
Functional Glial Activation Mediates Phenotypic Effects of APOEɛ4 and Sex in Alzheimer’s Disease
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Trial
2.2. Study Eligibility
2.3. Data Collection and Processing
2.4. Statistical Analysis
3. Results
3.1. Patient Cohort
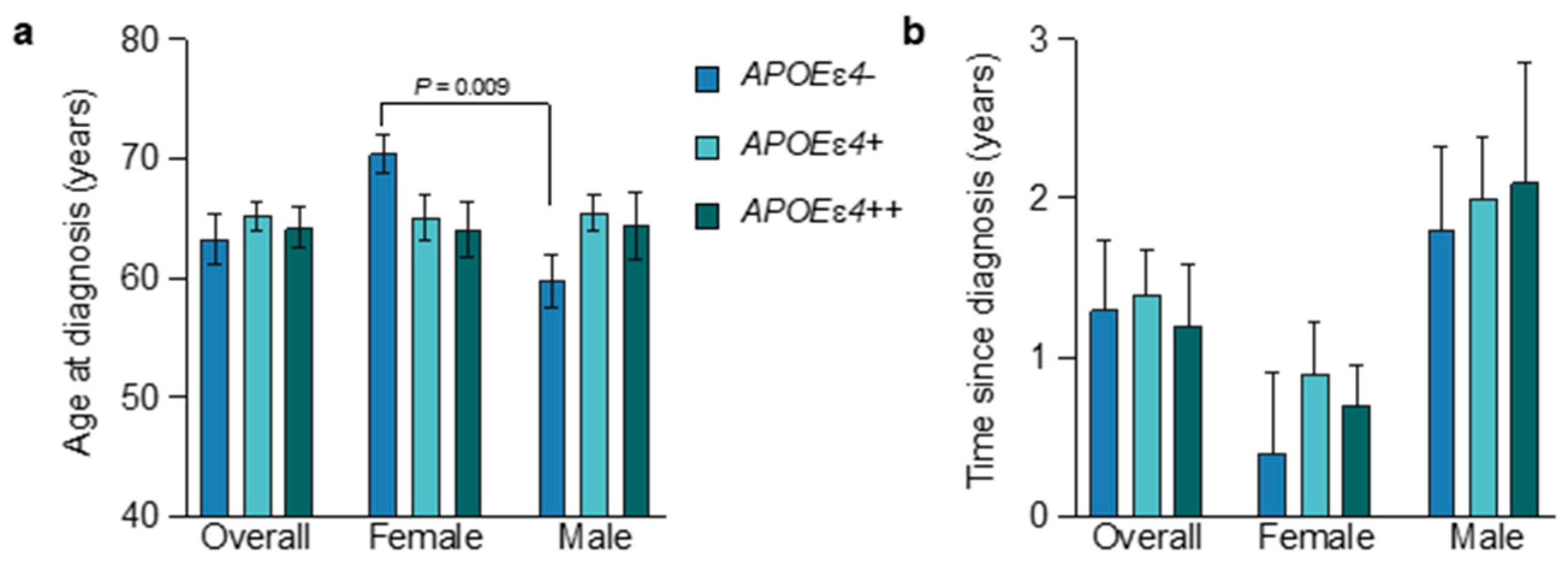
3.2. Age at Diagnosis and APOEɛ4 Allele Status
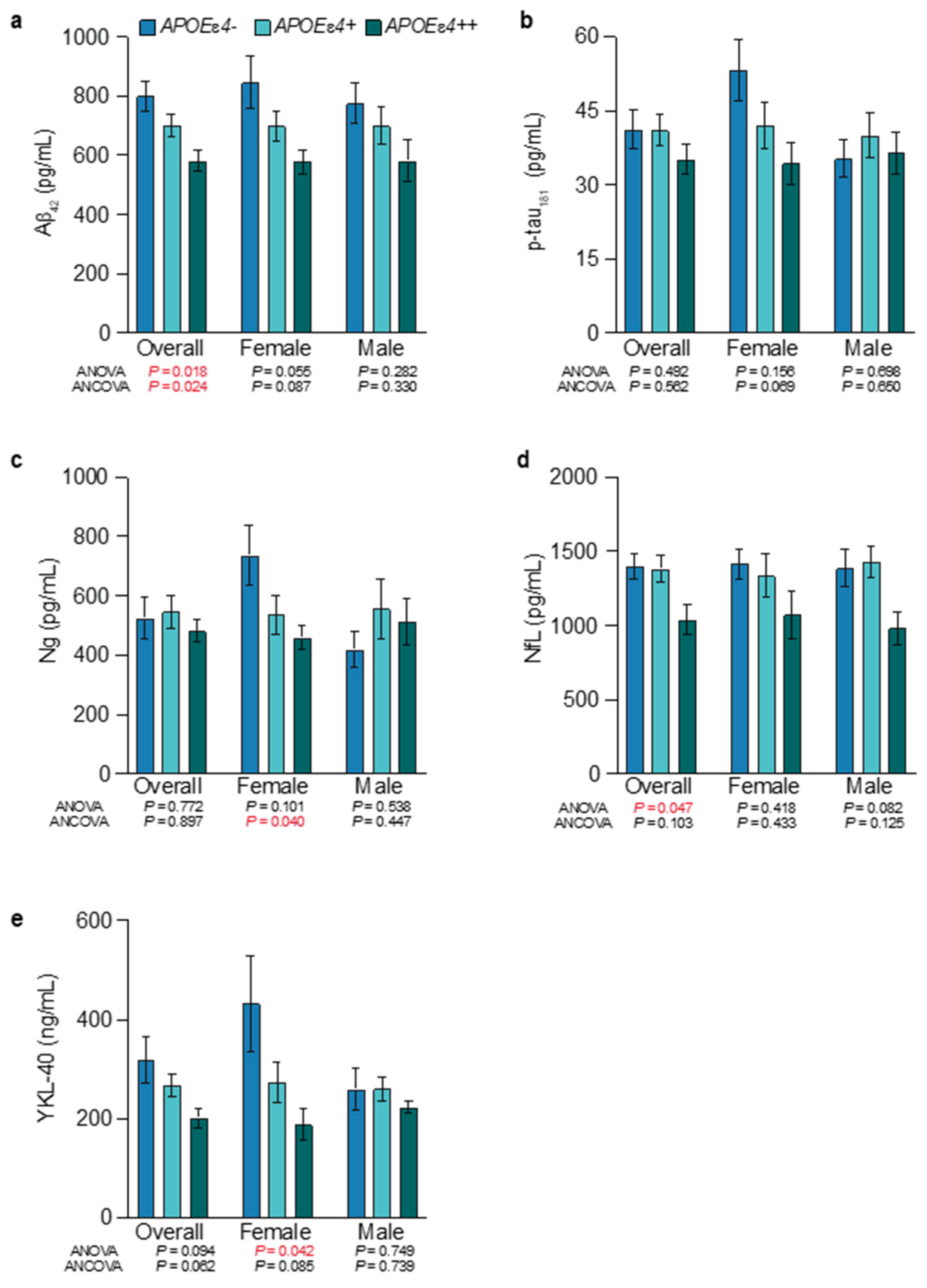
3.3. APOEɛ4-Allele-Frequency-Dependent Changes on CSF Markers
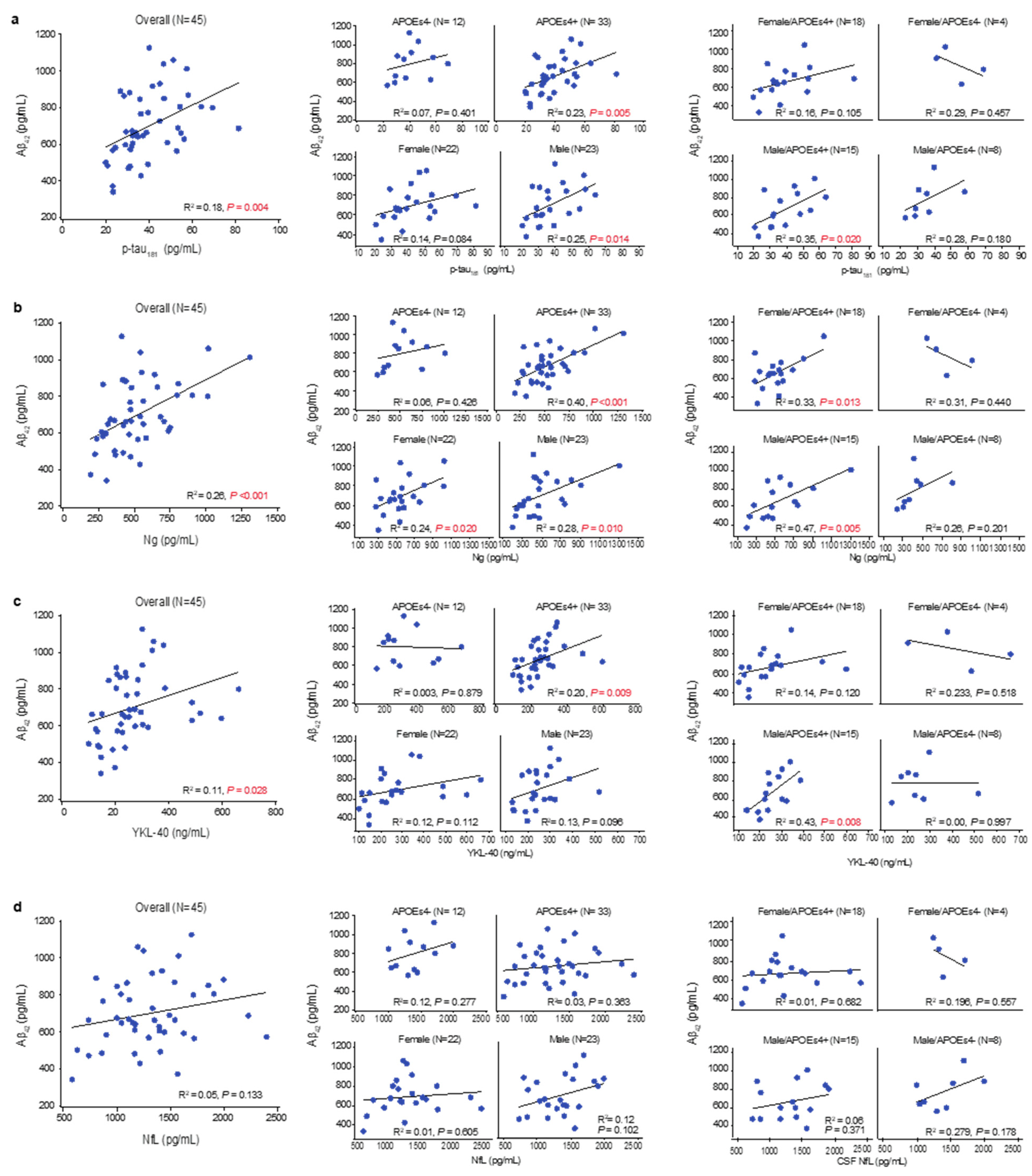
3.4. Amyloid Accumulation, Hypofunctional Glia-Mediated Clearance, and Neurodegenerative Pathology
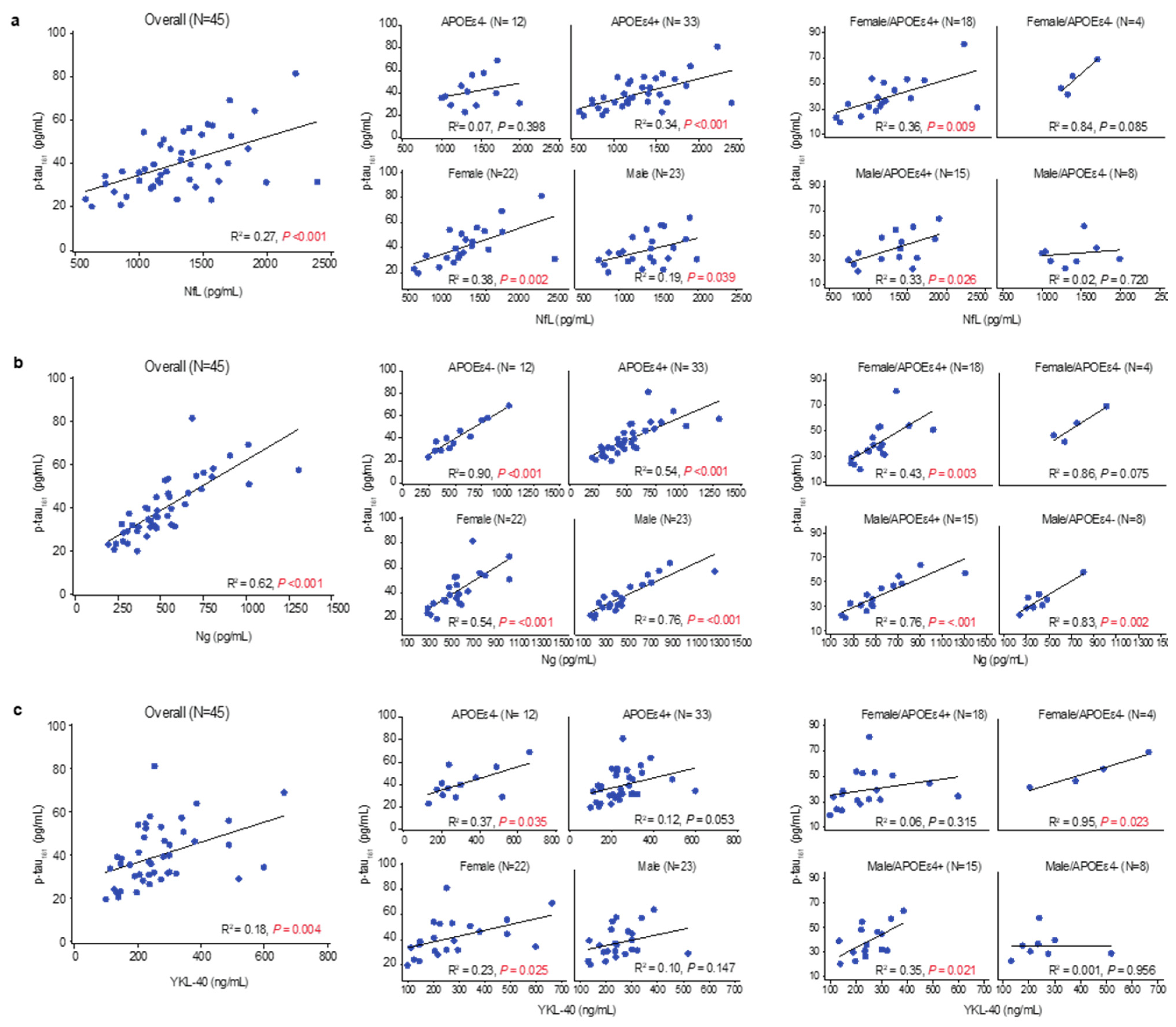
3.5. Tau Pathophysiology, Neurodegenerative Pathology, and Glial Activation
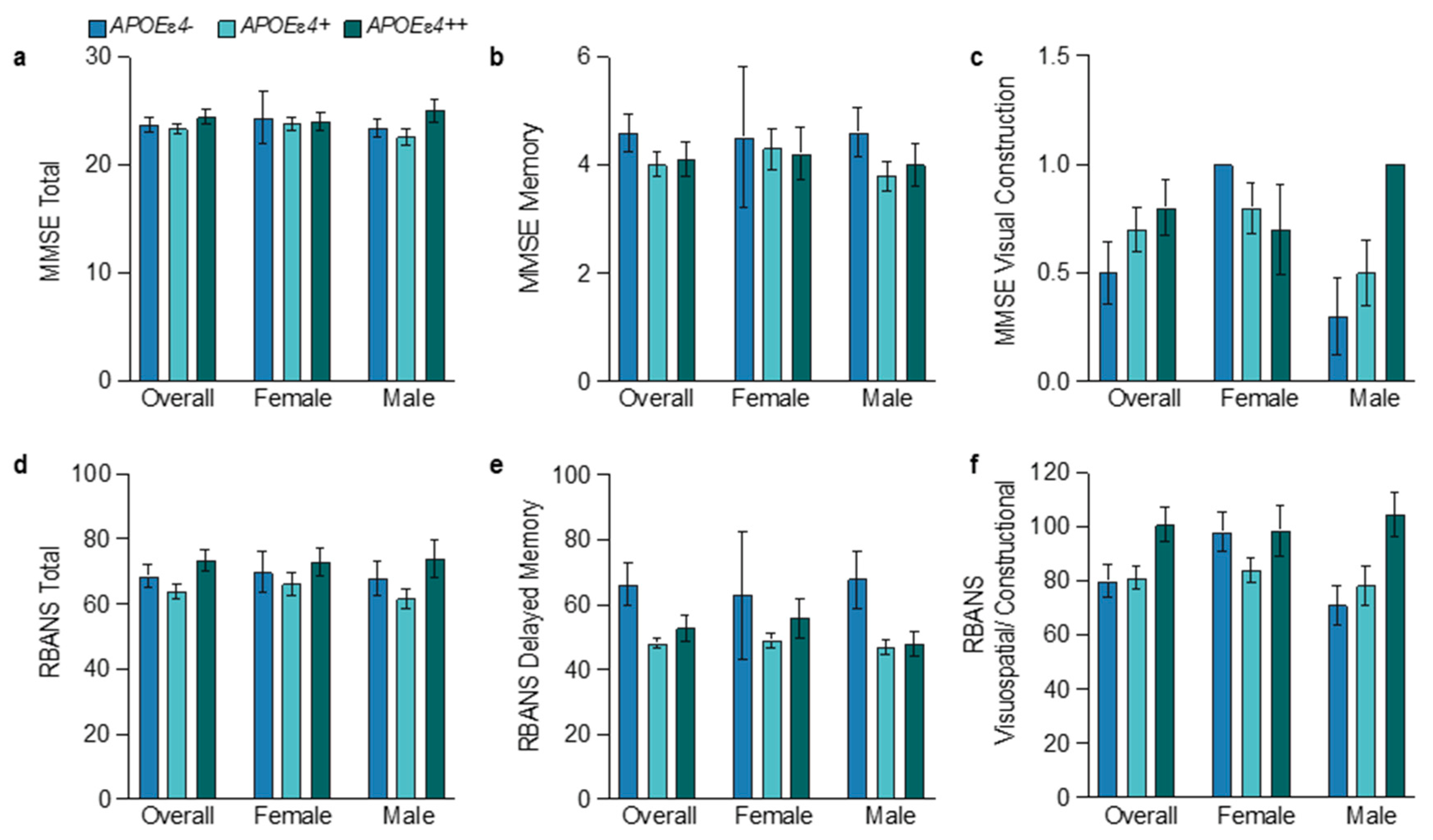
3.6. Sex and APOEɛ4 Allele Influences on Cognition
3.7. Brain Volumes in APOEɛ4 Noncarriers
3.8. APOEɛ4 Heterozygotes Exhibit Influences of Both Alleles
3.9. Hyperfunctional Glia in Female APOEɛ4 Noncarriers
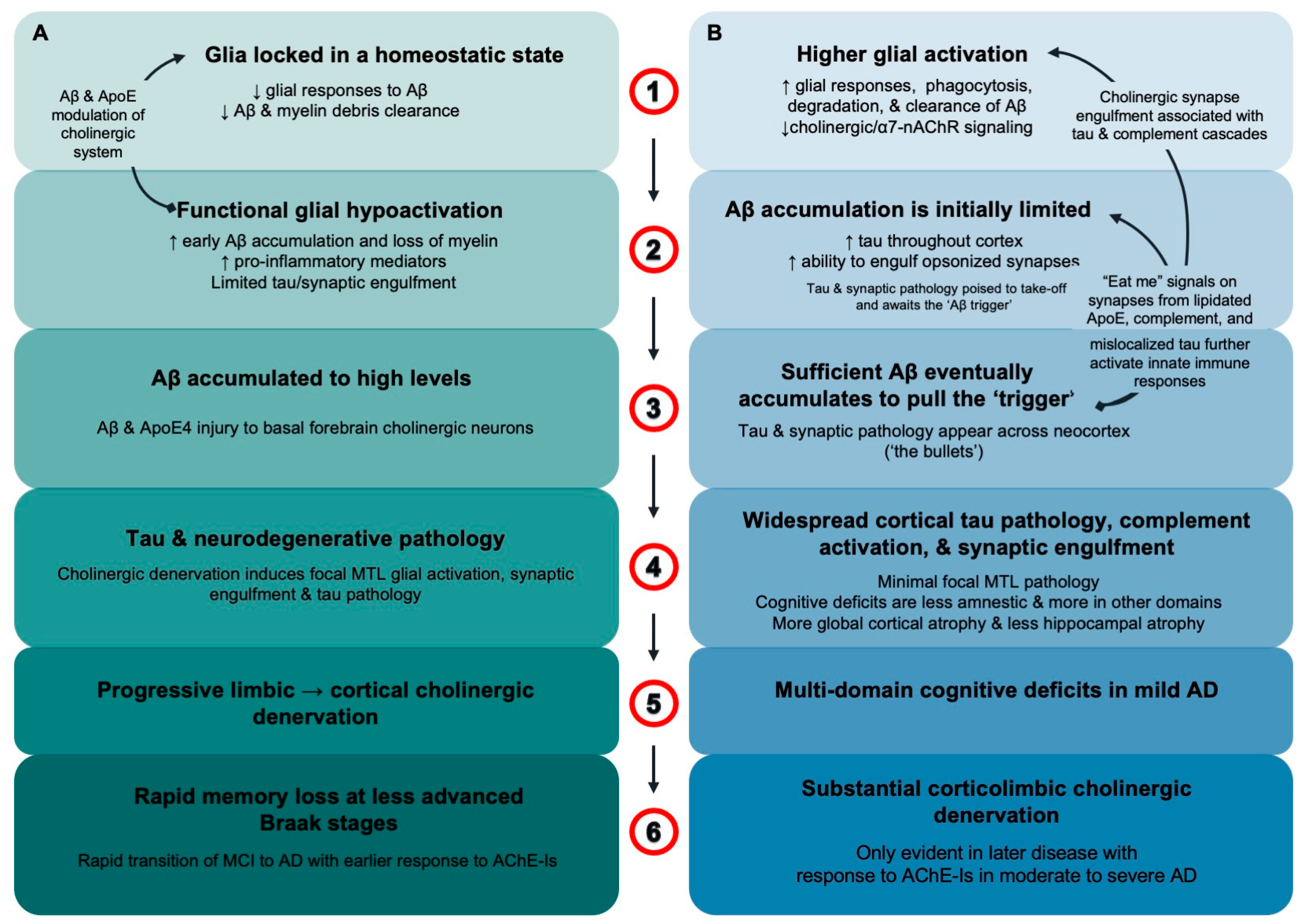
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Koutsodendris, N.; Nelson, M.R.; Rao, A.; Huang, Y. Apolipoprotein E and Alzheimer’s Disease: Findings, Hypotheses, and Potential Mechanisms. Annu. Rev. Pathol. 2022, 17, 73–99. [Google Scholar] [CrossRef] [PubMed]
- Moser, V.A.; Workman, M.J.; Hurwitz, S.J.; Lipman, R.M.; Pike, C.J.; Svendsen, C.N. Microglial transcription profiles in mouse and human are driven by APOE4 and sex. iScience 2021, 24, 103238. [Google Scholar] [CrossRef] [PubMed]
- Stephen, T.L.; Breningstall, B.; Suresh, S.; McGill, C.J.; Pike, C.J. APOE genotype and biological sex regulate astroglial interactions with amyloid plaques in Alzheimer’s disease mice. J. Neuroinflamm. 2022, 19, 286. [Google Scholar] [CrossRef]
- Sala Frigerio, C.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.T.; Woodbury, M.E.; Srivastava, G.; et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Abeta Plaques. Cell Rep. 2019, 27, 1293–1306.e6. [Google Scholar] [CrossRef]
- Mofrad, R.B.; Tijms, B.M.; Scheltens, P.; Barkhof, F.; van der Flier, W.M.; Sikkes, S.A.; Teunissen, C.E. Sex differences in CSF biomarkers vary by Alzheimer disease stage and APOE ε4 genotype. Neurology 2020, 95, e2378–e2388. [Google Scholar]
- Berchtold, N.C.; Cribbs, D.H.; Coleman, P.D.; Rogers, J.; Head, E.; Kim, R.; Beach, T.; Miller, C.; Troncoso, J.; Trojanowski, J.Q.; et al. Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc. Natl. Acad. Sci. USA 2008, 105, 15605–15610. [Google Scholar] [CrossRef]
- Casaletto, K.B.; Nichols, E.; Aslanyan, V.; Simone, S.M.; Rabin, J.S.; La Joie, R.; Brickman, A.M.; Dams-O’Connor, K.; Palta, P.; Kumar, R.G.; et al. Sex-specific effects of microglial activation on Alzheimer’s disease proteinopathy in older adults. Brain 2022, 145, 3536–3545. [Google Scholar] [CrossRef]
- Yin, Z.; Rosenzweig, N.; Kleemann, K.L.; Zhang, X.; Brandao, W.; Margeta, M.A.; Schroeder, C.; Sivanathan, K.N.; Silveira, S.; Gauthier, C.; et al. APOE4 impairs the microglial response in Alzheimer’s disease by inducing TGFbeta-mediated checkpoints. Nat. Immunol. 2023, 24, 1839–1853. [Google Scholar] [CrossRef]
- Machlovi, S.I.; Neuner, S.M.; Hemmer, B.M.; Khan, R.; Liu, Y.; Huang, M.; Zhu, J.D.; Castellano, J.M.; Cai, D.; Marcora, E.; et al. APOE4 confers transcriptomic and functional alterations to primary mouse microglia. Neurobiol. Dis. 2022, 164, 105615. [Google Scholar] [CrossRef]
- Podleśny-Drabiniok, A.; Marcora, E.; Goate, A.M. Microglial Phagocytosis: A Disease-Associated Process Emerging from Alzheimer’s Disease Genetics. Trends Neurosci. 2020, 43, 965–979. [Google Scholar] [CrossRef] [PubMed]
- Millet, A.; Ledo, J.H.; Tavazoie, S.F. An exhausted-like microglial population accumulates in aged and APOE4 genotype Alzheimer’s brains. Immunity 2024, 57, 153–170.e6. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154.e7. [Google Scholar] [CrossRef]
- Guillot-Sestier, M.V.; Doty, K.R.; Gate, D.; Rodriguez, J.; Jr Leung, B.P.; Rezai-Zadeh, K.; Town, T. Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron 2015, 85, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Calvet, M.; Morenas-Rodriguez, E.; Kleinberger, G.; Schlepckow, K.; Araque Caballero, M.A.; Franzmeier, N.; Capell, A.; Fellerer, K.; Nuscher, B.; Eren, E.; et al. Early increase of CSF sTREM2 in Alzheimer’s disease is associated with tau related-neurodegeneration but not with amyloid-beta pathology. Mol. Neurodegener. 2019, 14, 1. [Google Scholar] [CrossRef]
- Jain, N.; Lewis, C.A.; Ulrich, J.D.; Holtzman, D.M. Chronic TREM2 activation exacerbates Abeta-associated tau seeding and spreading. J. Exp. Med. 2023, 220, e20220654. [Google Scholar] [CrossRef]
- Gratuze, M.; Leyns, C.E.; Sauerbeck, A.D.; St-Pierre, M.K.; Xiong, M.; Kim, N.; Serrano, J.R.; Tremblay, M.E.; Kummer, T.T.; Colonna, M.; et al. Impact of TREM2R47H variant on tau pathology-induced gliosis and neurodegeneration. J. Clin. Investig. 2020, 130, 4954–4968. [Google Scholar] [CrossRef]
- Pascoal, T.A.; Benedet, A.L.; Ashton, N.J.; Kang, M.S.; Therriault, J.; Chamoun, M.; Savard, M.; Lussier, F.Z.; Tissot, C.; Karikari, T.K.; et al. Microglial activation and tau propagate jointly across Braak stages. Nat. Med. 2021, 27, 1592–1599. [Google Scholar] [CrossRef]
- Bellaver, B.; Povala, G.; Ferreira, P.C.L.; Ferrari-Souza, J.P.; Leffa, D.T.; Lussier, F.Z.; Benedet, A.L.; Ashton, N.J.; Triana-Baltzer, G.; Kolb, H.C.; et al. Astrocyte reactivity influences amyloid-beta effects on tau pathology in preclinical Alzheimer’s disease. Nat. Med. 2023, 29, 1775–1781. [Google Scholar] [CrossRef]
- Safaiyan, S.; Kannaiyan, N.; Snaidero, N.; Brioschi, S.; Biber, K.; Yona, S.; Edinger, A.L.; Jung, S.; Rossner, M.J.; Simons, M. Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci. 2016, 19, 995–998. [Google Scholar] [CrossRef]
- Hu, Y.; Fryatt, G.L.; Ghorbani, M.; Obst, J.; Menassa, D.A.; Martin-Estebane, M.; Muntslag, T.A.O.; Olmos-Alonso, A.; Guerrero-Carrasco, M.; Thomas, D.; et al. Replicative senescence dictates the emergence of disease-associated microglia and contributes to Abeta pathology. Cell Rep. 2021, 35, 109228. [Google Scholar] [CrossRef] [PubMed]
- Connolly, K.; Lehoux, M.; O’Rourke, R.; Assetta, B.; Erdemir, G.A.; Elias, J.A.; Lee, C.G.; Huang, Y.A. Potential role of chitinase-3-like protein 1 (CHI3L1/YKL-40) in neurodegeneration and Alzheimer’s disease. Alzheimer’s Dement. 2023, 19, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Yin, F. Lipid metabolism and Alzheimer’s disease: Clinical evidence, mechanistic link and therapeutic promise. FEBS J. 2023, 290, 1420–1453. [Google Scholar] [CrossRef] [PubMed]
- Leng, L.; Yuan, Z.; Pan, R.; Su, X.; Wang, H.; Xue, J.; Zhuang, K.; Gao, J.; Chen, Z.; Lin, H.; et al. Microglial hexokinase 2 deficiency increases ATP generation through lipid metabolism leading to beta-amyloid clearance. Nat. Metab. 2022, 4, 1287–1305. [Google Scholar] [CrossRef]
- Choi, H.; Mook-Jung, I. Lipid fuel for hungry-angry microglia. Nat. Metab. 2022, 4, 1223–1224. [Google Scholar] [CrossRef]
- Prakash, P.; Manchanda, P.; Paouri, E.; Bisht, K.; Sharma, K.; Wijewardhane, P.R.; Randolph, C.E.; Clark, M.G.; Fine, J.; Thayer, E.A.; et al. Amyloid beta Induces Lipid Droplet-Mediated Microglial Dysfunction in Alzheimer’s Disease. bioRxiv 2023. 2023.06.04.543525. [Google Scholar] [CrossRef]
- Baenziger, J.E.; Morris, M.L.; Darsaut, T.E.; Ryan, S.E. Effect of membrane lipid composition on the conformational equilibria of the nicotinic acetylcholine receptor. J. Biol. Chem. 2000, 275, 777–784. [Google Scholar] [CrossRef]
- Barrantes, F.J. Cholesterol effects on nicotinic acetylcholine receptor: Cellular aspects. Subcell. Biochem. 2010, 51, 467–487. [Google Scholar]
- Benfante, R.; Di Lascio, S.; Cardani, S.; Fornasari, D. Acetylcholinesterase inhibitors targeting the cholinergic anti-inflammatory pathway: A new therapeutic perspective in aging-related disorders. Aging Clin. Exp. Res. 2021, 33, 823–834. [Google Scholar] [CrossRef]
- Kumar, R.; Nordberg, A.; Darreh-Shori, T. Amyloid-beta peptides act as allosteric modulators of cholinergic signalling through formation of soluble BAbetaACs. Brain 2016, 139 Pt 1, 174–192. [Google Scholar] [CrossRef]
- Baidya, A.T.; Kumar, A.; Kumar, R.; Darreh-Shori, T. Allosteric Binding Sites of Abeta Peptides on the Acetylcholine Synthesizing Enzyme ChAT as Deduced by In Silico Molecular Modeling. Int. J. Mol. Sci. 2022, 23, 6073. [Google Scholar] [CrossRef] [PubMed]
- Darreh-Shori, T.; Siawesh, M.; Mousavi, M.; Andreasen, N.; Nordberg, A. Apolipoprotein epsilon4 modulates phenotype of butyrylcholinesterase in CSF of patients with Alzheimer’s disease. J. Alzheimer’s Dis. 2012, 28, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Darreh-Shori, T.; Vijayaraghavan, S.; Aeinehband, S.; Piehl, F.; Lindblom, R.P.; Nilsson, B.; Ekdahl, K.N.; Langstrom, B.; Almkvist, O.; Nordberg, A. Functional variability in butyrylcholinesterase activity regulates intrathecal cytokine and astroglial biomarker profiles in patients with Alzheimer’s disease. Neurobiol. Aging 2013, 34, 2465–2481. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.M.; Darreh-Shori, T.; Junge, C.; Li, D.; Yang, Q.; Edwards, A.L.; Graham, D.L.; Moore, K.; Mummery, C.J. Onset of Alzheimer disease in apolipoprotein varepsilon4 carriers is earlier in butyrylcholinesterase K variant carriers. BMC Neurol. 2024, 24, 116. [Google Scholar] [CrossRef]
- Lane, R.M.; Darreh-Shori, T. Understanding the beneficial and detrimental effects of donepezil and rivastigmine to improve their therapeutic value. J. Alzheimer’s Dis. 2015, 44, 1039–1062. [Google Scholar] [CrossRef]
- Savva, G.M.; Wharton, S.B.; Ince, P.G.; Forster, G.; Matthews, F.E.; Brayne, C.; Medical Research Council Cognitive Function and Ageing Study. Age, neuropathology, and dementia. N. Engl. J. Med. 2009, 360, 2302–2309. [Google Scholar] [CrossRef]
- Mummery, C.J.; Borjesson-Hanson, A.; Blackburn, D.J.; Vijverberg, E.G.B.; De Deyn, P.P.; Ducharme, S.; Jonsson, M.; Schneider, A.; Rinne, J.O.; Ludolph, A.C.; et al. Tau-targeting antisense oligonucleotide MAPT(Rx) in mild Alzheimer’s disease: A phase 1b, randomized, placebo-controlled trial. Nat. Med. 2023, 29, 1437–1447. [Google Scholar] [CrossRef]
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- Morris, J.C.; Ernesto, C.; Schafer, K.; Coats, M.; Leon, S.; Sano, M.; Thal, L.J.; Woodbury, P. Clinical dementia rating training and reliability in multicenter studies: The Alzheimer’s Disease Cooperative Study experience. Neurology 1997, 48, 1508–1510. [Google Scholar] [CrossRef]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef]
- Zeng, X.; Cheung, S.K.K.; Shi, M.; Or, P.M.Y.; Li, Z.; Liu, J.Y.H.; Ho, W.L.H.; Liu, T.; Lu, K.; Rudd, J.A.; et al. Astrocyte-specific knockout of YKL-40/Chi3l1 reduces Aβ burden and restores memory functions in 5xFAD mice. J. Neuroinflamm. 2023, 20, 290. [Google Scholar] [CrossRef] [PubMed]
- Budelier, M.M.; He, Y.; Barthelemy, N.R.; Jiang, H.; Li, Y.; Park, E.; Henson, R.L.; Schindler, S.E.; Holtzman, D.M.; Bateman, R.J. A map of neurofilament light chain species in brain and cerebrospinal fluid and alterations in Alzheimer’s disease. Brain Commun. 2022, 4, fcac045. [Google Scholar] [CrossRef]
- Arvidsson Rådestig, M.; Skoog, I.; Skillbäck, T.; Zetterberg, H.; Kern, J.; Zettergren, A.; Andreasson, U.; Wetterberg, H.; Kern, S.; Blennow, K. Cerebrospinal fluid biomarkers of axonal and synaptic degeneration in a population-based sample. Alzheimer’s Res. Ther. 2023, 15, 44. [Google Scholar] [CrossRef]
- Tarawneh, R.; D’Angelo, G.; Crimmins, D.; Herries, E.; Griest, T.; Fagan, A.M.; Zipfel, G.J.; Ladenson, J.H.; Morris, J.C.; Holtzman, D.M. Diagnostic and Prognostic Utility of the Synaptic Marker Neurogranin in Alzheimer Disease. JAMA Neurol. 2016, 73, 561–571. [Google Scholar] [CrossRef]
- Wang, X.; Ghayoor, A.; Novicki, A.; Holmes, S.; Seibyl, J.; Hesterman, J. [P4–266]: Application of a Multi-Atlas Segmentation Tool to Hippocampus, Ventricle and Whole Brain Segmentation. Alzheimer’s Dement. 2017, 13, P1385–P1386. [Google Scholar] [CrossRef]
- Graffelman, J. Exploring diallelic genetic markers: The HardyWeinberg package. J. Stat. Softw. 2015, 64, 1–23. [Google Scholar] [CrossRef]
- Huang, L.C.; Lee, M.Y.; Chien, C.F.; Chang, Y.P.; Li, K.Y.; Yang, Y.H. Age and sex differences in the association between APOE genotype and Alzheimer’s disease in a Taiwan Chinese population. Front. Aging Neurosci 2023, 15, 1246592. [Google Scholar] [CrossRef] [PubMed]
- Bellou, E.; Baker, E.; Leonenko, G.; Bracher-Smith, M.; Daunt, P.; Menzies, G.; Williams, J.; Escott-Price, V.; Alzheimer’s Disease Neuroimaging Initiative. Age-dependent effect of APOE and polygenic component on Alzheimer’s disease. Neurobiol Aging 2020, 93, 69–77. [Google Scholar] [CrossRef]
- Whitwell, J.L.; Tosakulwong, N.; Weigand, S.D.; Graff-Radford, J.; Ertekin-Taner, N.; Machulda, M.M.; Duffy, J.R.; Schwarz, C.G.; Senjem, M.L.; Jack, C.R.; et al. Relationship of APOE, age at onset, amyloid and clinical phenotype in Alzheimer disease. Neurobiol. Aging 2021, 108, 90–98. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, W.; Ye, T.; Lin, X.; Zhang, J.; Alzheimer’s Disease Neuroimaging Initiative. Sex Difference in the Association of APOE4 with Memory Decline in Mild Cognitive Impairment. J. Alzheimer’s Dis. 2019, 69, 1161–1169. [Google Scholar] [CrossRef]
- Neu, S.C.; Pa, J.; Kukull, W.; Beekly, D.; Kuzma, A.; Gangadharan, P.; Wang, L.S.; Romero, K.; Arneric, S.P.; Redolfi, A.; et al. Apolipoprotein E Genotype and Sex Risk Factors for Alzheimer Disease: A Meta-analysis. JAMA Neurol. 2017, 74, 1178–1189. [Google Scholar] [CrossRef]
- Lane, R.M.; He, Y. Butyrylcholinesterase genotype and gender influence Alzheimer’s disease phenotype. Alzheimer’s Dement. 2013, 9, e1–e73. [Google Scholar] [CrossRef] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of Age, Sex, and Ethnicity on the Association Between Apolipoprotein E Genotype and Alzheimer Disease: A Meta-analysis. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef]
- Fleisher, A.S.; Chen, K.; Liu, X.; Ayutyanont, N.; Roontiva, A.; Thiyyagura, P.; Protas, H.; Joshi, A.D.; Sabbagh, M.; Sadowsky, C.H.; et al. Apolipoprotein E epsilon4 and age effects on florbetapir positron emission tomography in healthy aging and Alzheimer disease. Neurobiol Aging 2013, 34, 1–12. [Google Scholar] [CrossRef]
- Groot, C.; Grothe, M.J.; Mukherjee, S.; Jelistratova, I.; Jansen, I.; van Loenhoud, A.C.; Risacher, S.L.; Saykin, A.J.; Mac Donald, C.L.; Mez, J.; et al. Differential patterns of gray matter volumes and associated gene expression profiles in cognitively-defined Alzheimer’s disease subgroups. NeuroImage Clin. 2021, 30, 102660. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Ossenkoppele, R.; Smith, R.; Strandberg, O.; Ohlsson, T.; Jogi, J.; Palmqvist, S.; Stomrud, E.; Hansson, O. Greater tau load and reduced cortical thickness in APOE epsilon4-negative Alzheimer’s disease: A cohort study. Alzheimer’s Res. Ther. 2018, 10, 77. [Google Scholar] [CrossRef]
- Reiman, E.M.; Arboleda-Velasquez, J.F.; Quiroz, Y.T.; Huentelman, M.J.; Beach, T.G.; Caselli, R.J.; Chen, Y.; Su, Y.; Myers, A.J.; Hardy, J.; et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5000-person neuropathological study. Nat. Commun. 2020, 11, 667. [Google Scholar] [CrossRef]
- Josephs, K.A.; Whitwell, J.L.; Ahmed, Z.; Shiung, M.M.; Weigand, S.D.; Knopman, D.S.; Boeve, B.F.; Parisi, J.E.; Petersen, R.C.; Dickson, D.W.; et al. Beta-amyloid burden is not associated with rates of brain atrophy. Ann. Neurol. 2008, 63, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Drose, S.; Brandt, U.; et al. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Argyrousi, E.K.; Staniszewski, A.; Zhang, H.; Calcagno, E.; Zuccarello, E.; Acquarone, E.; Fa, M.; Li Puma, D.D.; Grassi, C.; et al. Tau is not necessary for amyloid-beta-induced synaptic and memory impairments. J. Clin. Investig. 2020, 130, 4831–4844. [Google Scholar] [CrossRef]
- Costoya-Sánchez, A.; Moscoso, A.; Silva-Rodríguez, J.; Pontecorvo, M.J.; Devous, M.D.; Sr Aguiar, P.; Schöll, M.; Grothe, M.J.; Alzheimer’s Disease Neuroimaging Initiative and the Harvard Aging Brain Study. Increased Medial Temporal Tau Positron Emission Tomography Uptake in the Absence of Amyloid-β Positivity. JAMA Neurol. 2023, 80, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, R.; Binette, A.P.; Groot, C.; Smith, R.; Strandberg, O.; Palmqvist, S.; Stomrud, E.; Tideman, P.; Ohlsson, T.; Jögi, J.; et al. Amyloid and Tau PET positive cognitively unimpaired individuals are at high risk for future cognitive decline. Nat. Med. 2022, 28, 2381–2387. [Google Scholar] [CrossRef] [PubMed]
- Susanto, T.A.; Pua, E.P.; Zhou, J.; Alzheimer’s Disease Neuroimaging Initiative. Cognition, brain atrophy, and cerebrospinal fluid biomarkers changes from preclinical to dementia stage of Alzheimer’s disease and the influence of apolipoprotein e. J. Alzheimer’s Dis. 2015, 45, 253–268. [Google Scholar] [CrossRef]
- Schmitz, T.W.; Soreq, H.; Poirier, J.; Spreng, R.N. Longitudinal Basal Forebrain Degeneration Interacts with TREM2/C3 Biomarkers of Inflammation in Presymptomatic Alzheimer’s Disease. J. Neurosci. 2020, 40, 1931–1942. [Google Scholar] [CrossRef]
- Schmitz, T.W.; Spreng, R.N.; Alzheimer’s Disease Neuroimaging Initiative. Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer’s pathology. Nat. Commun. 2016, 7, 13249. [Google Scholar]
- Yu, M.C.; Chuang, Y.F.; Wu, S.C.; Ho, C.F.; Liu, Y.C.; Chou, C.J. White matter hyperintensities in cholinergic pathways are associated with dementia severity in e4 carriers but not in non-carriers. Front. Neurol. 2023, 14, 1100322. [Google Scholar] [CrossRef]
- Hu, L.; Wong, T.P.; Cote, S.L.; Bell, K.F.; Cuello, A.C. The impact of Abeta-plaques on cortical cholinergic and non-cholinergic presynaptic boutons in alzheimer’s disease-like transgenic mice. Neuroscience 2003, 121, 421–432. [Google Scholar] [CrossRef]
- Bell, K.F.; Cuello, A.C. Altered synaptic function in Alzheimer’s disease. Eur. J. Pharmacol. 2006, 545, 11–21. [Google Scholar] [CrossRef]
- Mesulam, M.; Shaw, P.; Mash, D.; Weintraub, S. Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Eur. J. Pharmacol. 2004, 55, 815–828. [Google Scholar] [CrossRef]
- Ballinger, E.C.; Ananth, M.; Talmage, D.A.; Role, L.W. Basal Forebrain Cholinergic Circuits and Signaling in Cognition and Cognitive Decline. Neuron 2016, 91, 1199–1218. [Google Scholar] [CrossRef]
- Fontana, I.C.; Kumar, A.; Nordberg, A. The role of astrocytic alpha7 nicotinic acetylcholine receptors in Alzheimer disease. Nat. Rev. Neurol. 2023, 19, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Ferrari-Souza, J.P.; Lussier, F.Z.; Leffa, D.T.; Therriault, J.; Tissot, C.; Bellaver, B.; Ferreira, P.C.L.; Malpetti, M.; Wang, Y.T.; Povala, G.; et al. APOEepsilon4 associates with microglial activation independently of Abeta plaques and tau tangles. Sci. Adv. 2023, 9, eade1474. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.M.; He, Y. Emerging hypotheses regarding the influences of butyrylcholinesterase-K variant, APOE epsilon 4, and hyperhomocysteinemia in neurodegenerative dementias. Med. Hypotheses 2009, 73, 230–250. [Google Scholar] [CrossRef] [PubMed]
- Steward, A.; Biel, D.; Dewenter, A.; Roemer, S.; Wagner, F.; Dehsarvi, A.; Rathore, S.; Otero Svaldi, D.; Higgins, I.; Brendel, M.; et al. ApoE4 and Connectivity-Mediated Spreading of Tau Pathology at Lower Amyloid Levels. JAMA Neurol. 2023, 80, 1295–1306. [Google Scholar] [CrossRef]
- Chuang, Y.F.; Varma, V.; An, Y.; Tanaka, T.; Davatzikos, C.; Resnick, S.M.; Thambisetty, M. Interaction between Apolipoprotein E and Butyrylcholinesterase Genes on Risk of Alzheimer’s Disease in a Prospective Cohort Study. J. Alzheimer’s Dis. 2020, 75, 417–427. [Google Scholar] [CrossRef]
- Lane, R.; Feldman, H.H.; Meyer, J.; He, Y.; Ferris, S.H.; Nordberg, A.; Darreh-Shori, T.; Soininen, H.; Pirttila, T.; Farlow, M.R.; et al. Synergistic effect of apolipoprotein E epsilon4 and butyrylcholinesterase K-variant on progression from mild cognitive impairment to Alzheimer’s disease. Pharmacogenet Genom. 2008, 18, 289–298. [Google Scholar] [CrossRef]
- Bigler, E.D.; Lowry, C.M.; Anderson, C.V.; Johnson, S.C.; Terry, J.; Steed, M. Dementia, quantitative neuroimaging, and apolipoprotein E genotype. Am. J. Neuroradiol. 2000, 21, 1857–1868. [Google Scholar]
- Yasuda, M.; Mori, E.; Kitagaki, H.; Yamashita, H.; Hirono, N.; Shimada, K.; Maeda, K.; Tanaka, C. Apolipoprotein E ε4 allele and whole brain atrophy in late-onset Alzheimer’s disease. Am. J. Psychiatry 1998, 155, 779–784. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Petersen, R.C.; Xu, Y.C.; O’Brien, P.C.; Waring, S.C.; Tangalos, E.G.; Smith, G.E.; Ivnik, R.J.; Thibodeau, S.N.; Kokmen, E. Hippocampal atrophy and apolipoprotein E genotype are independently associated with Alzheimer’s disease. Ann. Neurol. 1998, 43, 303–310. [Google Scholar] [CrossRef]
- Tijms, B.M.; Vromen, E.M.; Mjaavatten, O.; Holstege, H.; Reus, L.M.; van der Lee, S.; Wesenhagen, K.E.J.; Lorenzini, L.; Vermunt, L.; Venkatraghavan, V.; et al. Cerebrospinal fluid proteomics in patients with Alzheimer’s disease reveals five molecular subtypes with distinct genetic risk profiles. Nat. Aging 2024, 4, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Sauty, B.; Durrleman, S. Impact of sex and APOE-epsilon4 genotype on patterns of regional brain atrophy in Alzheimer’s disease and healthy aging. Front. Neurol. 2023, 14, 1161527. [Google Scholar] [CrossRef] [PubMed]
- Laws, K.R.; Irvine, K.; Gale, T.M. Sex differences in cognitive impairment in Alzheimer’s disease. World J. Psychiatry 2016, 6, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, M.T.; Iulita, M.F.; Cavedo, E.; Chiesa, P.A.; Schumacher Dimech, A.; Santuccione Chadha, A.; Baracchi, F.; Girouard, H.; Misoch, S.; Giacobini, E.; et al. Sex differences in Alzheimer disease—The gateway to precision medicine. Nat. Rev. Neurol. 2018, 14, 457–469. [Google Scholar] [CrossRef]
- Dubal, D.B. Sex difference in Alzheimer’s disease: An updated, balanced and emerging perspective on differing vulnerabilities. Handb. Clin. Neurol. 2020, 175, 261–273. [Google Scholar]
- Beam, C.R.; Kaneshiro, C.; Jang, J.Y.; Reynolds, C.A.; Pedersen, N.L.; Gatz, M. Differences Between Women and Men in Incidence Rates of Dementia and Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, 1077–1083. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lane, R.M.; Li, D.; Darreh-Shori, T. Functional Glial Activation Mediates Phenotypic Effects of APOEɛ4 and Sex in Alzheimer’s Disease. Neuroglia 2024, 5, 323-343. https://doi.org/10.3390/neuroglia5030022
Lane RM, Li D, Darreh-Shori T. Functional Glial Activation Mediates Phenotypic Effects of APOEɛ4 and Sex in Alzheimer’s Disease. Neuroglia. 2024; 5(3):323-343. https://doi.org/10.3390/neuroglia5030022
Chicago/Turabian StyleLane, Roger M., Dan Li, and Taher Darreh-Shori. 2024. "Functional Glial Activation Mediates Phenotypic Effects of APOEɛ4 and Sex in Alzheimer’s Disease" Neuroglia 5, no. 3: 323-343. https://doi.org/10.3390/neuroglia5030022
APA StyleLane, R. M., Li, D., & Darreh-Shori, T. (2024). Functional Glial Activation Mediates Phenotypic Effects of APOEɛ4 and Sex in Alzheimer’s Disease. Neuroglia, 5(3), 323-343. https://doi.org/10.3390/neuroglia5030022