
Phosphine Functionalized CpC Ligands and Their Metal Complexes


Abstract
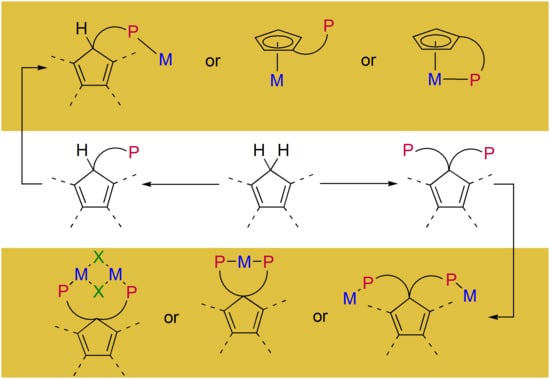
1. Introduction
2. Materials and Methods
3. Results
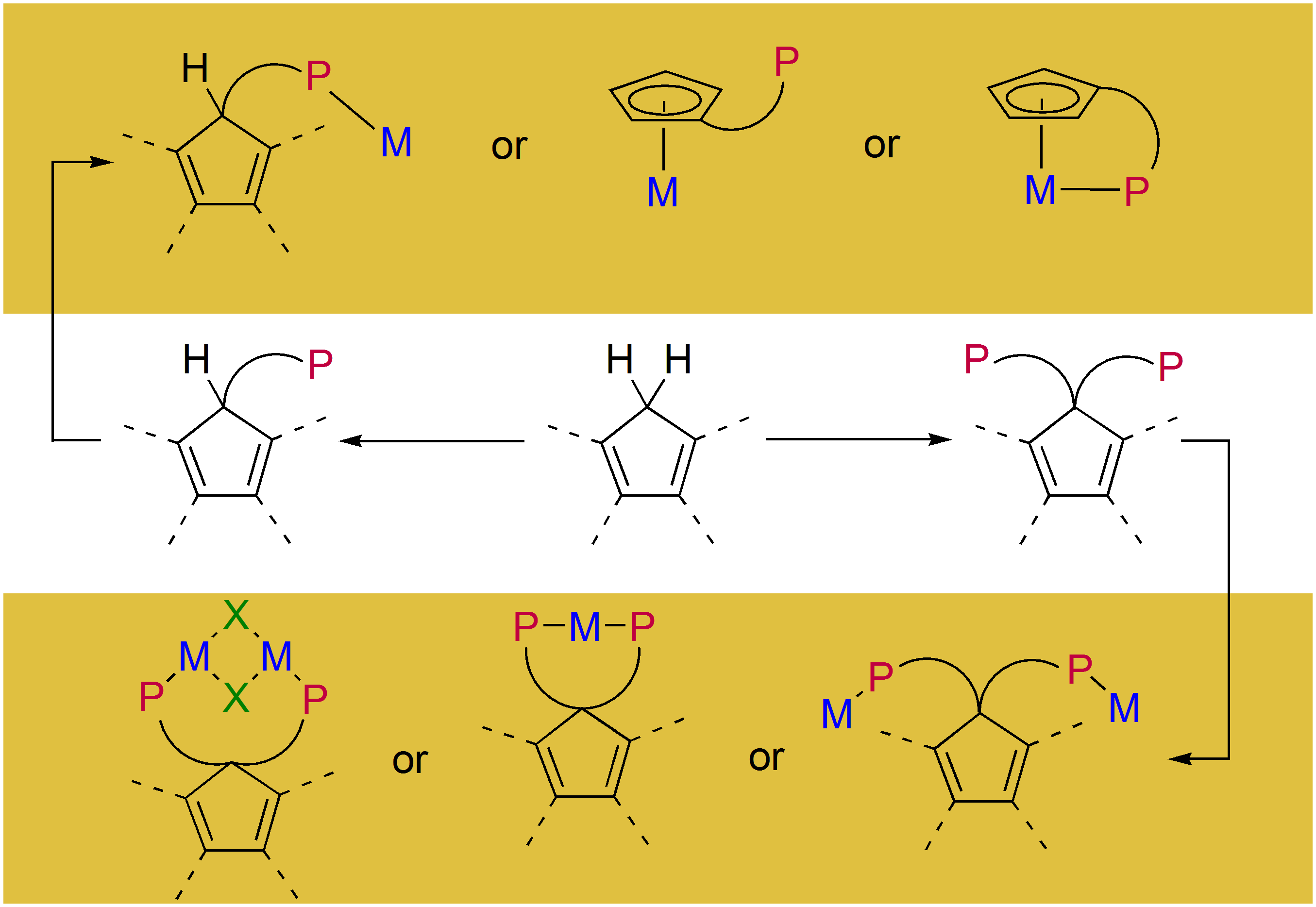
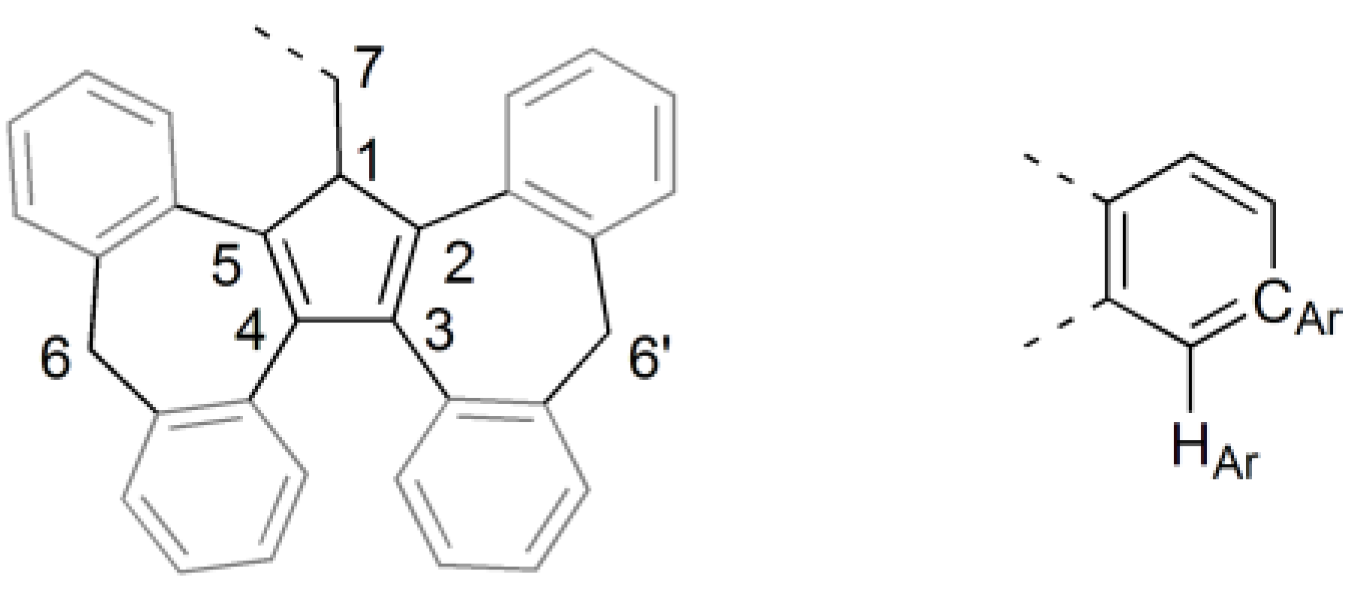
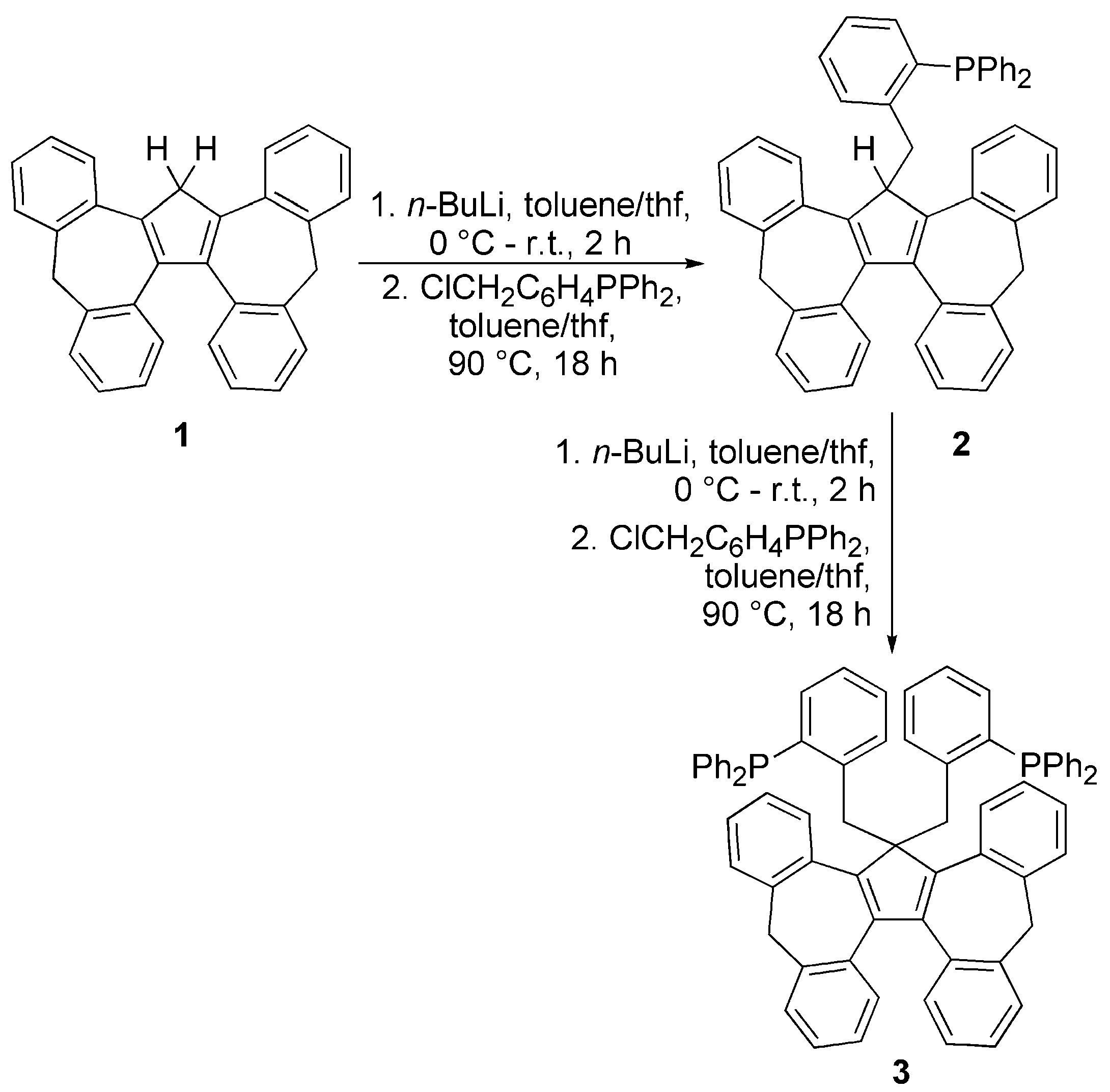
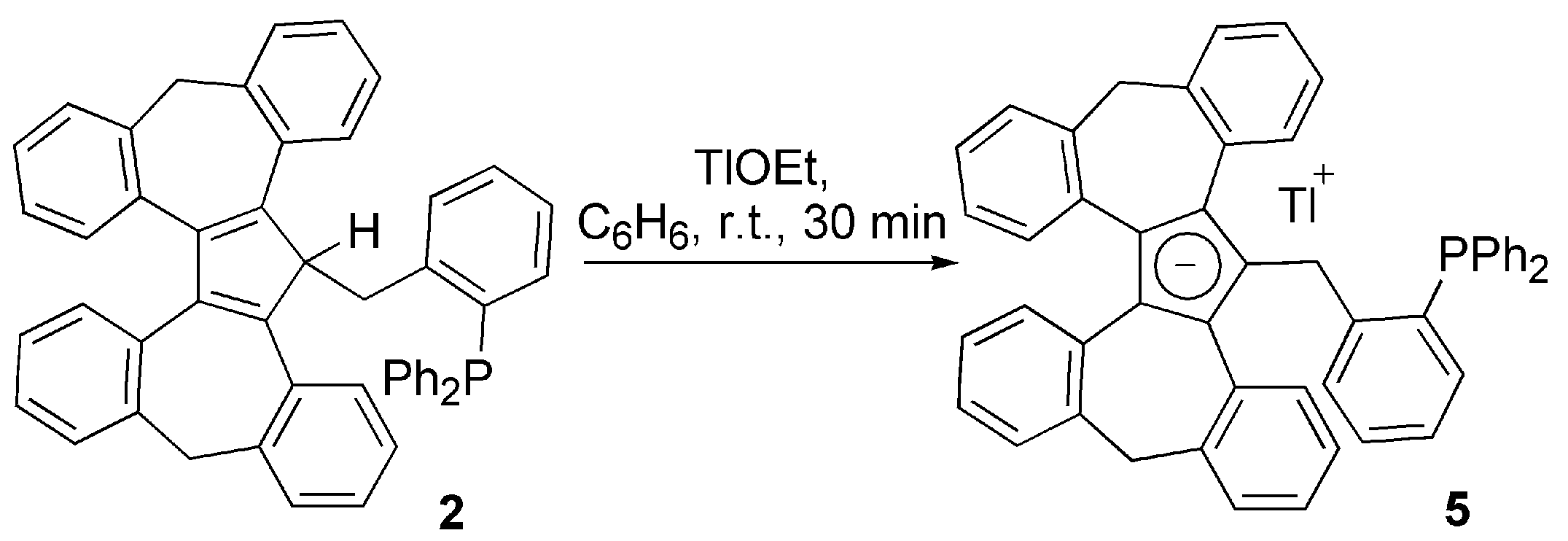
3.1. Ligand Synthesis and Characterization
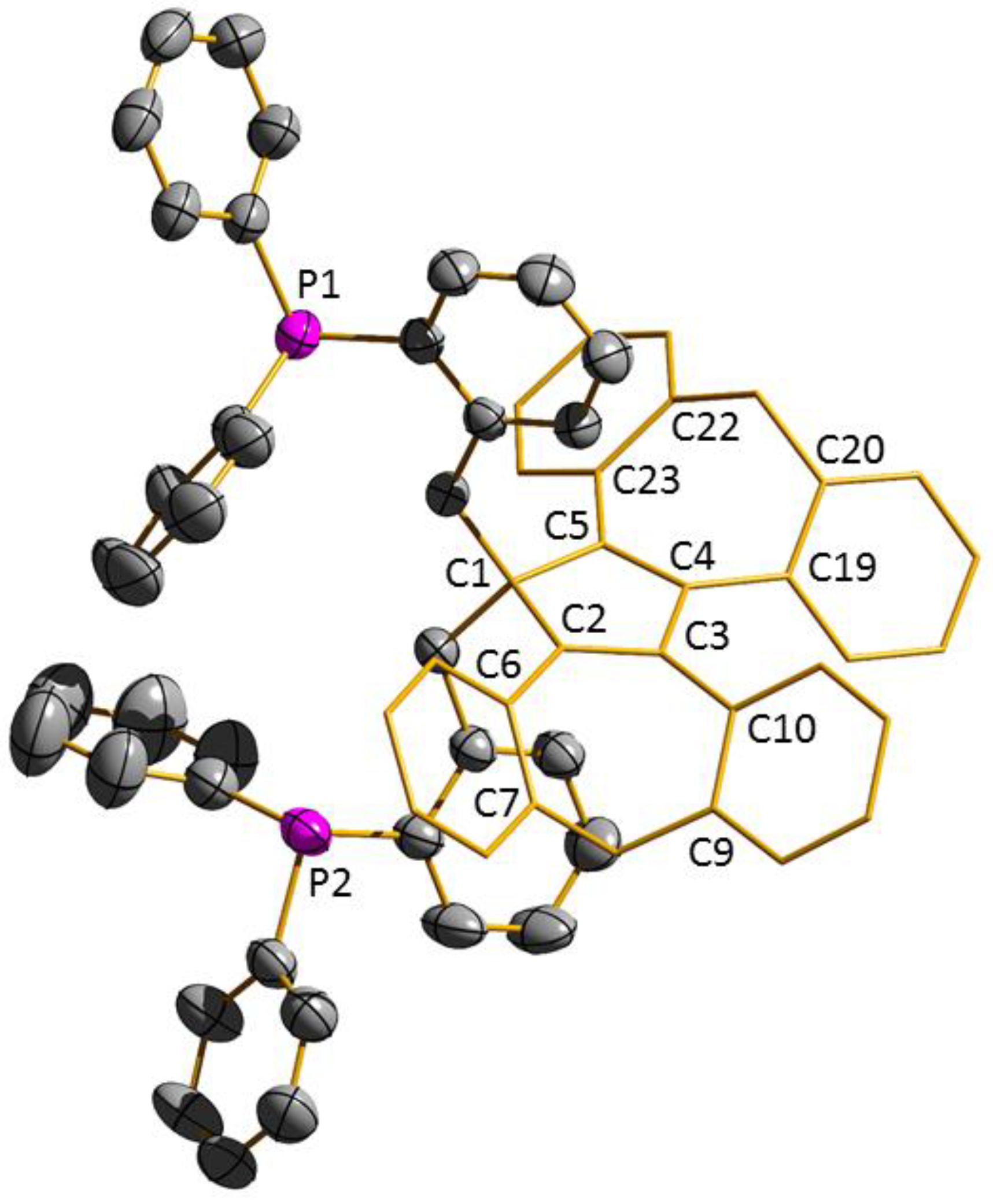
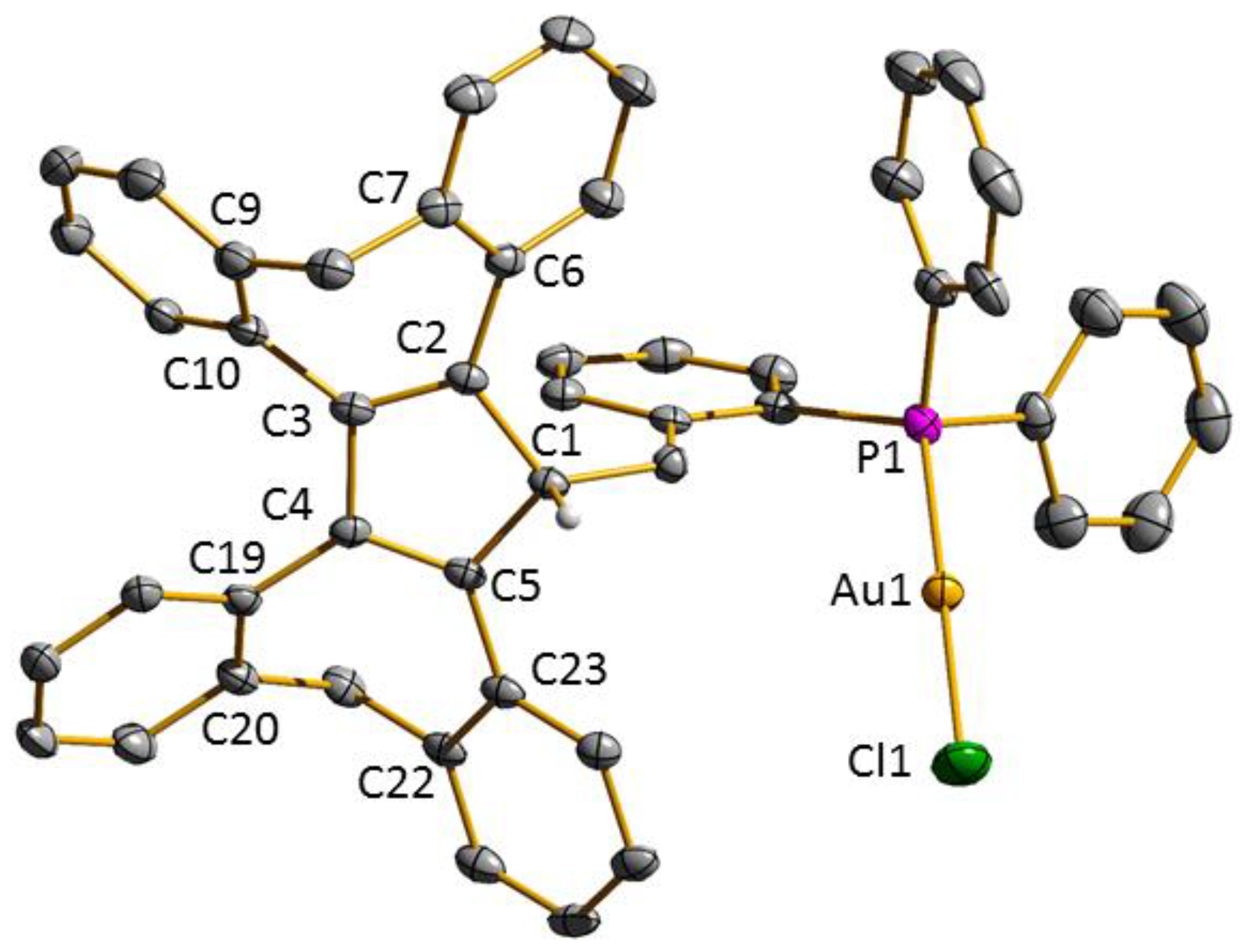
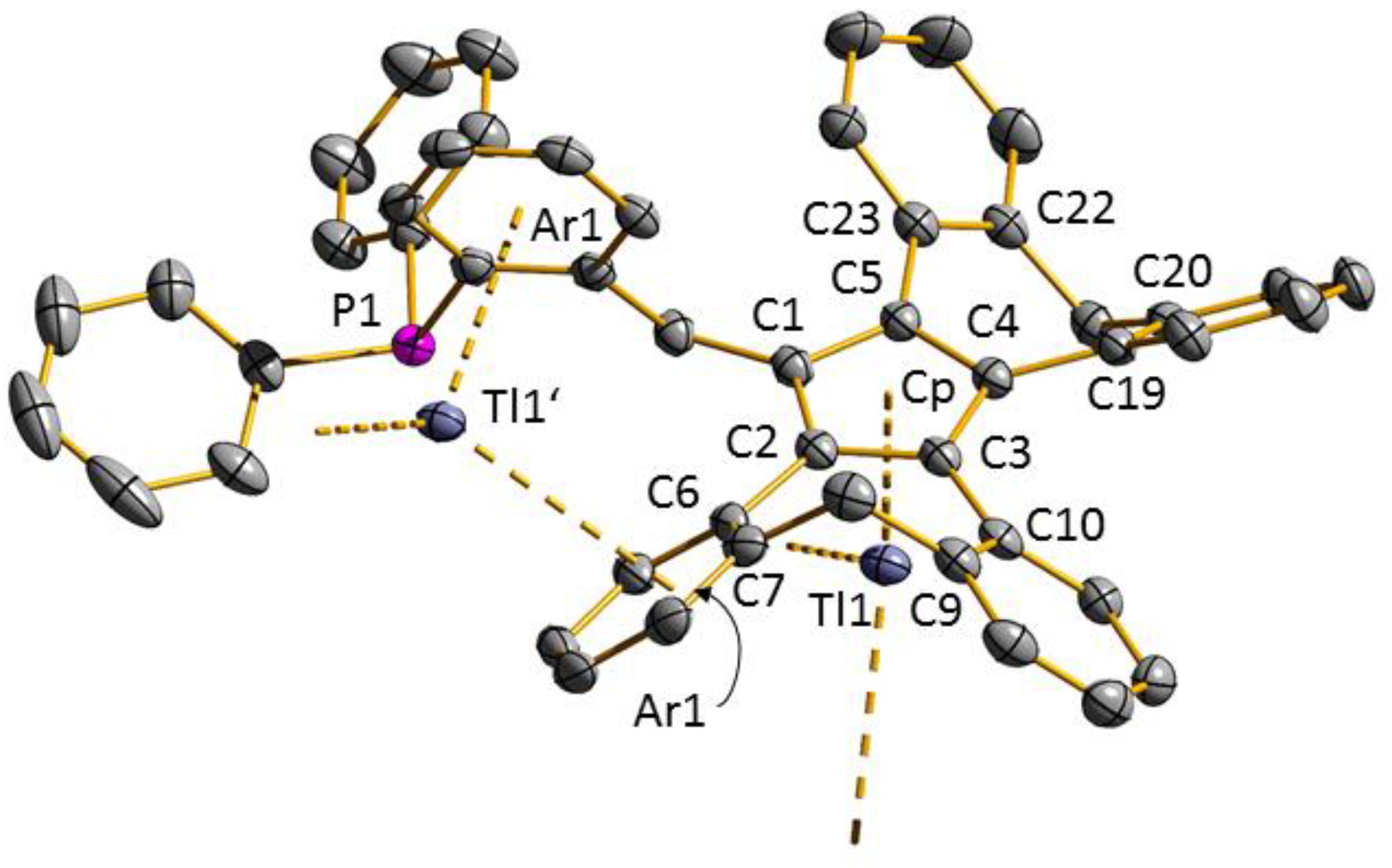
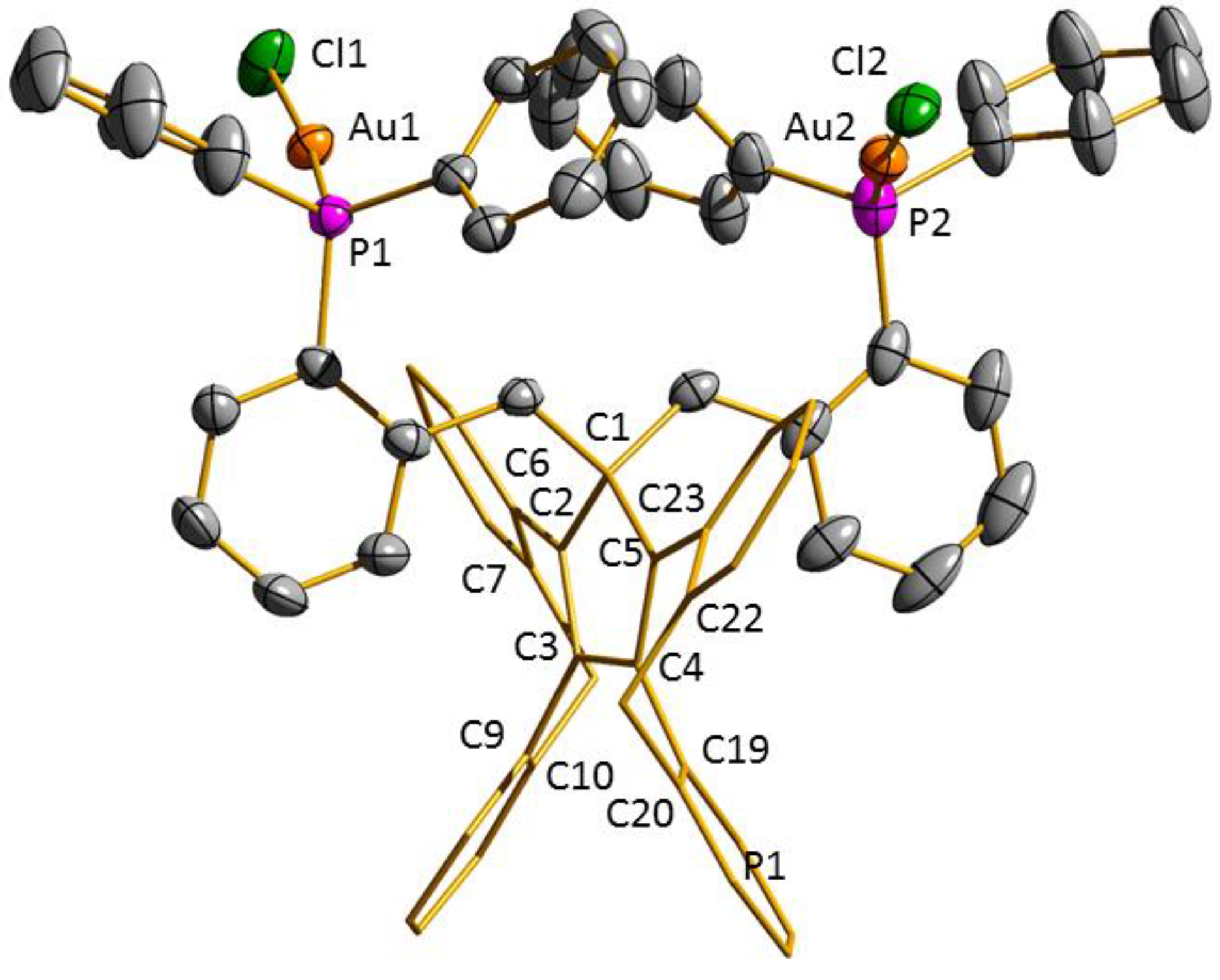
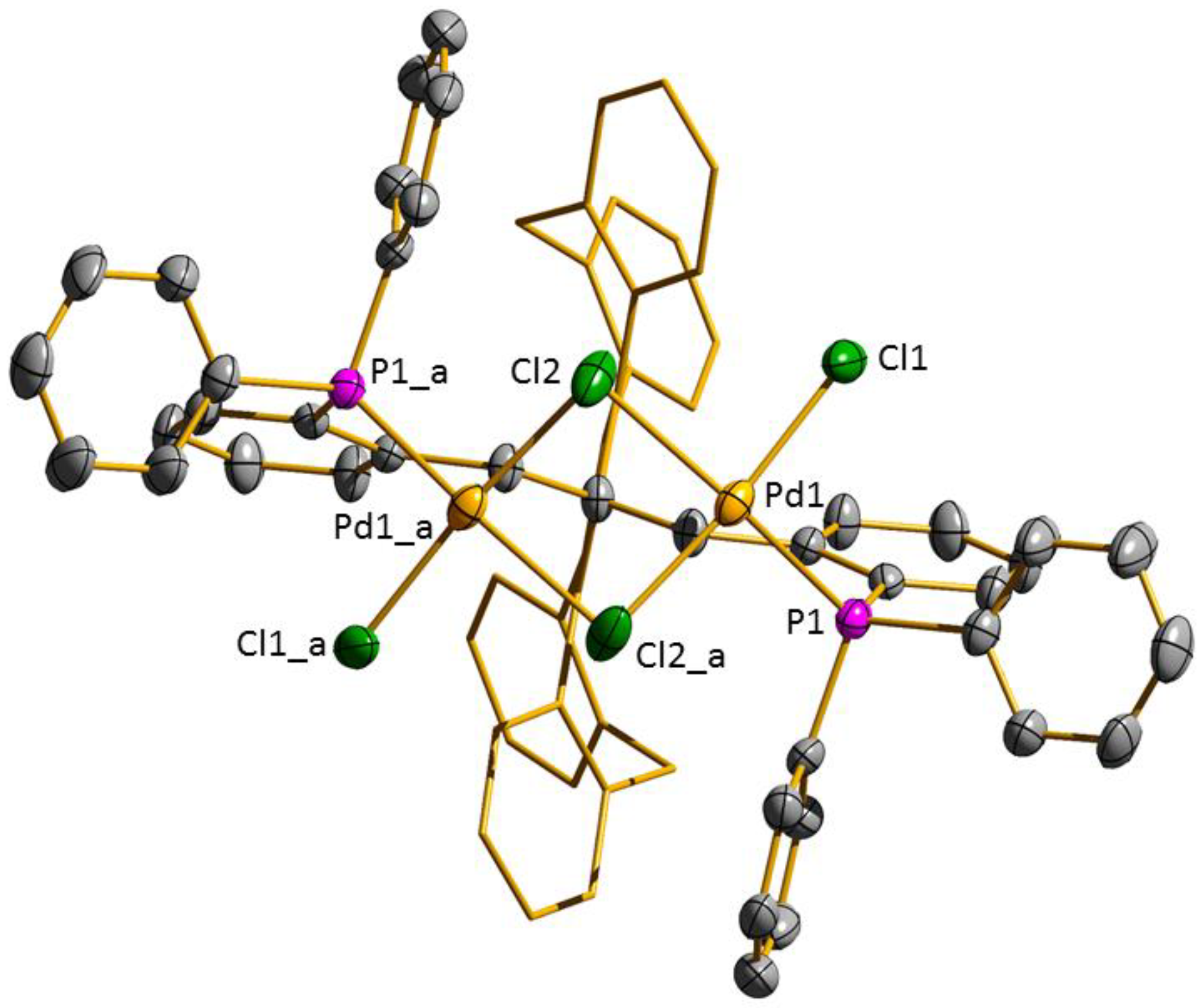
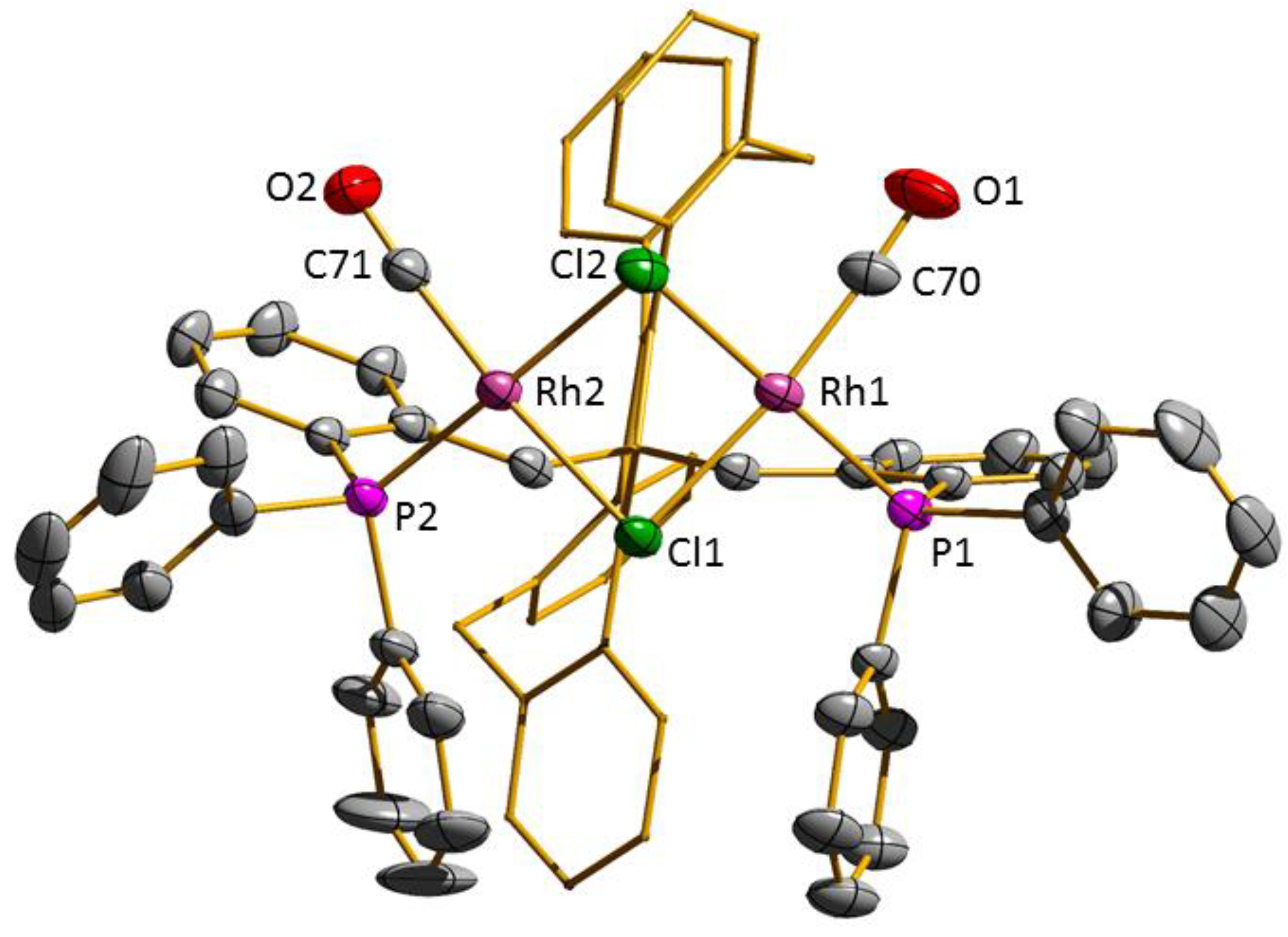
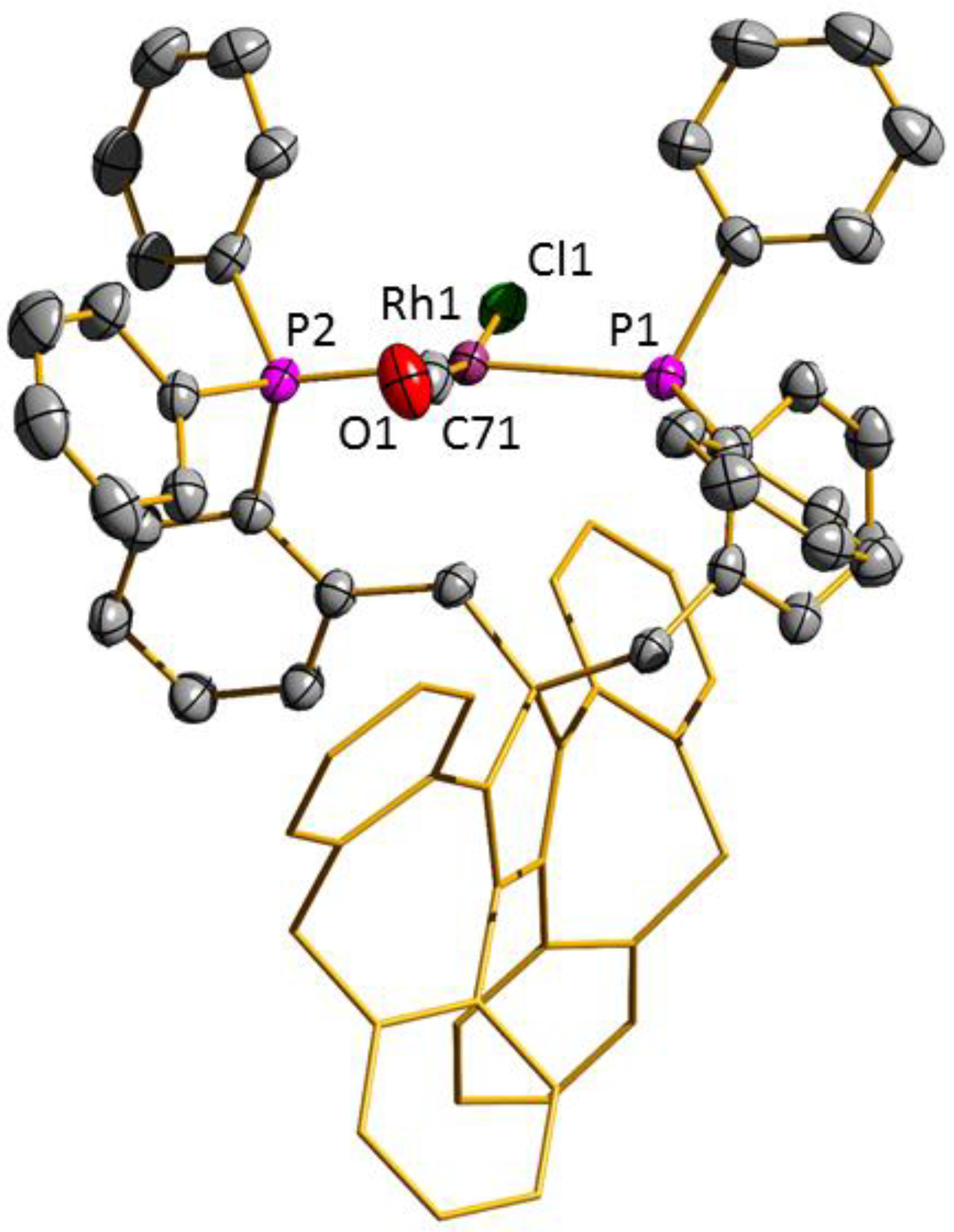
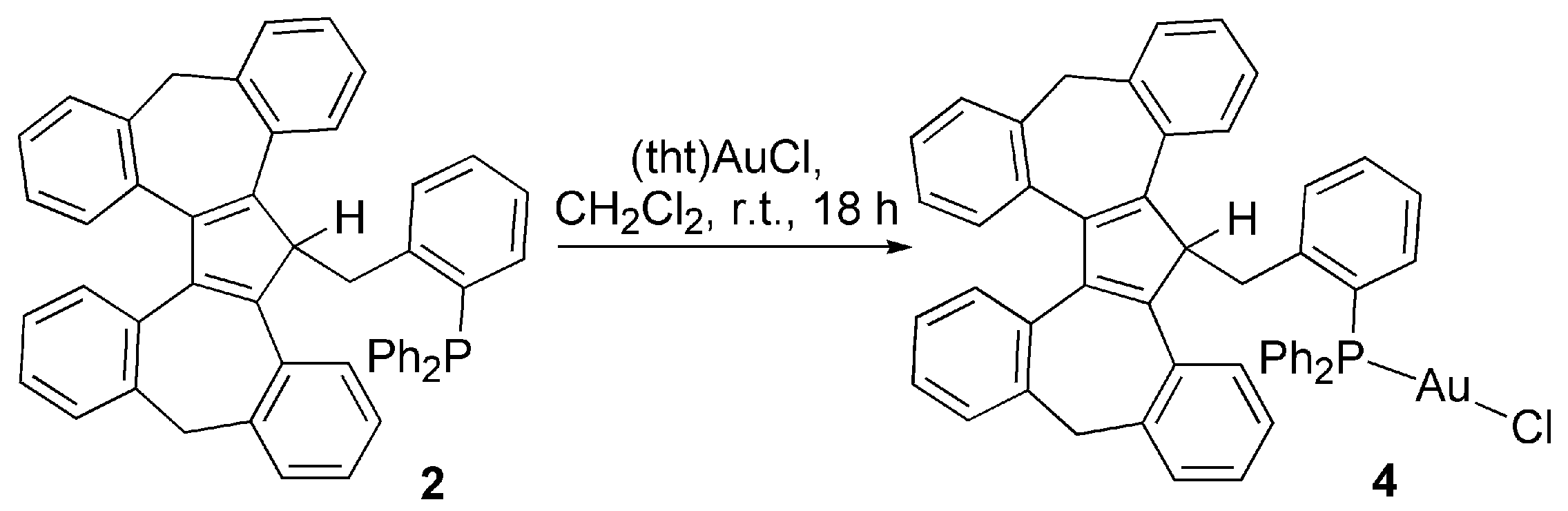
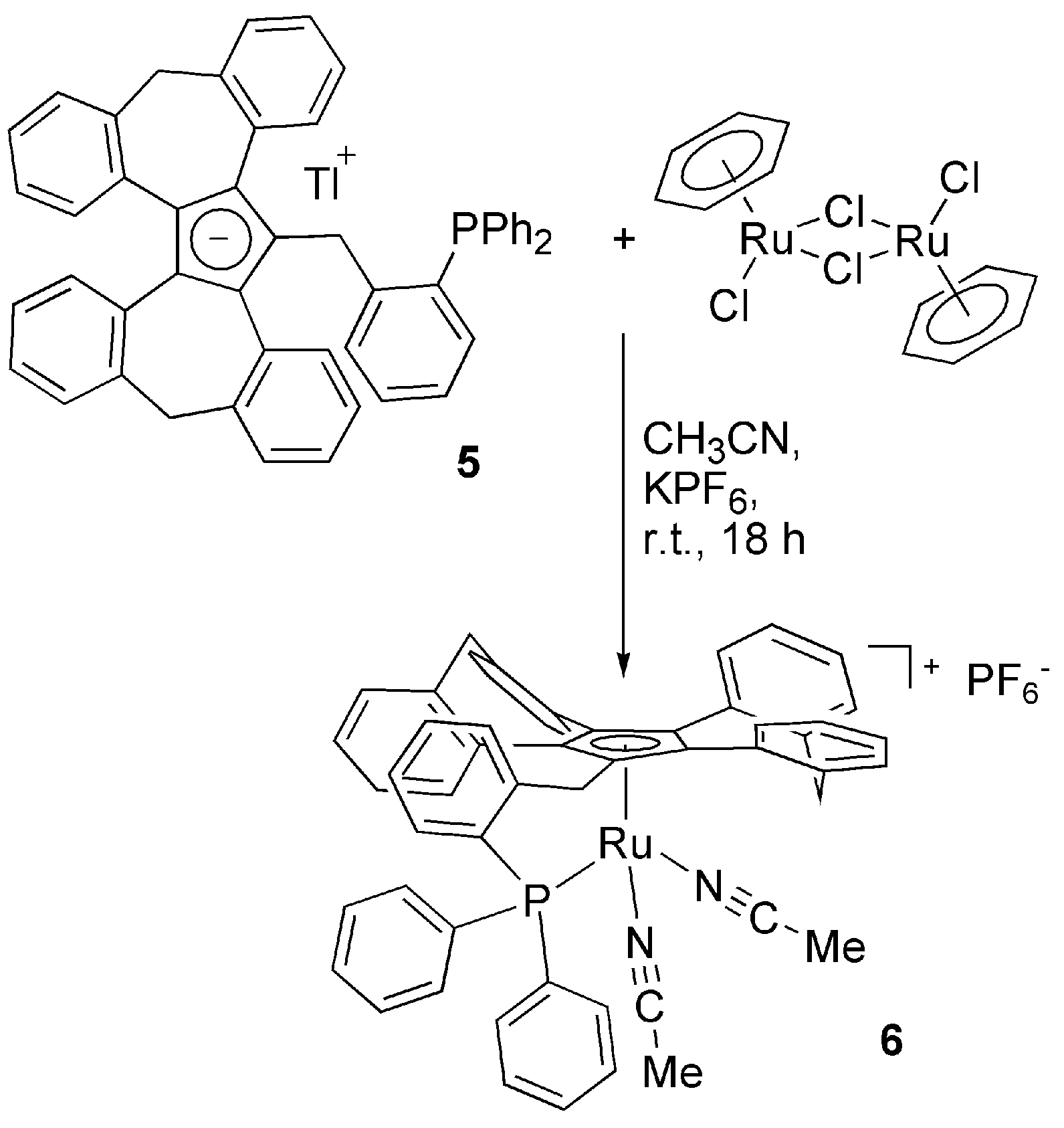
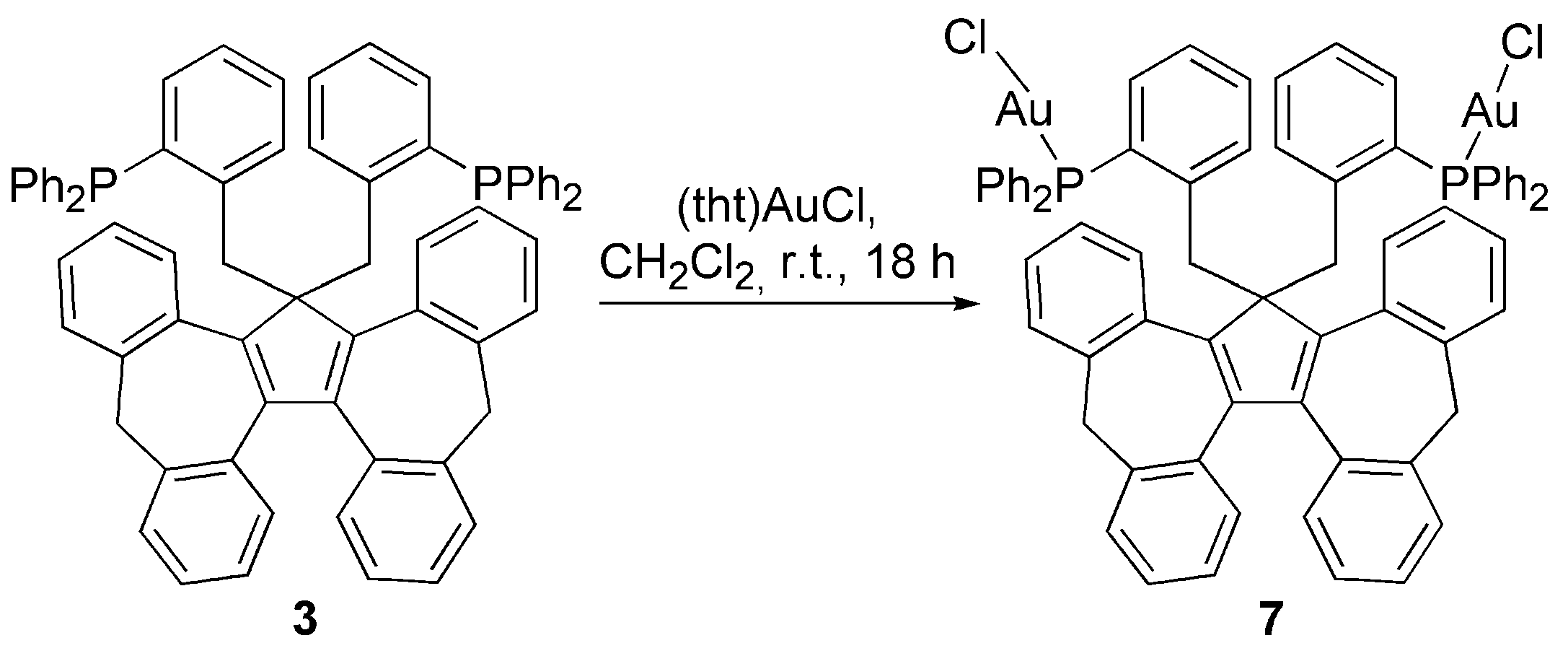
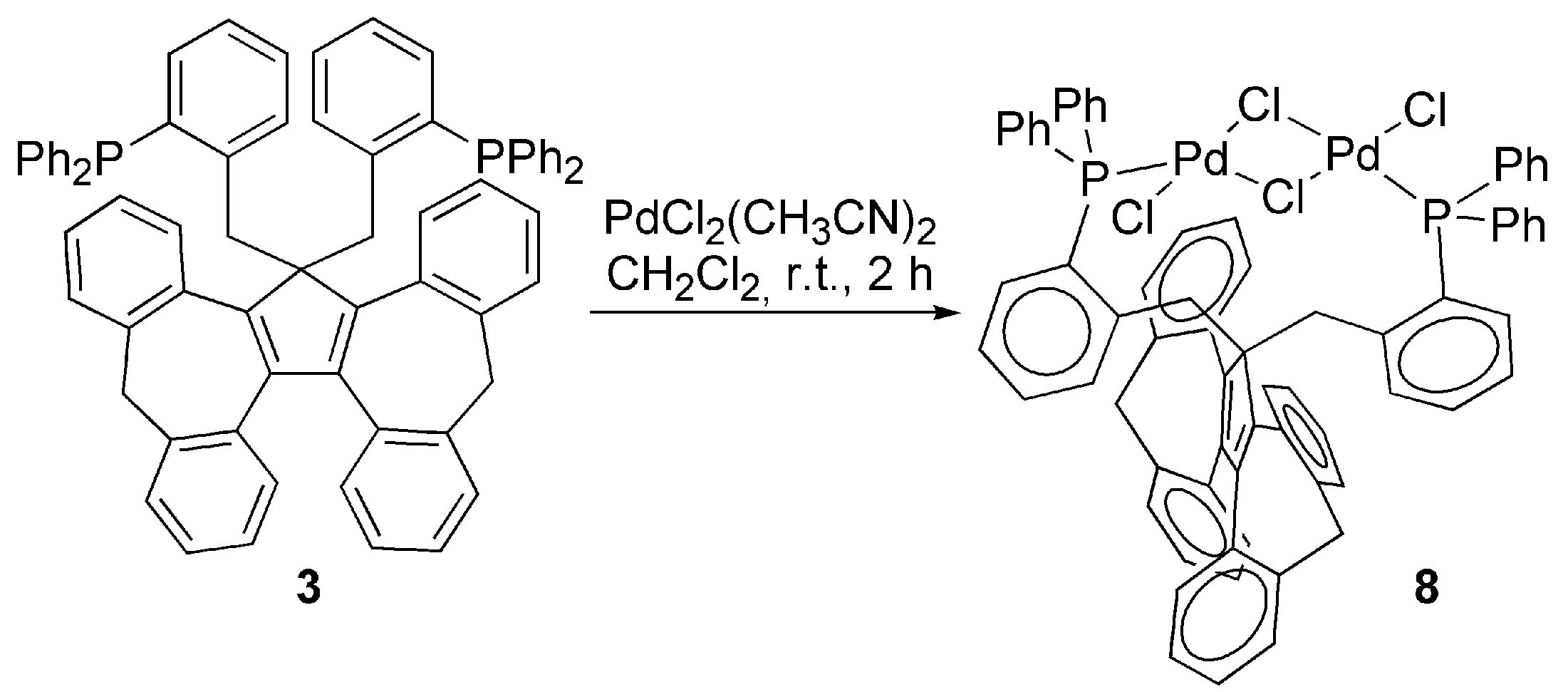
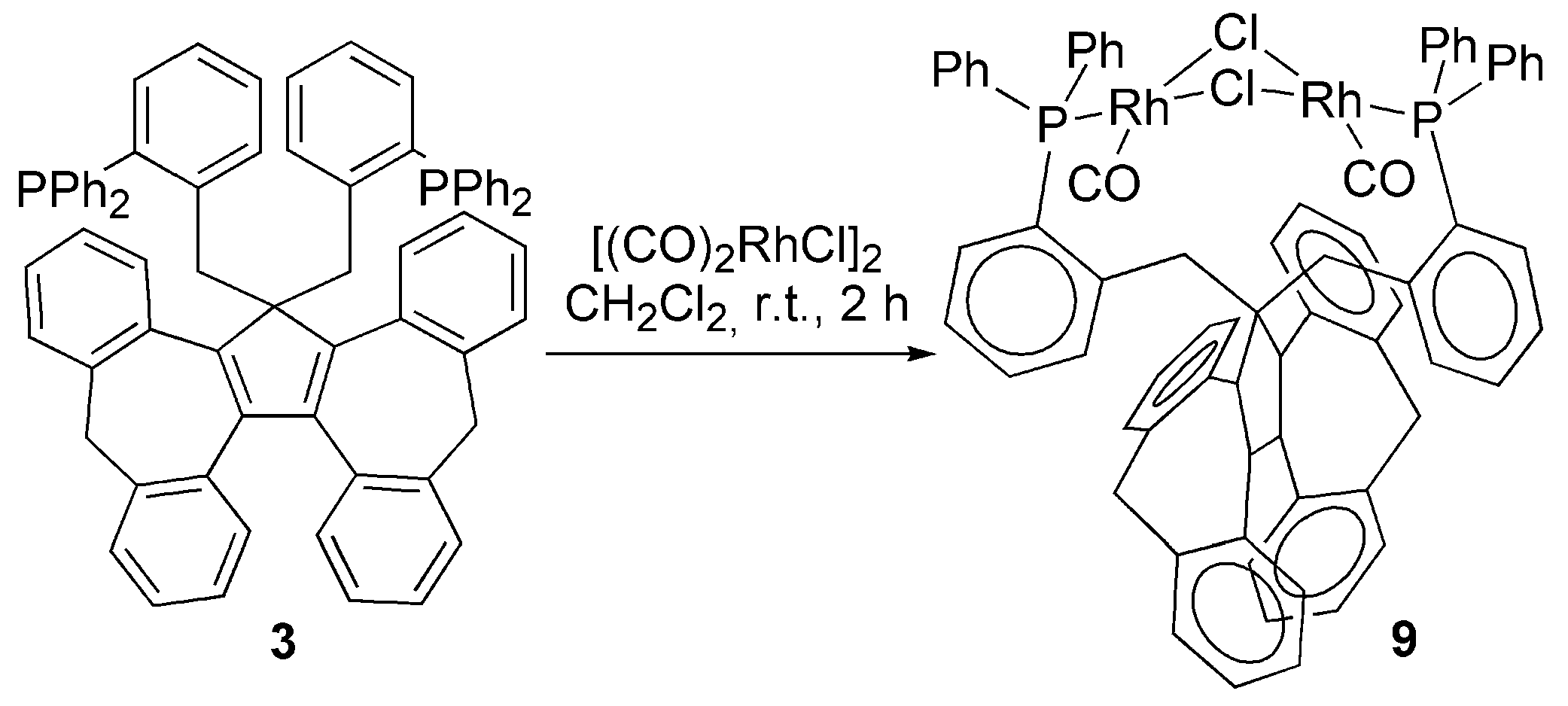
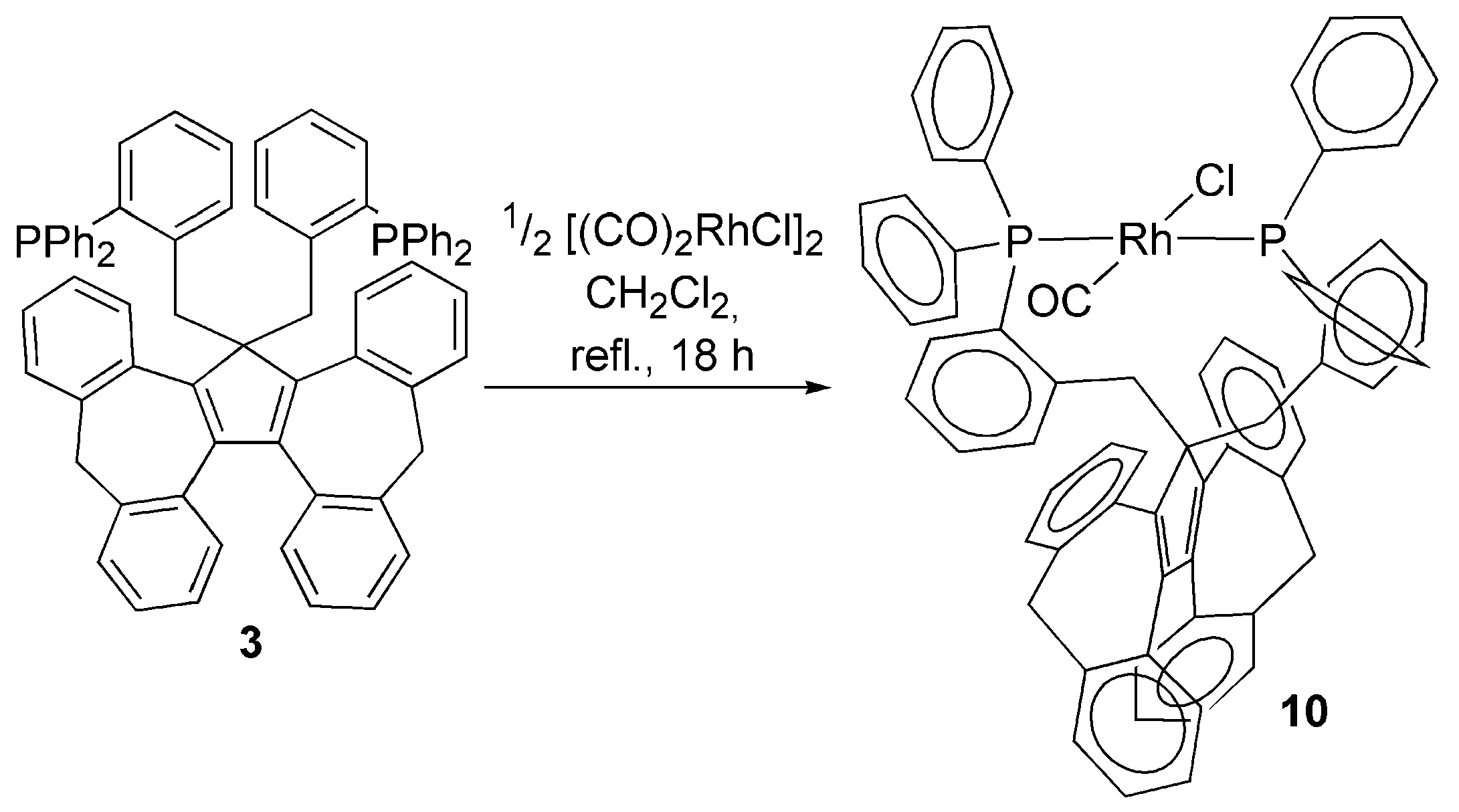
3.2. Transition Metal Complexes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Charrier, C.; Mathey, F. La diphenyl-cyclopentadienylmethyl-phosphine et ses complexesferroceniques et cymantreniques. Tetrahedron Lett. 1978, 19, 2407–2410. [Google Scholar] [CrossRef]
- Kettenbach, R.T.; Bonrath, W.; Butenschön, H. [ω-(Phosphanyl)alkyl]cyclopentadienyl Complexes. Chem. Ber. 1993, 126, 1657–1669. [Google Scholar] [CrossRef]
- Butenschön, H. Cyclopentadienylmetal Complexes Bearing Pendant Phosphorus, Arsenic, and Sulfur Ligands. Chem. Rev. 2000, 100, 1527–1564. [Google Scholar] [CrossRef] [PubMed]
- Bensley, D.M.; Mintz, E.A.; Sussangkarn, S.J. Synthesis of [C5(CH3)4H]CH2CH2CH2P(C6H5)2: A novel heterodifunctional ligand possessing both a tetramethylcyclopentadiene and a remote diphenylphosphine functionality. J. Org. Chem. 1988, 53, 4417–4419. [Google Scholar] [CrossRef]
- Bensley, D.M.; Mintz, E. 1,2,3,4,6-Pentamethylfulvene: A convenient precursor to substituted tetramethylcyclopentadienyl transition metal complexes. J. Organomet. Chem. 1988, 353, 93–102. [Google Scholar] [CrossRef]
- Hayashi, T.; Kumada, M. Asymmetric synthesis catalyzed by chiral ferrocenylphosphine-transition metal complexes. 2. Nickel- and palladium-catalyzed asymmetric Grignard cross-coupling. Acc. Chem. Res. 1982, 15, 395–401. [Google Scholar] [CrossRef]
- Fihri, A.; Meunier, P.; Hierso, J.-C. Performances of symmetrical achiral ferrocenylphosphine ligands in palladium-catalyzed cross-coupling reactions: A review of syntheses, catalytic applications and structural properties. Coord. Chem. Rev. 2007, 251, 2017–2055. [Google Scholar] [CrossRef]
- Kataoka, N.; Shelby, Q.; Stambuli, J.P.; Hartwig, J.F. Air Stable, Sterically Hindered Ferrocenyl Dialkylphosphines for Palladium-Catalyzed C−C, C−N, and C−O Bond-Forming Cross-Couplings. J. Org. Chem. 2002, 67, 5553–5566. [Google Scholar] [CrossRef]
- Ito, Y.; Sawamura, M.; Hayashi, T. Catalytic asymmetric aldol reaction: Reaction of aldehydes with isocyanoacetate catalyzed by a chiral ferrocenylphosphine-gold(I) complex. J. Am. Chem. Soc. 1986, 108, 6405–6406. [Google Scholar] [CrossRef]
- Hayashi, T.; Mise, T.; Fukushima, M.; Kagotani, M.; Nagashima, N.; Hamada, Y.; Matsumoto, A.; Kawakami, S.; Konishi, M. Asymmetric Synthesis Catalyzed by Chiral Ferrocenylphosphine–Transition Metal Complexes. I. Preparation of Chiral Ferrocenylphosphines. Bull. Chem. Soc. Jpn. 1980, 53, 1138–1151. [Google Scholar] [CrossRef]
- Hayashi, T.; Yamamoto, A.; Ito, Y.; Nishioka, E.; Miura, H.; Yanagi, K. Asymmetric Synthesis Catalyzed by Chiral Ferrocenylphosphine-Transition-Metal Complexes. 8. Palladium-Catalyzed Asymmetric Allylic Amination. J. Am. Chem. Soc. 1989, 111, 6301–6311. [Google Scholar] [CrossRef]
- Hayashi, T.; Hayashizaki, K.; Kiyoi, T.; Ito, Y. Asymmetric synthesis catalyzed by chiral ferrocenylphosphine-transition-metal complexes. 6. Practical asymmetric synthesis of 1,1′-binaphthyls via asymmetric cross-coupling with a chiral [(alkoxyalkyl)ferrocenyl]monophosphine/nickel catalyst. J. Am. Chem. Soc. 1988, 110, 8153–8156. [Google Scholar] [CrossRef]
- Chen, Y.; Yi, X.; Cheng, Y.; Huang, A.; Yang, Z.; Zhao, X.; Ling, F.; Zhong, W. Rh-Catalyzed Highly Enantioselective Hydrogenation of Functionalized Olefins with Chiral Ferrocenylphosphine-Spiro Phosphonamidite Ligands. J. Org. Chem. 2022, 87, 7864–7874. [Google Scholar] [CrossRef] [PubMed]
- Barth, E.L.; Davis, R.M.; Mohadjer Beromi, M.; Walden, A.G.; Balcells, D.; Brudvig, G.W.; Dardir, A.H.; Hazari, N.; Lant, H.M.C.; Mercado, B.Q.; et al. Bis(dialkylphosphino)ferrocene-Ligated Nickel(II) Precatalysts for Suzuki–Miyaura Reactions of Aryl Carbonates. Organometallics 2019, 38, 3377–3387. [Google Scholar] [CrossRef]
- Škoch, K.; Schulz, J.; Císařová, I.; Štěpnička, P. Pd(II) Complexes with Chelating Phosphinoferrocene Diaminocarbene Ligands: Synthesis, Characterization, and Catalytic Use in Pd-Catalyzed Borylation of Aryl Bromides. Organometallics 2019, 38, 3060–3073. [Google Scholar] [CrossRef]
- Škoch, K.; Císařová, I.; Štěpnička, P. Synthesis of a Polar Phosphinoferrocene Amidosulfonate Ligand and Its Application in Pd-Catalyzed Cross-Coupling Reactions of Aromatic Boronic Acids and Acyl Chlorides in an Aqueous Medium. Organometallics 2016, 35, 3378–3387. [Google Scholar] [CrossRef]
- Chung, J.-Y.; Schulz, C.; Bauer, H.; Sun, Y.; Sitzmann, H.; Auerbach, H.; Pierik, A.J.; Schünemann, V.; Neuba, A.; Thiel, W.R. Cyclopentadienide Ligand CpC– Possessing Intrinsic Helical Chirality and Its Ferrocene Analogues. Organometallics 2015, 34, 5374–5382. [Google Scholar] [CrossRef]
- Chung, J.-Y.; Sun, Y.; Thiel, W.R. Titanium(IV) complexes bearing the (CpC)− ligand. J. Organometal. Chem. 2017, 829, 31–36. [Google Scholar] [CrossRef]
- Nährig, F.; Gemmecker, G.; Chung, J.-Y.; Hütchen, P.; Lauk, S.; Klein, M.; Sun, Y.; Niedner-Schatteburg, G.; Sitzmann, H.; Thiel, W.R. Complexes of Platinum Group Elements Containing the Intrinsically Chiral Cyclopentadienide Ligand (CpC)−1. Organometallics 2020, 39, 1934–1944. [Google Scholar] [CrossRef]
- Nährig, F.; Nunheim, N.; Salih, K.S.M.; Chung, J.-Y.; Gond, D.; Sun, Y.; Becker, S.; Thiel, W.R. A Novel Cyclopentadienone and its Ruthenium and Iron Tricarbonyl Complexes. Eur. J. Inorg. Chem. 2021, 4832–4841. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 6th ed.; Butterworth-Heinemann: Oxford, UK, 2009. [Google Scholar]
- Herrmann, J.; Pregosin, P.S.; Salzmann, R.; Albinati, A. Palladium π-Allyl Chemistry of New P,S Bidentate Ligands. Selective but Variable Dynamics in the Isomerization of the η3-C3H5 and η3-PhCHCHCHPh π-Allyl Ligands. Organometallics 1995, 14, 3311–3318. [Google Scholar] [CrossRef]
- Rauchfuss, T.B.; Patino, F.T.; Roundhill, D.M. Platinum Metal Complexes of Amine- and Ether-Substituted Phosphines. Inorg. Chem. 2002, 14, 652–656. [Google Scholar] [CrossRef]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Burla, M.C.; Polidori, G.; Camalli, M. SIR92—A program for automatic solution of crystal structures by direct methods. J. Appl. Cryst. 1994, 27, 435. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction; Version 1.171.38.46; CrysAlisPro: Seattle, WA, USA, 2015.
- Li, J.; Yin, J.; Wang, G.-X.; Yin, Z.-B.; Zhang, W.-X.; Xi, Z. Synthesis and reactivity of asymmetric Cr(i) dinitrogen complexes supported by cyclopentadienyl–phosphine ligands. Chem. Commun. 2019, 55, 9641–9644. [Google Scholar] [CrossRef] [PubMed]
- Ireland, T.; Tappe, K.; Grossheimann, G.; Knochel, P. Synthesis of a New Class of Chiral 1,5-Diphosphanylferrocene Ligands and Their Use in Enantioselective Hydrogenation. Chem. Eur. J. 2002, 8, 843–852. [Google Scholar] [CrossRef]
- Trampert, J.; Sun, Y.; Thiel, W.R. The reactivity of [{2-(diphenylphosphino)phenyl}methyl]-3-imidazol-2-ylidenes towards group VIII element precursors. J. Organometal. Chem. 2020, 915, 121222. [Google Scholar] [CrossRef]
- Keough, P.T.; Grayson, M. Phosphonioethylation. Michael Addition to Vinylphosphonium Salts. J. Org. Chem. 1964, 29, 631–635. [Google Scholar] [CrossRef]
- Matsusaka, Y.; Shitaya, S.; Nomura, K.; Inagaki, A. Synthesis of Mono-, Di-, and Trinuclear Rhodium Diphosphine Complexes Containing Light-Harvesting Fluorene Backbones. Inorg. Chem. 2017, 56, 1027–1030. [Google Scholar] [CrossRef]
- Xie, L.; Zhao, S.; Zhang, M.; Wu, C.; Li, T.; Gao, M. Compound of Fluorene Class Containing Dual Hetero Atoms as well as Its Synthetic Method and Application. CN Patent 1480453A, 10 March 2004. [Google Scholar]
- Liu, H.; Xie, L.; Jing, J.M. Solid Catalyst Composition for Olefinic Polymerization and Catalyst Thereof. CN Patent 1508159A, 30 June 2004. [Google Scholar]
- Eger, T.R.; Munstein, I.; Steiner, A.; Sun, Y.; Niedner-Schatteburg, G.; Thiel, W.R. New cationic organometallic phosphane ligands and their coordination to gold(I). J. Organomet. Chem. 2016, 810, 51–56. [Google Scholar] [CrossRef]
- Batchelor, L.K.; Păunescu, E.; Soudani, M.; Scopelliti, R.; Dyson, P.J. Influence of the Linker Length on the Cytotoxicity of Homobinuclear Ruthenium(II) and Gold(I) Complexes. Inorg. Chem. 2017, 56, 9617–9633. [Google Scholar] [CrossRef] [PubMed]
- Borissova, A.O.; Korlyukov, A.A.; Antipin, M.Y.; Lyssenko, K.A. Estimation of Dissociation Energy in Donor−Acceptor Complex AuCl·PPh3 via Topological Analysis of the Experimental Electron Density Distribution Function. J. Phys. Chem. A 2008, 112, 11519–11522. [Google Scholar] [CrossRef] [PubMed]
- Zi, W.; Dean Toste, F. Recent advances in enantioselective gold catalysis. Chem. Soc. Rev. 2016, 45, 4567–4589. [Google Scholar] [CrossRef]
- Johnson, B.F.G. The chemistry of gold. Gold Bull. 1971, 4, 9–11. [Google Scholar] [CrossRef]
- Scherbaum, F.; Grohmann, A.; Huber, B.; Krüger, C.; Schmidbaur, H. Use of the CH Acidity of 2,4,4-Trimethyl-4,5-dihydrooxazole to Synthesize Triauriomethanes and Novel Gold Clusters. Angew. Chem. Int. Ed. Engl. 1988, 27, 1544–1546. [Google Scholar] [CrossRef]
- Janiak, C. (Organo)thallium (I) and (II) chemistry: Syntheses, structures, properties and applications of subvalent thallium complexes with alkyl, cyclopentadienyl, arene or hydrotris(pyrazolyl)borate ligands. Coord. Chem. Rev. 1997, 163, 107–216. [Google Scholar] [CrossRef]
- Schumann, H.; Janiak, C.; Khani, H. Cyclopentadienylthallium(I) compounds with bulky cyclopentadienyl ligands. J. Organomet. Chem. 1987, 330, 347–355. [Google Scholar] [CrossRef]
- Schumann, H.; Janiak, C.; Khan, M.A.; Zuckerman, J.J. Eine zweite ungewöhnliche Kristallmodifikation von pentabezylcyclopentadienylthallium(I), (PhCH2)5C5Tl. J. Organomet. Chem. 1988, 354, 7–13. [Google Scholar] [CrossRef]
- Frasson, E.; Menegus, F.; Panattoni, C. Chain Structure of the Cyclopentadienily of Monovalent Indium and Thallium. Nature 1963, 199, 1087–1089. [Google Scholar] [CrossRef]
- Werner, H.; Otto, H.; Kraus, H.J. Die Kristallstruktur von TlC5Me5. J. Organomet. Chem. 1986, 315, C57–C60. [Google Scholar] [CrossRef]
- Schmidbaur, H.; Bublak, W.; Riede, J.; Müller, G. [{1,3,5-(CH3)3H3C6}6TI4]·[GaBr4]4—Synthese und Struktur eines gemischten Mono- und Bis(aren)thallium-Komplexes. Angew. Chem. 1985, 97, 402–403. [Google Scholar] [CrossRef]
- Schmidbaur, H. Arenkomplexe von einwertigem Gallium, Indium und Thallium. Angew. Chem. 1985, 97, 893–904. [Google Scholar] [CrossRef]
- Nakayama, H.; Nishijima, C.; Tachiyashiki, S. Ion-Molecule Reactions Betwee Thallium(I) and Various Ligands: Formation of 1:1 Complexes in Gas Phase. Chem. Lett. 1974, 3, 733–736. [Google Scholar] [CrossRef]
- Betley, T.A.; Peters, J.C. The Strong-Field Tripodal Phosphine Donor, [PhB(CH2PiPr2)3]−, Provides Access to Electronically and Coordinatively Unsaturated Transition Metal Complexes. Inorg. Chem. 2003, 42, 5074–5084. [Google Scholar] [CrossRef]
- Szlosek, R.; Ackermann, M.T.; Marquardt, C.; Seidl, M.; Timoshkin, A.Y.; Scheer, M. Coordination of Pnictogenylboranes Towards Tl(I) Salts and a Tl- Mediated P-P Coupling. Chem. Eur. J. 2023, 29, e202202911. [Google Scholar] [CrossRef]
- Doppiu, A.; Englert, U.; Salzer, A. Cationic half-sandwich ruthenium(II) complexes with cyclopentadienyl–phosphine ligands. Inorg. Chim. Acta 2003, 350, 435–441. [Google Scholar] [CrossRef]
- Doppiu, A.; Salzer, A. A New Route to Cationic Half-Sandwich Ruthenium(II) Complexes with Chiral Cyclopentadienylphos phane Ligands. Eur. J. Inorg. Chem. 2004, 2244–2252. [Google Scholar] [CrossRef]
- Azerraf, C.; Cohen, S.; Gelman, D. Roof-Shaped Halide-Bridged Bimetallic Complexes via Ring Expansion Reaction. Inorg. Chem. 2006, 45, 7010–7017. [Google Scholar] [CrossRef] [PubMed]
- Azerraf, C.; Grossman, O.; Gelman, D. Rigid trans-spanning triptycene-based ligands: How flexible they can be? J. Organomet. Chem. 2007, 692, 761–767. [Google Scholar] [CrossRef]
- Noskowska, M.; Śliwińska, E.; Duczmal, W. Simple fast preparation of neutral palladium (II) complexes with SnCl−3 and Cl− ligands. Trans. Met. Chem. 2003, 28, 756–759. [Google Scholar] [CrossRef]
- Aullón, G.; Ujaque, G.; Lledós, A.; Alvarez, S.; Alemany, P. To Bend or Not To Bend: Dilemma of the Edge-Sharing Binuclear Square Planar Complexes of d8 Transition Metal Ions. Inorg. Chem. 1998, 37, 804–813. [Google Scholar] [CrossRef]
- Bunten, K.A.; Farrar, D.H.; Poë, A.J.; Lough, A. Stoichiometric and Catalytic Oxidation of BINAP by Dioxygen in a Rhodium(I) Complex. Organometallics 2002, 21, 3344–3350. [Google Scholar] [CrossRef]
- Mondal, J.U.; Young, K.G.; Blake, D.M. Enthalpy changes in oxidative addition reactions of iodine with monomeric and dimeric rhodium(I) complexes. J. Organomet. Chem. 1982, 240, 447–451. [Google Scholar] [CrossRef]
- Dyer, G.; Wharf, R.M.; Hill, W.E. 31P NMR studies of cis-[RhCl(CO)(bis-phosphine)] complexes. Inorg. Chim. Acta 1987, 133, 137–140. [Google Scholar] [CrossRef]
- Mann, B.E.; Masters, C.; Shaw, B.L. Nuclear magnetic resonance studies on metal complexes. Part VII. The 31P n.m.r. spectra of some complexes of the type trans-RhCl-(CO)L2 [L = tertiary phosphine or P(OMe3)]. J. Chem. Soc. A 1971, 1104–1106. [Google Scholar] [CrossRef]
- Vallarino, L. Carbonyl complexes of rhodium. Part I. Complexes with triarylphosphines, triarylarsines, and triarylstibines. J. Chem. Soc. 1957, 2287–2292. [Google Scholar] [CrossRef]
- Marty, W.; Kapoor, P.N.; Bürgi, H.-B.; Fischer, E. Complexes of 3,3′-Oxybis[(diphenylphosphino)methylbenzene] with Ni(II), Pd(II), Pt(II), Rh(I), and Ag(I). How Important is Backbone Rigidity in the Formation of trans-Spanning Bidenatate Chelates? Helv. Chim. Acta 1987, 70, 158–170. [Google Scholar] [CrossRef]
- Eberhard, M.R.; Heslop, K.M.; Orpen, A.G.; Pringle, P.G. Nine-Membered Trans Square-Planar Chelates Formed by a bisbi Analogue. Organometallics 2005, 24, 335–337. [Google Scholar] [CrossRef]
- Reed, F.J.S.; Venanzi, L.M. Transition metal complexes with bidentate ligands spanning trans-positions. IV. Preparation and properties of some rhodium and iridium complexes of 2,11-bis(diphenylphosphinomethyl)benzo[c]phenanthrene. Helv. Chim. Acta 1977, 60, 2804–2814. [Google Scholar] [CrossRef]
- López-Valbuena, J.M.; Escudero-Adan, E.C.; Benet-Buchholz, J.; Freixa, Z.; van Leeuwen, P.W.N.M. An approach to bimetallic catalysts by ligand design. Dalton Trans. 2010, 39, 8560–8574. [Google Scholar] [CrossRef]
- Hierso, J.-C.; Lacassin, F.; Broussier, R.; Amardeil, R.; Meunier, P. Synthesis and characterisation of a new class of phosphine-phosphonite ferrocenediyl dinuclear rhodium complexes. J. Organomet. Chem. 2004, 689, 766–769. [Google Scholar] [CrossRef]
- Meeuwissen, J.; Sandee, A.J.; de Bruin, B.; Siegler, M.A.; Spek, A.L.; Reek, J.N.H. Phosphinoureas: Cooperative Ligands in Rhodium-Catalyzed Hydroformylation? On the Possibility of a Ligand-Assisted Reductive Elimination of the Aldehyde. Organometallics 2010, 29, 2413–2421. [Google Scholar] [CrossRef]
- Sanger, A.R.J. Reactions of di-µ-chloro-bis[cyclo-octa-1,5-dienerhodium(I)] with carbon mono-oxide and mono-, di-, or tri-tertiary phosphines, or 1,2-bis(diphenylarsino)ethane. Chem. Soc. Dalton Trans. 1977, 120–129. [Google Scholar] [CrossRef]
- Thurner, C.L.; Barz, M.; Spiegler, M.; Thiel, W.R. Ligands with cycloalkane backbones II. Chelate ligands from 2-(diphenylphosphinyl)cyclohexanol: Syntheses and transition metal complexes. J. Organomet. Chem. 1997, 541, 39–49. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3 | 4 | 5 | |
|---|---|---|---|
| empirical formula | C83H68P | C57H45AuClP | C50H36PTl |
| formula weight | 1127.31 | 993.31 | 872.13 |
| crystal size [mm] | 0.408 × 0.307 × 0.239 | 0.244 × 0.106 × 0.069 | 0.313 × 0.093 × 0.063 |
| T [K] | 293(2) | 150(2) | 150(2) |
| λ [Å] | 1.54184 | 1.54184 | 1.54184 |
| crystal system | triclinic | triclinic | monoclinic |
| space group | P21/n | ||
| a [Å] | 14.1471(5) | 10.3433(3) | 18.7154(2) |
| b [Å] | 14.5119(4) | 15.2162(6) | 12.2606(1) |
| c [Å] | 17.7514(6) | 15.5558(7) | 20.4489(2) |
| α [°] | 107.920(3) | 106.743(4) | 90 |
| β [°] | 109.661(3) | 107.515(3) | 97.058(1) |
| γ [°] | 93.028(3) | 96.040(3) | 90 |
| V [Å3] | 3215.3(2) | 2185.84(16) | 4656.69(8) |
| Z | 2 | 2 | 4 |
| ρ calcd. [g·cm−3] | 1.164 | 1.509 | 1.244 |
| μ [mm−1] | 0.950 | 7.519 | 7.198 |
| Θ-range [°] | 3.251–62.736 | 3.608–62.729 | 3.417–62.775 |
| refl. coll. | 24,391 | 15,048 | 35,775 |
| indep. refl. | 10,246 [Rint = 0.0229] | 6947 [Rint = 0.0481] | 7447 [Rint = 0.0257] |
| data/restr./param. | 10246/0/768 | 6947/0/542 | 7447/0/469 |
| final R indices [I > 2σ(I)] a | 0.0522, 0.1455 | 0.0367, 0.0946 | 0.0171, 0.0402 |
| R indices (all data) | 0.0573, 0.1524 | 0.0381, 0.0961 | 0.0179, 0.0407 |
| GooF b | 1.036 | 1.082 | 1.029 |
| Δρmax/min (e·Å−3) | 1.687/−0.283 | 1.444/−1.808 | 0.391/−0.266 |
| 7 | 8 | 9 | 10 | |
|---|---|---|---|---|
| empirical formula | C69H52Au2Cl2P2 | C69H52Cl4P2Pd2 | C71H52Cl2O2P2Rh2 | C70H52ClOP2Rh |
| formula weight | 1407.88 | 1297.64 | 1275.78 | 1109.41 |
| crystal size [mm] | 0.216 × 0.095 × 0.079 | 0.199 × 0.101 × 0.044 | 0.272 × 0.151 × 0.137 | 0.233 × 0.121 × 0.059 |
| T [K] | 150(2) | 150(2) | 150(2) | 150(2) |
| λ [Å] | 1.54184 | 1.54184 | 1.54184 | 1.54184 |
| crystal system | triclinic | monoclinic | triclinic | monoclinic |
| space group | P2/n | P21/n | ||
| a [Å] | 13.1508(3) | 21.0446(7) | 13.2483(3) | 13.3016(3) |
| b [Å] | 13.5694(4) | 13.2823(4) | 14.1374(3) | 21.0013(4) |
| c [Å] | 22.2637(5) | 23.4004(9) | 20.4213(4) | 20.0864(4) |
| α [°] | 76.934(2) | 90 | 89.220(2) | 90 |
| β [°] | 87.525(2) | 107.894(4) | 73.556(2) | 109.243(2) |
| γ [°] | 64.608(3) | 90 | 75.287(2) | 90 |
| V [Å3] | 3489.67(17) | 6224.5(4) | 3541.10(14) | 5297.7(2) |
| Z | 2 | 4 | 2 | 4 |
| ρ calcd. [g·cm−3] | 1.340 | 1.385 | 1.197 | 1.391 |
| μ [mm−1] | 9.186 | 7.028 | 5.189 | 3.997 |
| Θ-range [°] | 3.705–62.698 | 3.327–62.748 | 3.238–62.724 | 3.140–62.754 |
| refl. coll. | 27,287 | 24,022 | 49,439 | 21,589 |
| indep. refl. | 11,102 [Rint = 0.0308] | 9919 [Rint = 0.0550] | 11,257 [Rint = 0.0564] | 8461 [Rint = 0.0294] |
| data/restr./param. | 11102/0/676 | 9919/0/695 | 11257/0/713 | 8461/0/676 |
| final R indices [I > 2σ(I)] a | 0.0296, 0.0721 | 0.0510, 0.1388 | 0.0310, 0.0790 | 0.0321, 0.0852 |
| R indices (all data) | 0.0349, 0.0739 | 0.0623, 0.1466 | 0.0335, 0.0807 | 0.0362, 0.0886 |
| GooF b | 1.037 | 1.082 | 1.023 | 1.032 |
| Δρmax/min (e·Å−3) | 1.310/−1.262 | 1.253/−0.718 | 0.734/−0.840 | 0.713/−0.518 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nährig, F.; Sun, Y.; Thiel, W.R. Phosphine Functionalized CpC Ligands and Their Metal Complexes. Chemistry 2023, 5, 912-933. https://doi.org/10.3390/chemistry5020062
Nährig F, Sun Y, Thiel WR. Phosphine Functionalized CpC Ligands and Their Metal Complexes. Chemistry. 2023; 5(2):912-933. https://doi.org/10.3390/chemistry5020062
Chicago/Turabian StyleNährig, Florian, Yu Sun, and Werner R. Thiel. 2023. "Phosphine Functionalized CpC Ligands and Their Metal Complexes" Chemistry 5, no. 2: 912-933. https://doi.org/10.3390/chemistry5020062
APA StyleNährig, F., Sun, Y., & Thiel, W. R. (2023). Phosphine Functionalized CpC Ligands and Their Metal Complexes. Chemistry, 5(2), 912-933. https://doi.org/10.3390/chemistry5020062