ-iminophosphonate and phosphine oxide derivatives. J. Struct. Chem. 2015, 56, 34–41. [Google Scholar] [CrossRef]
- Ony‘skoP, P.; Chudakova, T.I.; Pirozhenko, V.V.; Rozhenko, A.B. α-Ketophosphonates in the Synthesis of α-iminophosphonates. Curr. Green Chem. 2020, 7, 226–238. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian: Wallingford, CT, USA, 2013. [Google Scholar]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Foster, J.P.; Weinhold, F. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F. Natural localized molecular orbitals. J. Chem. Phys. 1985, 83, 1736–1740. [Google Scholar] [CrossRef]
- Scalmani, G.; Frisch, M.J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 2010, 132, 114110. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Jmol: An Open-Source Java Viewer for Chemical Structures in 3D. Available online: http://www.jmol.org/ (accessed on 21 April 2021).
- Canepa, P.; Hanson, R.M.; Ugliengo, P.; Alfredsson, M. J-ICE: A newJmolinterface for handling and visualizing crystallographic and electronic properties. J. Appl. Crystallogr. 2010, 44, 225–229. [Google Scholar] [CrossRef]
- TURBOMOLE GmbH. TURBOMOLE V6.3, 2011, University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007. TURBOMOLE GmbH, Since 2007. Available online: http://www.turbomole.com (accessed on 21 April 2021).
- Furche, F.; Ahlrichs, R.; Hättig, C.; Klopper, W.; Sierka, M.; Weigend, F. Turbomole. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 91–100. [Google Scholar] [CrossRef]
- Weigend, F.; Häser, M. RI-MP2: First derivatives and global consistency. Theor. Chem. Acc. 1997, 97, 331–340. [Google Scholar] [CrossRef]
- Haase, F.; Ahlrichs, R. Semidirect MP2 gradient evaluation on workstation computers: The MPGRAD program. J. Comput. Chem. 1993, 14, 907–912. [Google Scholar] [CrossRef]
- Ghahremanpour, M.M.; Van Maaren, P.J.; Ditz, J.C.; Lindh, R.; Van Der Spoel, D. Large-scale calculations of gas phase thermochemistry: Enthalpy of formation, standard entropy, and heat capacity. J. Chem. Phys. 2016, 145, 114305. [Google Scholar] [CrossRef]
- Grimme, S. Improved second-order Møller–Plesset perturbation theory by separate scaling of parallel- and antiparallel-spin pair correlation energies. J. Chem. Phys. 2003, 118, 9095–9102. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Weigend, F.; Häser, M.; Patzelt, H.; Ahlrichs, R. RI-MP2: Optimized auxiliary basis sets and demonstration of efficiency. Chem. Phys. Lett. 1998, 294, 143–152. [Google Scholar] [CrossRef]
- Hättig, C.; Schmitz, G.; Kossmann, J. Auxiliary basis sets for density-fitted correlated wavefunction calculations: Weighted core-valence and ECP basis sets for post-d elements. Phys. Chem. Chem. Phys. 2012, 14, 6549–6555. [Google Scholar] [CrossRef] [PubMed]
- Bushmarinov, I.; Lyssenko, K.A.; Antipin, M.Y. Atomic energy in the ’Atoms in Molecules’ theory and its use for solving chemical problems. Russ. Chem. Rev. 2009, 78, 283–302. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Hanson, R.M. Jmol– a paradigm shift in crystallographic visualization. J. Appl. Crystallogr. 2010, 43, 1250–1260. [Google Scholar] [CrossRef]
- Ramasubbu, N.; Parthasarathy, R.; Murray-Rust, P. Angular preferences of intermolecular forces around halogen centers: Preferred directions of approach of electrophiles and nucleophiles around carbon-halogen bond. J. Am. Chem. Soc. 1986, 108, 4308–4314. [Google Scholar] [CrossRef]
- Lommerse, J.P.M.; Stone, A.J.; Taylor, R.; Allen, F.H. The Nature and Geometry of Intermolecular Interactions between Halogens and Oxygen or Nitrogen. J. Am. Chem. Soc. 1996, 118, 3108–3116. [Google Scholar] [CrossRef]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef]
- Erdélyi, M. Halogen bonding in solution. Chem. Soc. Rev. 2012, 41, 3547–3557. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef]
- Metrangolo, P.; Murray, J.S.; Pilati, T.; Politzer, P.; Resnati, G.; Terraneo, G. The fluorine atom as a halogen bond donor, viz. a positive site. CrystEngComm 2011, 13, 6593–6596. [Google Scholar] [CrossRef]
- Bader, R.F.W. Bond Paths Are Not Chemical Bonds. J. Phys. Chem. A 2009, 113, 10391–10396. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, S. Why Bond Critical Points Are Not “Bond” Critical Points. Chem. A Eur. J. 2018, 24, 5401–5405. [Google Scholar] [CrossRef] [PubMed]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
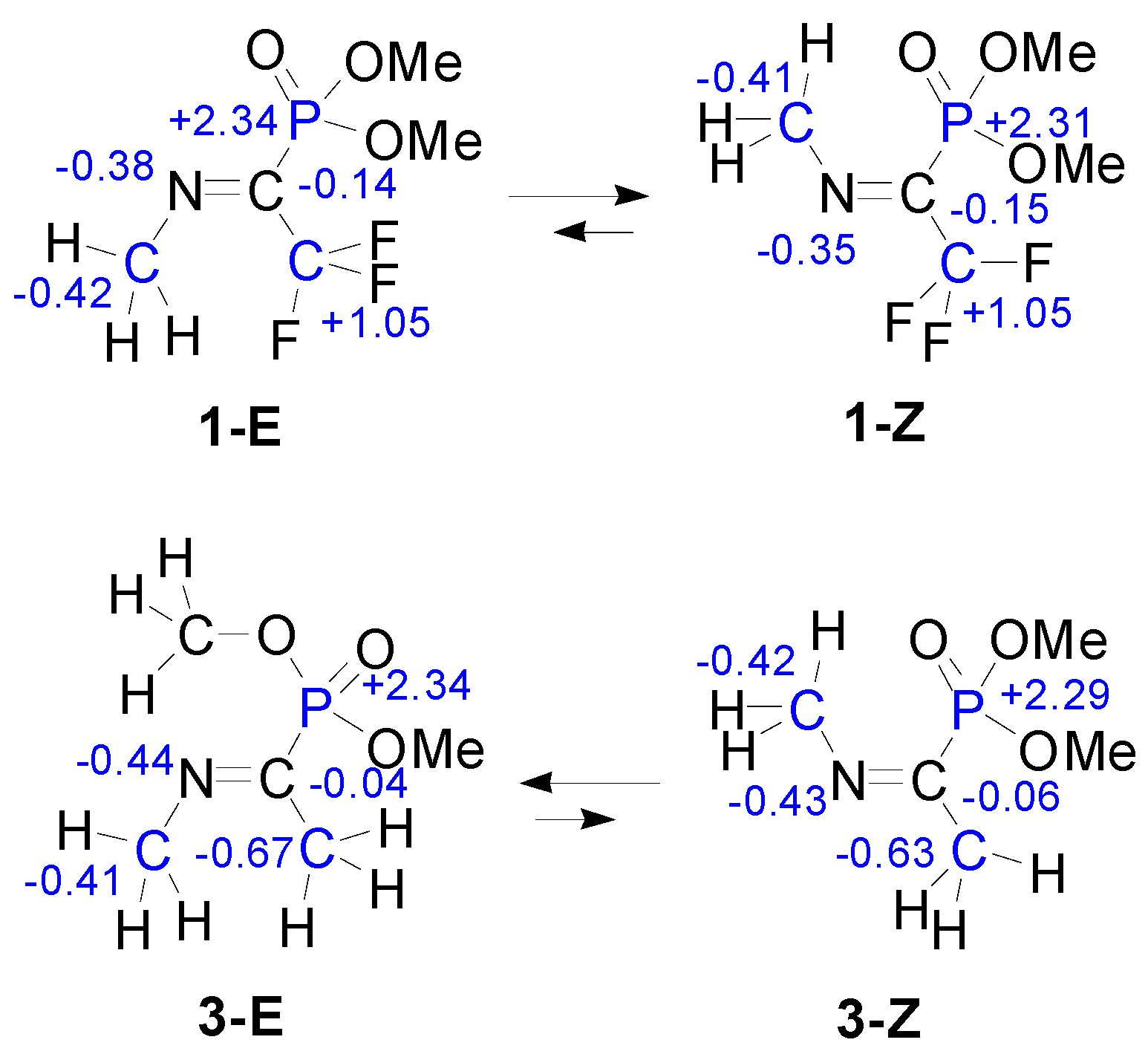{kind=link}
{kind=link}
{kind=link}
{kind=link}
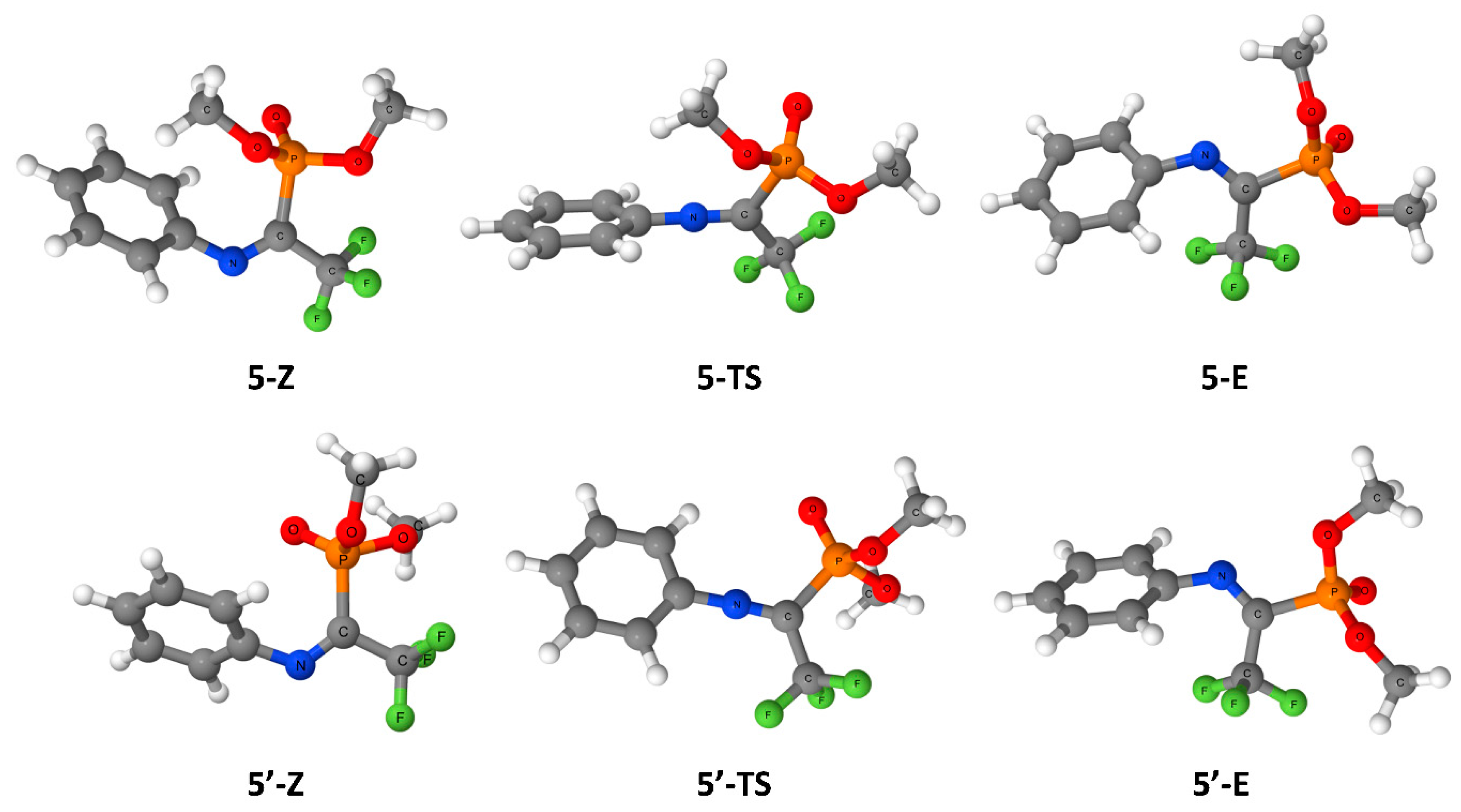{kind=link}
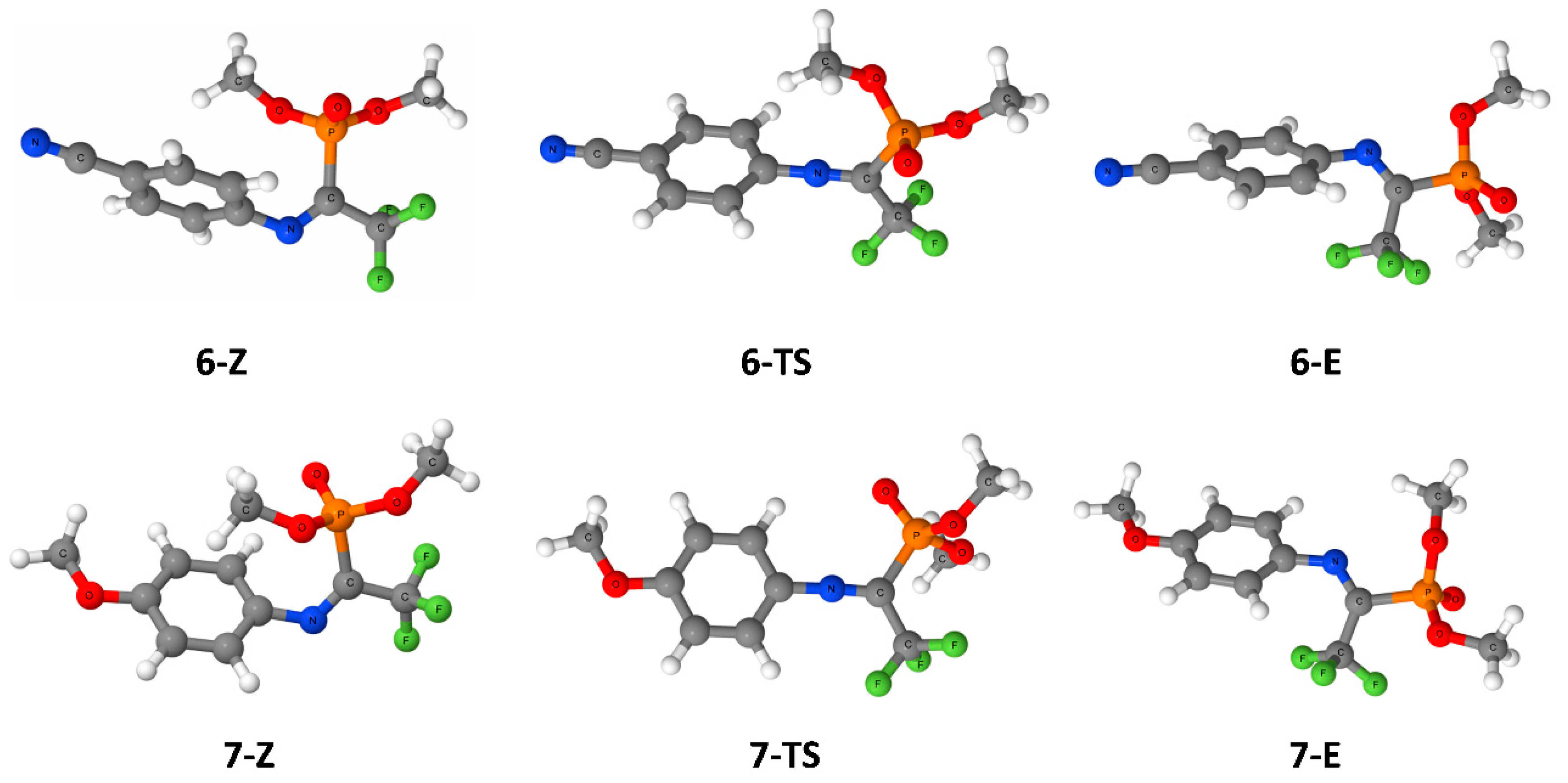{kind=link}
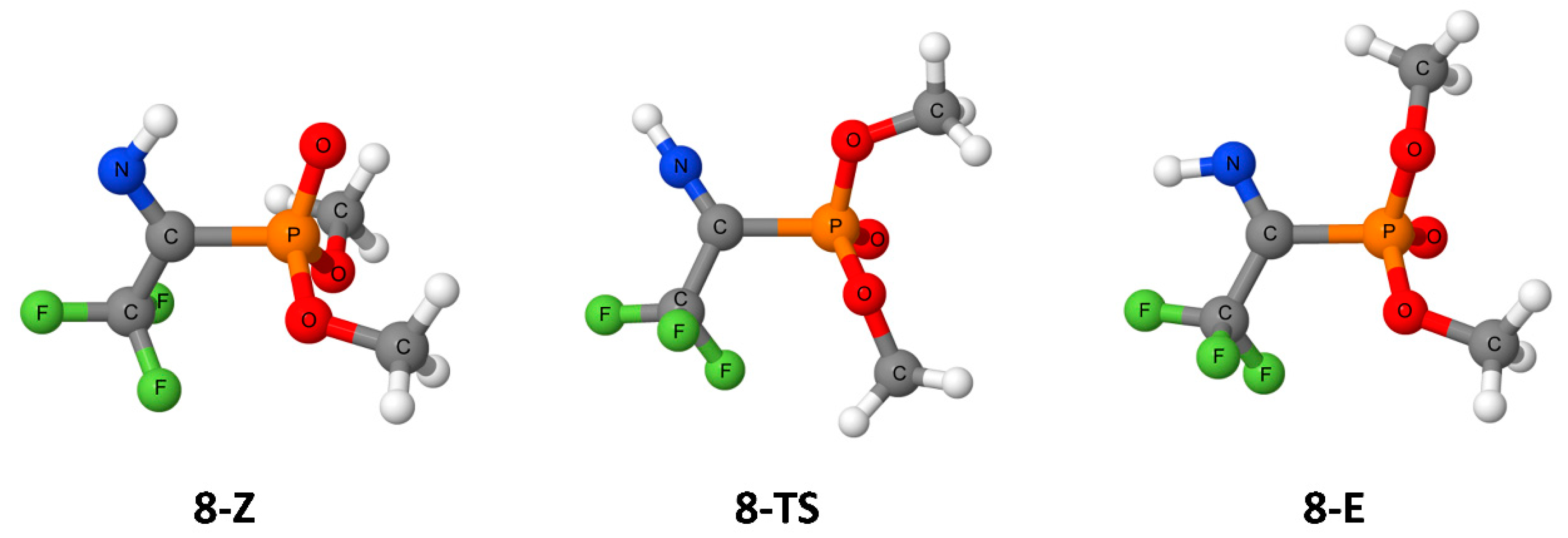{kind=link}
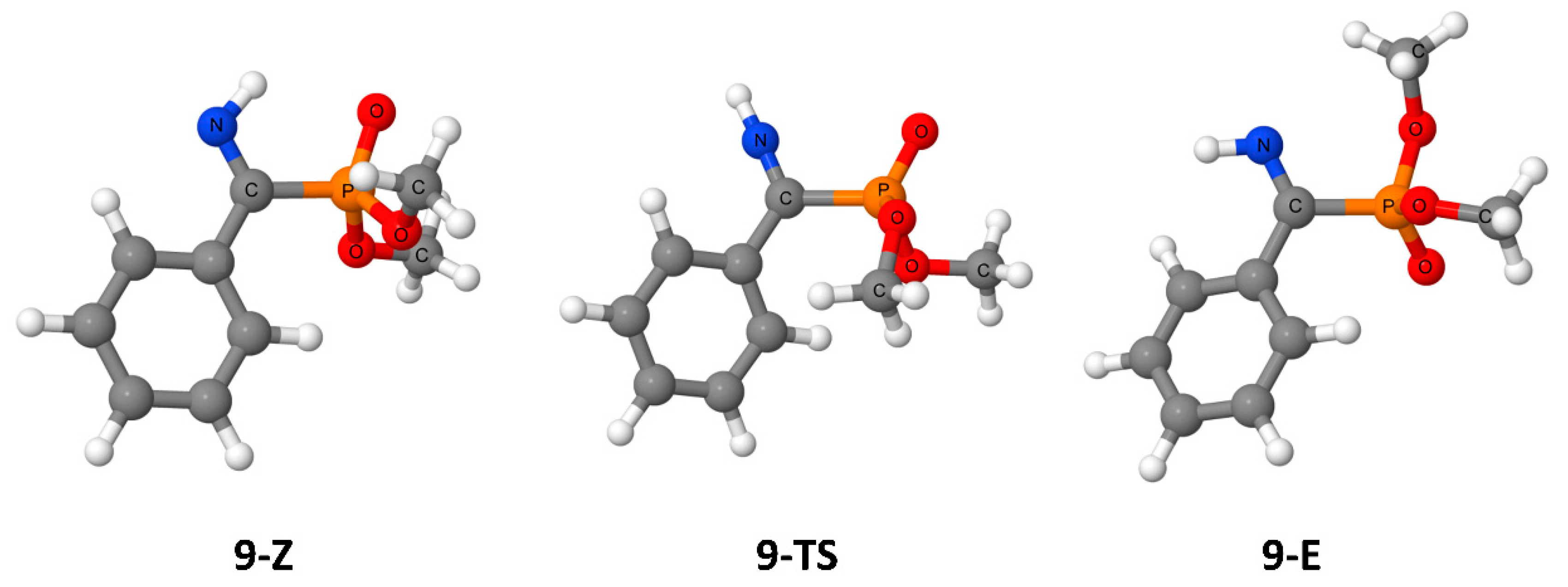{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
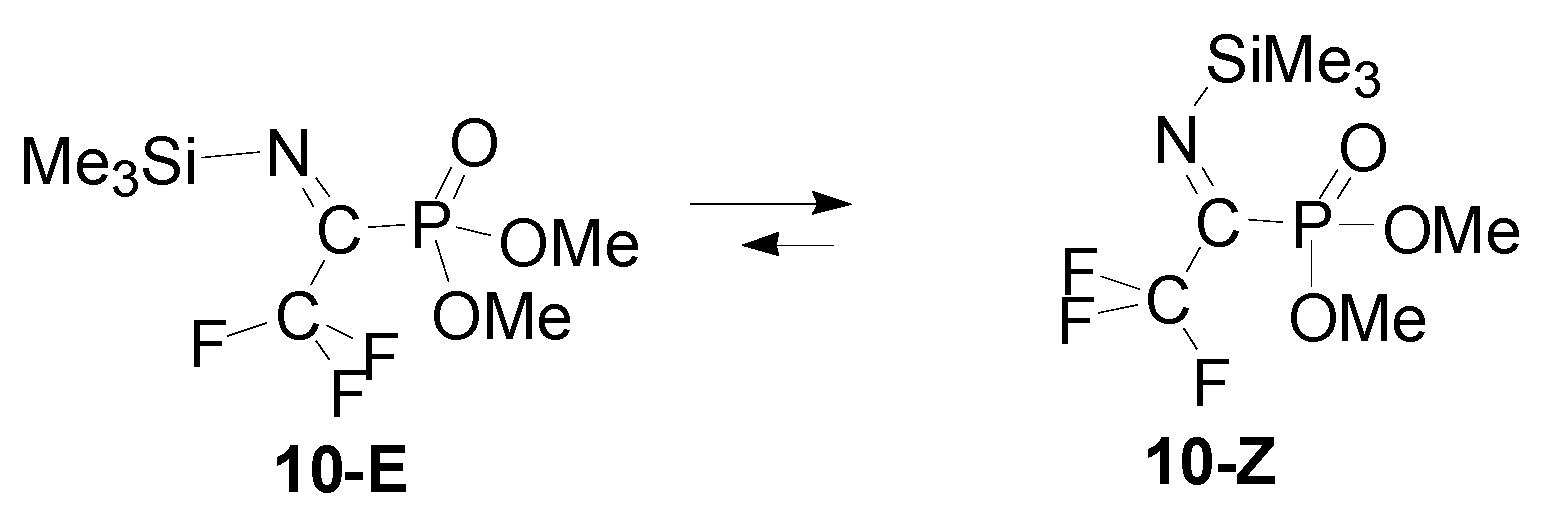{kind=link}
{kind=link}
| Entry | R1 | R2 | R3 | Z/E | ΔG(Z/E) a |
|---|---|---|---|---|---|
| 1 | H | CF3 | Et | 10:1 [12] | |
| 2 | H | CHF2 | Et | 5:1 | |
| 3 | H | C3F7 | Et | 20:1 | |
| 4 | Ph | CF3 | Et | 7:1 | −16.0 |
| 5 | 4-MeOC6H4 | CF3 | Me | 5:1 [9] | −14.4 |
| 6 | 4-CNC6H4 | CF3 | Me | 7:1 [9] | −19.7 |
| 7 | Me | CF3 | Et | 10:1 | |
| 8 | cyc-Pr | CF3 | Et | 11:1 [11] | −5.9 |
| 9 | Me | Ph | Et | 1:17 [9] | |
| 10 | CHMe2 | Ph | Et | 1:20 | |
| 11 | 4-MeOC6H4 | Ph | Et | 1:13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rozhenko, A.B.; Kyrylchuk, A.A.; Lapinska, Y.O.; Rassukana, Y.V.; Trachevsky, V.V.; Pirozhenko, V.V.; Leszczynski, J.; Onysko, P.P. Z,E-Isomerism in a Series of Substituted Iminophosphonates: Quantum Chemical Research. Organics 2021, 2, 84-97. https://doi.org/10.3390/org2020008
Rozhenko AB, Kyrylchuk AA, Lapinska YO, Rassukana YV, Trachevsky VV, Pirozhenko VV, Leszczynski J, Onysko PP. Z,E-Isomerism in a Series of Substituted Iminophosphonates: Quantum Chemical Research. Organics. 2021; 2(2):84-97. https://doi.org/10.3390/org2020008
Chicago/Turabian StyleRozhenko, Alexander B., Andrey A. Kyrylchuk, Yuliia O. Lapinska, Yuliya V. Rassukana, Vladimir V. Trachevsky, Volodymyr V. Pirozhenko, Jerzy Leszczynski, and Petro P. Onysko. 2021. "Z,E-Isomerism in a Series of Substituted Iminophosphonates: Quantum Chemical Research" Organics 2, no. 2: 84-97. https://doi.org/10.3390/org2020008
APA StyleRozhenko, A. B., Kyrylchuk, A. A., Lapinska, Y. O., Rassukana, Y. V., Trachevsky, V. V., Pirozhenko, V. V., Leszczynski, J., & Onysko, P. P. (2021). Z,E-Isomerism in a Series of Substituted Iminophosphonates: Quantum Chemical Research. Organics, 2(2), 84-97. https://doi.org/10.3390/org2020008