


Stabilization of 2-Pyridyltellurium(II) Derivatives by Oxidorhenium(V) Complexes

 , and
, and
Abstract
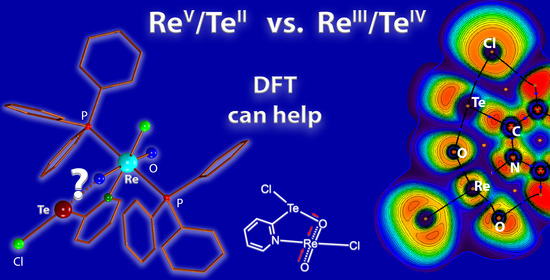
1. Introduction
2. Materials and Methods
2.1. Syntheses
2.2. X-ray Crystallography
2.3. Computational Details
3. Results and Discussions
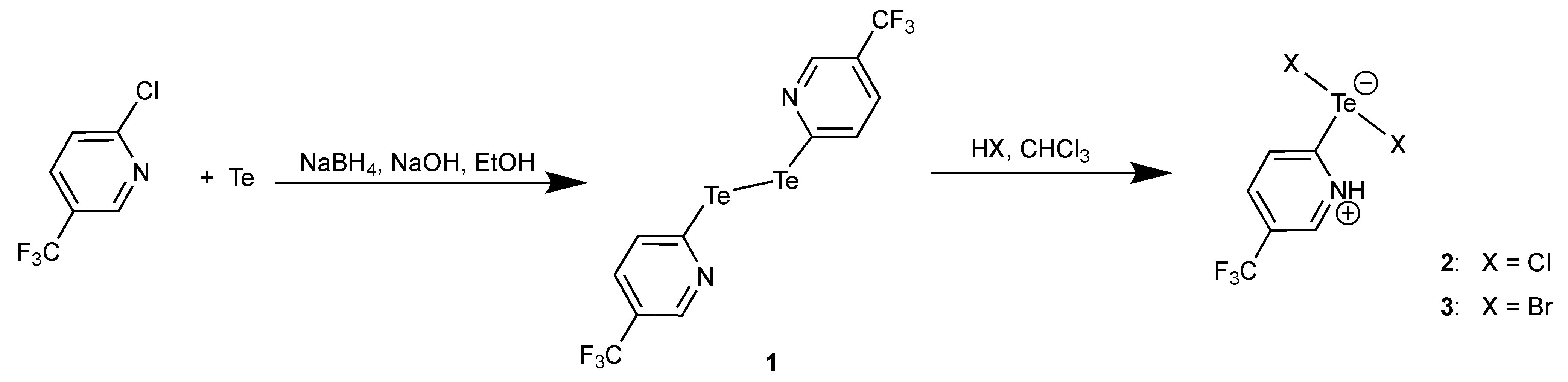
3.1. Bis(5-Trifluoromethyl-2-pyridyl)ditellane and the Zwitterions [HCF3pyTeX2] (X = Cl, Br)
3.2. [ReO2Cl(CF3pyTeCl)(PPh3)2] (4) and [ReO2Cl(pyTeCl)(PPh3)2] (5)
4. Computational Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gysling, H.J. The ligand chemistry of tellurium. Coord. Chem. Rev. 1982, 42, 133–244. [Google Scholar] [CrossRef]
- Sudha, N.; Singh, H.B. Intramolecular coordination in tellurium chemistry. Coord. Chem. Rev. 1994, 135–136, 469–515. [Google Scholar] [CrossRef]
- Chivers, T. Tellurium compounds of the main-group elements: Progress and prospects. J. Chem. Soc. Dalton Trans. 1996, 1185–1194. [Google Scholar] [CrossRef]
- Singh, A.K.; Sharma, S. Recent developments in the ligand chemistry of tellurium. Coord. Chem. 2000, 209, 49–98. [Google Scholar] [CrossRef]
- Chivers, T.; Laitinen, R.S. Tellurium: A maverick among the chalcogens. Chem. Soc. Rev. 2015, 44, 1725–1739. [Google Scholar] [CrossRef]
- Jones, J.S.; Gabbaï, F.P. Coordination and redox non-innocent behavior of hybrid ligands containing tellurium. Chem. Lett. 2016, 45, 376–384. [Google Scholar] [CrossRef]
- Jain, V.K.; Chauhan, R.S. New vistas in the chemistry of platinum group metals with tellurium ligands. Coord. Chem. Rev. 2016, 306, 270–301. [Google Scholar] [CrossRef]
- Arora, A.; Oswal, P.; Datta, A.; Kumar, A. Complexes of metals with organotellurium compounds and nanosized metal tellurides for catalysis, electrocatalysis and photocatalysis. Coord. Chem. Rev. 2022, 459, 214406. [Google Scholar] [CrossRef]
- Gysling, H.J.; Luss, H.R. Synthesis and properties of the hybrid tellurium-phosphorus ligand phenyl o-(diphenylphosphino)phenyl telluride. X-ray structure of [Pt[PhTe(o-(PPh2C6H4)]2][Pt(SCN)4].2DMF. Organometallics 1984, 3, 596–598. [Google Scholar] [CrossRef]
- Do, T.G.; Hupf, E.; Lork, E.; Mebs, S.; Beckmann, J. Bis(6-diphenylphosphinoacenaphth-5-yl)telluride as a ligand toward coinage metal chlorides. Dalton Trans. 2019, 48, 2635–2645. [Google Scholar] [CrossRef]
- Nordheider, A.; Hupf, E.; Chalmers, B.A.; Knight, F.R.; Buhl, M.; Mebs, S.; Checinska, L.; Lork, E.; Camacho, P.S.; Ashbrook, E.S.; et al. Peri-substituted phosphorus–tellurium systems–An experimental and theoretical investigation of the P···Te through-space interaction. Inorg. Chem. 2015, 54, 2435–2446. [Google Scholar] [CrossRef] [PubMed]
- Do, T.G.; Hupf, E.; Lork, E.; Beckmann, J. Bis(6-diphenylphosphinoacenaphth-5-yl)telluride as a ligand toward manganese and rhenium carbonyls. Molecules 2018, 23, 2805. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lin, T.-P.; Gabbaï, F.P. Telluroether to telluroxide conversion in the coordination sphere of a metal: Oxidation-induced umpolung of a Te–Au bond. Organometallics 2014, 33, 4368–4373. [Google Scholar] [CrossRef]
- Lin, T.-P.; Gabbaï, F.P. Two-electron redox chemistry at the dinuclear core of a TePt platform: Chlorine photoreductive elimination and isolation of a TeVPtI complex. J. Am. Chem. Soc. 2012, 134, 12230–12238. [Google Scholar] [CrossRef]
- Gupta, A.; Deka, R.; Srivastava, K.; Singh, H.B.; Butcher, R.J. Synthesis of Pd(II) complexes of unsymmetrical, hybrid selenoether and telluroether ligands: Isolation of tellura-palladacycles by fine tuning of intramolecular chalcogen bonding in hybrid telluroether ligands. Polyhedron 2019, 172, 95–103. [Google Scholar] [CrossRef]
- Gupta, A.K.; Deka, R.; Singh, H.B.; Butcher, R.J. Reactivity of bis[{2,6-(dimethylamino)methyl}phenyl]telluride with Pd(II) and Hg(II): Isolation of the first Pd(II) complex of an organotellurenium cation as a ligand. New J. Chem. 2019, 43, 13225–13233. [Google Scholar] [CrossRef]
- Ji, B.; Ding, K. Synthesis and crystallographic characterization of a palladium(II) complex with the ligand (4-ethoxyphenyl)[(2-amino-5-methyl)phenyl] telluride. Inorg. Chem. Commun. 1999, 2, 347–350. [Google Scholar] [CrossRef]
- Panda, S.; Singh, H.B.; Butcher, R.J. Contrasting coordination behaviour of 22-membered chalcogenaaza (Se, Te) macrocylces towards Pd(II) and Pt( II): Isolation and structural characterization of the first metallamacrocyle with a C–Pt–Se linkage. Chem. Commun. 2004, 322–323. [Google Scholar] [CrossRef]
- Panda, S.; Zade, S.S.; Singh, H.B.; Butcher, R.J. The ligation properties of some reduced Schiff base selena/telluraaza macrocycles: Versatile structural trends. Eur. J. Inorg. Chem. 2006, 172–184. [Google Scholar] [CrossRef]
- Menon, S.C.; Panda, A.; Singh, H.B.; Patel, R.P.; Kulshreshtha, S.K.; Darby, W.L.; Butcher, R.J. Tellurium azamacrocycles: Synthesis, characterization and coordination studies. J. Organomet. Chem. 2004, 689, 1452–1463. [Google Scholar] [CrossRef]
- Menon, S.C.; Panda, A.; Singh, H.B.; Butcher, R.J. Synthesis and single crystal X-ray structure of the first cationic Pd(II) complex of a tellurium-containing polyaza macrocycle: Contrasting reactions of Pd(II) and Pt(II) with a 22-membered macrocyclic Schiff base. Chem. Commun. 2000, 143–144. [Google Scholar] [CrossRef]
- Nakayama, Y.; Watanabe, K.; Ueyama, N.; Nakamura, A.; Harada, A.; Okuda, J. Titanium complexes having chelating diaryloxo ligands bridged by tellurium and their catalytic behavior in the polymerization of ethylene. Organometallics 2000, 19, 2498–2503. [Google Scholar] [CrossRef]
- Takashima, Y.; Nakayama, Y.; Yasuda, H.; Nakamura, A.; Harada, A. Synthesis of cis-dichloride complexes of Group 6 transition metals bearing alkyne and chalcogen-bridged chelating bis(aryloxo) ligands as catalyst precursors for ring-opening metathesis polymerization. J. Organomet. Chem. 2002, 654, 74–82. [Google Scholar] [CrossRef]
- Baranov, A.V.; Matsulevich, Z.V.; Fukin, G.F.; Baranov, E.V. Synthesis and structure of pyridine-2-tellurenyl chloride. Russ. Chem. Bull. 2010, 59, 581–583. [Google Scholar]
- Chauhan, R.S.; Kedarnath, G.; Wadawale, A.; Slawin, A.M.Z.; Jain, V.K. Reactivity of 2-chalcogenopyridines with palladium–phosphine complexes: Isolation of different complexes. Dalton Trans. 2013, 42, 259–269. [Google Scholar] [CrossRef]
- Cechin, C.N.; Razera, G.F.; Tirloni, B.; Piquini, P.C.; de Carvalho, L.M.; Abram, U.; Lang, E.S. Oxidation of crude palladium powder by a diiodine adduct of (2-PyTe)2 to obtain the novel PdII complex [PdI(TePy-2)(I2TePy-2)2]. Inorg. Chem. Commun. 2020, 118, 107966. [Google Scholar] [CrossRef]
- da Silva, F.D.; Simoes, C.A.D.P.; dos Santos, S.S.; Lang, E.S. Versatility of bis(2-pyridyl)ditellane. ChemistrySelect 2017, 2, 2708–2712. [Google Scholar] [CrossRef]
- Parshall, G.W.; Shive, L.W.; Cotton, F.A. Phosphine complexes of rhenium. Inorg. Synth. 1977, 17, 110–112. [Google Scholar]
- Coppens, P. The Evaluation of Absorption and Extinction in Single-Crystal Structure Analysis; Crystallographic Computing: Copenhagen, Denmark, 1979. [Google Scholar]
- Sheldrick, G.M. SADABS; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, 71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Putz, H.; Brandenburg, K. DIAMOND, Crystal and Molecular Structure Visualization Crystal Impact; Version 4.6.5; Brandenburg GbR: Bonn, Germany, 2021. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView; Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Hay, P.J.; Wadt, W.R.; Willard, R.J. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Check, C.E.; Faust, T.O.; Bailey, J.M.; Wright, B.J.; Gilbert, T.M.; Sunderlin, L.S.J. Addition of Polarization and Diffuse Functions to the LANL2DZ Basis Set for P-Block Elements. Phys. Chem. A 2001, 105, 8111–8116. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Spitznagel, G.W.; Clark, T.; von Rague Schleyer, P.; Hehre, W.J. An evaluation of the performance of diffuse function-augmented basis sets for second row elements, Na-Cl. J. Comput. Chem. 1987, 8, 1109–1116. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L.J. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm. J. Mol. Graph. Model. 2012, 38, 314–323. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics 2012, 4, 1–17. [Google Scholar] [CrossRef]
- Bhasin, K.K.; Arora, V.; Klapötke, T.M.; Crawford, M.-J. One-Pot Synthesis of Pyridyltellurium Derivatives from a Reaction with Isopropylmagnesium Chloride and X-ray Crystal Structures of Various Pyridyl Ditellurides. Eur. J. Inorg. Chem. 2004, 4781–4788. [Google Scholar] [CrossRef]
- Hauge, S.; Vikane, O. Three-coordinated Divalent Tellurium Complexes: The Crystal Structures of Tetraphenylarsonium Diiodophenyltellurate(II) and Tetraphenylarsonium Bromoiodophenyltellurate(II). Acta Chem. Scand. 1983, A37, 723–728. [Google Scholar] [CrossRef]
- Du Mont, W.-W.; Meyer, H.-U.; Kubiniok, S.; Pohl, S.; Saak, W. Spaltung sperriger Diarylditelluride mit Brom und Iod; Strukturbestimmung an Et4N+ 2,4,6-(i-C3H7)3C6H2TeI−2. Chem. Ber. 1992, 125, 761–766. [Google Scholar] [CrossRef]
- Faoro, E.; de Oliveira, G.M.; Schulz Lang, E. Synthesis and Structural Characterization of the novel T-shaped Organotellurium(II) Dihalides (PyH)[mesTeClBr] and (PyH)[mesTeX2] (Py = pyridine; mes = mesityl; X = Cl, Br). Z. Anorg. Allg. Chem. 2006, 632, 2049–2052. [Google Scholar] [CrossRef]
- Schulz Lang, E.; de Oliveira, G.M.; Casagrande, G.A. Synthesis of new T-shaped hypervalent complexes of tellurium showing Te–π-aryl interactions: X-ray characterization of [(mes)XTe(μ-X)Te(mes)(etu)] (X = Br, I) and [Ph(etu)Te(μ-I)Te(etu)Ph][PhTeI4] (mes = mesityl; etu = ethylenethiourea). J. Organomet. Chem. 2006, 691, 59–64. [Google Scholar] [CrossRef]
- Faoro, E.; Oliveira, G.M.; Schulz Lang, E.; Pereira, C.B. Synthesis and structural features of new aryltellurenyl iodides. J. Organomet. Chem. 2010, 695, 1480–1486. [Google Scholar] [CrossRef]
- Dance, H.S.; McWhinnie, W.R. Isotopic studies by vibrational spectroscopy of the tellurium–carbon bond in diaryltellurium dihalides. J. Chem. Soc. Dalton Trans. 1975, 43–45. [Google Scholar] [CrossRef]
- Lee, H.; Kim, I.-Y.; Han, S.-S.; Bae, B.-S.; Choi, M.K.; Yang, I.-S. Spectroscopic ellipsometry and Raman study of fluorinated nanocrystalline carbon thin films. J. Appl. Phys. 2001, 90, 813–818. [Google Scholar] [CrossRef]
- Sandmann, D.J.; Li, L.; Tripathy, S.; Stark, J.C.; Acampora, L.A.; Foxman, B.M. Conformational polymorphism of di-2-naphthyl ditelluride. Organometallics 1994, 13, 348–353. [Google Scholar] [CrossRef]
- Khrustalev, V.N.; Matsulevich, Z.V.; Lukiyanova, J.M.; Aysin, R.R.; Peregudov, A.S.; Leites, L.A.; Borisov, A.V. A Facile Route for Stabilizing Highly Reactive ArTeCl Species Through the Formation of T-Shaped Tellurenyl Chloride Adducts: Quasi-Planar Zwitterionic [HPy*]TeCl2 and [HPm*]TeCl2; Py* = 2-pyridyl, Pm* = 2-(4,6-dimethyl)pyrimidyl. Eur. J. Inorg. Chem. 2014, 3582–3586. [Google Scholar] [CrossRef]
- Beckmann, J.; Hesse, M.; Poleschner, H.; Seppelt, K. Formation of mixed-valent aryltellurenyl halides RX2TeTeR. Angew. Chem. Int. Ed. 2007, 46, 8277–8280. [Google Scholar] [CrossRef] [PubMed]
- Benz, S.; Poblador-Bahamonde, A.I.; Low-Ders, N.; Matile, S. Catalysis with pnictogen, chalcogen, and halogen bonds. Angew. Chem. Int. Ed. 2018, 57, 5408–5412. [Google Scholar] [CrossRef]
- Vogel, L.; Wonner, P.; Huber, S.M. Chalcogen bonding: An overview. Angew. Chem. Int Ed. 2019, 58, 1880–1891. [Google Scholar] [CrossRef]
- Bamberger, J.; Ostler, F.; Mancheno, O.G. Frontiers in Halogen and chalcoge-bond donor organocatalysis. ChemCatChem 2019, 11, 5198–5211. [Google Scholar] [CrossRef]
- Ho, P.C.; Wang, J.Z.; Meloni, F.; Vargas-Baca, I. Chalcogen bonding in materials chemistry. Coord. Chem. Rev. 2020, 422, 213464. [Google Scholar] [CrossRef]
- Da Silva, F.D.; Bortolotto, T.; Tirloni, B.; de Freitas Daudt, N.; Schulz Lang, E.; Cargnelutti, R. Bis(2-pyridyl)ditellane as a precursor to CoII, CuI and CuII complex formation: Structural characterization and photocatalytic studies. New J. Chem. 2022, 46, 18165–18172. [Google Scholar] [CrossRef]
- Noschang Cabral, B.; Fonseca, J.R.; Roca Jungfer, M.; Krebs, A.; Hagenbach, A.; Schulz Lang, E.; Abram, U. Oxidorhenium(V) and Rhenium(III) Complexes with Arylselenolato and -tellurolato Ligands. Eur. J. Inorg. Chem. 2022, e202300023. [Google Scholar] [CrossRef]
- Abram, U. Rhenium. In Comprehensive Coordination Chemistry II; McCleverty, J.A., Meyer, T.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2003; Volume 5, pp. 271–403. [Google Scholar]
- Smeltz, J.L.; Lilly, C.P.; Boyle, P.D.; Ison, E.A. The electronic nature of terminal oxo ligands in transition-metal complexes: Ambiphilic reactivity of oxorhenium species. J. Am. Chem. Soc. 2013, 135, 9433–9441. [Google Scholar] [CrossRef] [PubMed]
- Lambic, N.S.; Sommer, R.D.; Ison, E.A. Transition-metal oxos as the Lewis basic component of frustrated Lewis pairs. J. Am. Chem. Soc. 2016, 138, 4832–4842. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.J.M.; Nabinger, J.A.; Bercaw, J.E. Homogeneous CO hydrogenation: Ligand effects on the Lewis acid-assisted reductive coupling of carbon monoxide. Organometallics 2010, 29, 4499–4516. [Google Scholar] [CrossRef]
- Lambic, N.S.; Brown, C.A.; Sommer, R.D.; Ison, E.A. Dramatic increase in the rate of olefin insertion by coordination of Lewis acids to the oxo ligand in oxorhenium(V) hydrides. Organometallics 2017, 36, 2042–2051. [Google Scholar] [CrossRef]
- Belanger, S.; Beauchamp, A.L. Oxo ligand reactivity in the [ReO2L4]+ complex of 1-methylimidazole. Preparation and crystal structures of salts containing the ReOL43+ core and apical CH3O-, BF3O2-, and (CH3O)2PO2- groups. Inorg. Chem. 1997, 36, 3640–3647. [Google Scholar] [CrossRef]
- Massaaki, A.; Tsuyoshi, M.; Hideki, S.; Akira, N.; Yoichi, S. Lewis acid trifluoroboron coordination to trans-dioxorhenium(V) moiety: Structural and spectroscopic characterization of trans-[ReV(O)(OBF3)(1-MeIm)4](BF4)(1-MeIm=1-methylimidazole). Chem. Lett. 1997, 26, 1073–1074. [Google Scholar]
- Hupf, E.; Do, T.G.; Nordheider, A.; Wehrhahn, M.; Sanz Camacho, P.; Ashbrook, S.E.; Lork, E.; Slawin, A.M.Z.; Mebs, S.; Woollins, J.D.; et al. Selective oxidation and functionalization of 6-diphenylphosphinoacenaphthyl-5-tellurenyl species 6-Ph2P-Ace-5-TeX (X = Mes, Cl, O3SCF3). Various types of P−E···Te(II,IV) bonding situations (E = O, S, Se). Organometallics 2017, 36, 1566–1579. [Google Scholar] [CrossRef]
- Deka, R.; Sarkar, A.; Butcher, R.J.; Junk, P.C.; Turner, D.R.; Deacon, G.B.; Singh, H.B. Isolation of the novel example of a monomeric organotelllurinic acid. Dalton Trans. 2020, 49, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Pietrasiak, E.; Gordon, C.P.; Coperet, C.; Togni, A. Understanding 125Te NMR chemical shifts in dissymmetric organo-telluride compounds from natural chemical shift analysis. Phys. Chem. Chem. Phys. 2020, 22, 2319–2326. [Google Scholar] [CrossRef] [PubMed]
- Cabeza, J.A.; van der Maelen, J.F.; García-Granda, S. Topological Analysis of the Electron Density in the N-Heterocyclic Carbene Triruthenium Cluster [Ru3(μ-H)2(μ3-MeImCH)(CO)9] (Me2Im = 1,3-dimethylimidazol-2-ylidene). Organometallics 2009, 28, 3666–3672. [Google Scholar] [CrossRef]
- Matito, E.; Solà, M. The role of electronic delocalization in transition metal complexes from the electron localization function and the quantum theory of atoms in molecules viewpoints. Coord. Chem. Rev. 2009, 253, 647–665. [Google Scholar] [CrossRef]
- Poater, J.; Duran, M.; Solà, M.; Silvi, B. Theoretical Evaluation of Electron Delocalization in Aromatic Molecules by Means of Atoms in Molecules (AIM) and Electron Localization Function (ELF) Topological Approaches. Coord. Chem. Rev. 2005, 105, 3911–3947. [Google Scholar] [CrossRef]
- Bruker. APEX2, SAINT and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2009. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Re-O1 | Re-O2 | O2-Te | Te-Cl2 | O1-Re-O2 | Re-O2-Te | O2-Te-Cl2 | O2-Te-C1 | |
|---|---|---|---|---|---|---|---|---|
| 4 | 1.721(4) | 1.822(3) | 2.102(4) | 2.578(2) | 165.9(2) | 135.2(2) | 171.6(1) | 81.0(2) |
| 5 | 1.730(2) | 1.824(2) | 2.102(2) | 2.5736(9) | 168.42(9) | 134.3(1) | 169.99(6) | 81.23(9) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Silva, F.D.; Roca Jungfer, M.; Hagenbach, A.; Lang, E.S.; Abram, U. Stabilization of 2-Pyridyltellurium(II) Derivatives by Oxidorhenium(V) Complexes. Chemistry 2023, 5, 934-947. https://doi.org/10.3390/chemistry5020063
da Silva FD, Roca Jungfer M, Hagenbach A, Lang ES, Abram U. Stabilization of 2-Pyridyltellurium(II) Derivatives by Oxidorhenium(V) Complexes. Chemistry. 2023; 5(2):934-947. https://doi.org/10.3390/chemistry5020063
Chicago/Turabian Styleda Silva, Felipe Dornelles, Maximilian Roca Jungfer, Adelheid Hagenbach, Ernesto Schulz Lang, and Ulrich Abram. 2023. "Stabilization of 2-Pyridyltellurium(II) Derivatives by Oxidorhenium(V) Complexes" Chemistry 5, no. 2: 934-947. https://doi.org/10.3390/chemistry5020063
APA Styleda Silva, F. D., Roca Jungfer, M., Hagenbach, A., Lang, E. S., & Abram, U. (2023). Stabilization of 2-Pyridyltellurium(II) Derivatives by Oxidorhenium(V) Complexes. Chemistry, 5(2), 934-947. https://doi.org/10.3390/chemistry5020063