Abstract
Platax teira is a marine fish species with both ornamental and economic value, but it faces challenges in aquaculture due to environmental stress and disease. Genetic research on P. teira has been limited due to the limitations of the partially incomplete reference genome and the lack of a complete transcriptome. In this study, we utilized PacBio SMRT sequencing to generate a full-length transcriptome for P. teira, obtaining 39,770 isoforms, including 32,265 known gene-related transcripts and 4730 novel transcripts from 3455 new genes. All novel genes were annotated, and enrichment analysis revealed significant associations between immune-related pathways, such as cAMP, MAPK, PI3K-Akt, and Wnt. We also identified 14,398 alternative splicing events, 2754 alternative polyadenylation events, 42,250 SSRs, 1569 transcription factors, and 2067 long non-coding RNAs. Additionally, protein–protein interaction (PPI) analysis of immune-related pathways predicted chemokines as key immune factors among novel genes. Domain prediction analysis highlighted the diverse functional potential of immune factors such as NLRC3, tyrosine kinase 2, and A2M in different alternative splicing events. Overall, the characterization of the full-length transcriptome dataset of P. teira lays the foundation for further studies on its genetic analysis and immune regulation.
Key Contribution:
This article generated the first full-length transcriptome of Platax teira using PacBio SMRT sequencing and identified 39,770 isoforms, including 4730 novel transcripts from 3455 new genes.
1. Introduction
Ornamental fish farming originated in the 18th century and has since developed into a global industry encompassing over 6000 freshwater and marine species. The industry continues to grow in scale and has become an important economic sector in many countries, particularly in developing nations []. To ensure the sustainable use of resources, the industry is increasingly shifting from wild capture to artificial domestication, thereby reducing its reliance on natural populations. Platax teira, commonly known as the longfin batfish, is native to the Indo-Pacific region and is widely distributed in tropical and subtropical coastal waters. It is a docile, slow-swimming, omnivorous species that primarily feeds on algae and small benthic invertebrates []. Although P. teira is of significant economic value as an ornamental fish, its susceptibility to diseases and immune response mechanisms in complex marine environments remain insufficiently studied. Particularly in aquaculture settings, investigating the immune-related mechanisms of P. teira is crucial for enhancing its disease resistance, adaptability, and survival ability in dynamic marine environments [,].
The immune system is the primary mechanism by which fish adapt to external stress, resist pathogen infections, and maintain homeostasis. In recent years, as aquaculture faces increasing disease and environmental challenges, studying the identification and functional elucidation of immune genes has become crucial to improving disease resistance and adaptability in cultured populations []. As a representative of third-generation sequencing technology, full-length transcriptome sequencing has significant advantages such as an ultra-long read length, no assembly, and coverage of complete transcripts []. It has been widely used in gene structure analysis, novel gene identification, simple sequence repeats (SSRs) detection, alternative splicing (AS) and alternative polyadenylation (APA) event identification, and non-coding RNA identification in animals, plants, and microorganisms [], and it has played a crucial role in the screening and functional study of immune-related genes. In non-model species without reference genomes, Single-Molecule Real-Time (SMRT) sequencing technology can realize the direct sequencing of complete RNA molecules, which significantly improves the accuracy of genome annotation and the depth of functional research []. Alternative splicing enables a single gene to produce diverse mRNA isoforms by varying splice patterns, leading to protein variants with distinct or sometimes opposing structures and functions []. It serves as a key regulatory mechanism of gene expression at the RNA level and contributes significantly to phenotypic diversity and biological complexity. AS exhibits tissue- and stage-specific patterns, and the generation of specific splice variants under certain conditions may have a substantial impact on phenotypic traits []. Therefore, the full-length transcriptome data of P. teira provide valuable resources for a deeper understanding of the alternative splicing patterns and their functions in its immune mechanisms.
Currently, most studies on P. teira have mainly focused on mitochondrial genomics, temperature-dependent embryonic development, and muscle nutritional composition [,,,]. However, studies on its immune system are still limited. Previous research has indicated that the fish immune system plays a crucial role in defending against pathogens and responding to environmental changes and stress. P. teira exhibits immune responses to environmental stressors such as water quality changes and temperature fluctuations, but these mechanisms have not yet been comprehensively explored at the molecular level. Therefore, in this study, we applied PacBio SMRT sequencing to perform a multiplex analysis of pooled RNA samples from ten different tissues, aiming to capture a broad spectrum of transcript isoforms and construct the full-length transcriptome of P. teira. The resulting dataset provides the first comprehensive resource for the analysis of AS, APA, SSR events in this species, along with a complete set of lncRNA transcripts. Additionally, we performed a protein–protein interaction analysis of transcripts of immune-related pathways of novel genes and predicted key immune factors. Furthermore, we conducted a basic molecular analysis of key immune factors involved in alternative splicing. Overall, this high-quality full-length transcriptome resource offers essential molecular data for refining the genome and exploring immune regulatory mechanisms in P. teira.
2. Materials and Methods
2.1. Sample Collection and RNA Extraction
A mature female Platax teira was collected from the experimental station, located in Lingshui, Hainan Province, China. For full-length transcriptome analysis, ten different tissues were collected, including black skin, light skin, fin, black scale, light scale, liver, muscle, intestine, gill, and heart samples. These tissues were pooled before sequencing, which ensures comprehensive transcript coverage but limits the ability to resolve tissue-specific gene expression. Tissues were immediately frozen in liquid nitrogen after dissection and stored at −80 °C until RNA extractions. Total RNA was extracted using the RNeasy Kit (Qiagen, Hilden, Germany). RNA purity was assessed using a NanoPhotometer spectrophotometer (IMPLEN, Westlake Village, CA, USA), and concentrations were quantified with the Qubit RNA Assay Kit on a Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, CA, USA). RNA integrity was evaluated with the RNA Nano 6000 Assay Kit using a Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA). RNA samples with an RNA Integrity Number (RIN) > 7.0 and an OD 260/280 ratio between 2.0 and 2.2 were deemed of sufficient quality for downstream applications. These high-quality RNA samples were used for PacBio library construction and subsequent sequencing. All sequencing procedures were conducted by Novogene Co., Ltd. (Beijing, China).
2.2. PacBio Iso-Seq Library Preparation and Sequencing
Total RNA was extracted from ten tissues of P. teira (black skin, light skin, fin, black scale, light scale, liver, muscle, intestine, gill, and heart tissue) and pooled to generate a single RNA sample for library preparation. The PacBio sequencing library was constructed following the Iso-Seq protocol (Pacific Biosciences, Menlo Park, CA, USA). Briefly, 2 μg of purified polyadenylated RNA was reverse-transcribed into cDNA using the SMARTer PCR cDNA Synthesis Kit (Takara Biotechnology, Dalian, China) with Oligo (dT) primers. The cDNA was amplified by PCR and size-selected using the BluePippin™ Size Selection System (Sage Science, Beverly, MA, USA). The SMRT bell library was prepared from the size-selected cDNA with the Pacific Biosciences DNA Template Prep Kit 2.0, followed by DNA polymerase binding using the DNA/Polymerase Binding Kit. Sequencing was performed on the PacBio Sequel platform by Novogene Co., Ltd. (Beijing, China).
2.3. Sequencing Data Processing
Raw subreads in BAM format were processed using the Circular Consensus Sequencing (CCS) module in SMRT Link v11.0 to generate high-accuracy CCS reads, using the following parameters: min length = 50; max length = 15,000; min passes = 3; min snr = 2.5; and min predicted accuracy = 0.99. CCS reads were classified as full-length non-chimeric (FLNC) or non-full-length (NFL) based on primer and poly-A tail presence. FLNC reads were clustered using a hierarchical nlog(n) algorithm, and high-quality consensus sequences were generated and polished for accuracy. The polished consensus reads were mapped to the reference genome using GMAP []. The reference genome used in this study is the one published by our research group in 2024 [].
2.4. Functional Annotation
For functional annotation, transcript sequences were compared to multiple public databases, including NCBI NT, NR, Pfam, KOG/COG, Swiss-Prot, GO, and KEGG. BLASTN (v2.14.0) searches against the NT database were performed with BLAST using an E-value cutoff of 1 × 10−5. Functional annotation for NR, KOG, Swiss-Prot, and KEGG was performed using Diamond blastx (v0.8.36) [] with the same E-value threshold. Pfam domain annotation was conducted using hmmscan against the Pfam database.
2.5. Gene Structure Analysis
Full-length transcripts were classified and characterized using the TAPIS pipeline (https://bitbucket.org/comp_bio/tapis, accessed on 3 July 2024), and the polished consensus was aligned to the reference genome. Reads aligned to unannotated regions of the genome GTF file were considered novel genes, while those aligned to different exons of known genes were defined as novel transcripts. Simple sequence repeats (SSRs) were identified using the MISA tool (http://pgrc.ipk-gatersleben.de/misa/, accessed on 10 July 2024) on transcripts longer than 500 bp. Seven types of SSRs were used: mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides and compound SSRs []. The detection criteria included a minimum of 10, 6, 5, 5, 5, and 5 repeats for each respective motif type. AS events (alternative 5′/3′ splice site (A5/A3), skipping exon (SE), alternative first/last exons (AF/AL), mutually exclusive exons (MX), and retained intron (RI)) were detected using the SUPPA tool [,]. SUPPA2 generated AS and transcript events from an annotation file (GFF/GTF format), producing two files: an ioe file listing AS events and their corresponding transcript and an ioi file providing the set of all transcripts for each gene to calculate relative transcript abundance. Alternative polyadenylation (APA) analysis was performed using TAPIS by aligning reads to annotated genes and identifying unique poly(A) sites through a greedy algorithm []. A greedy algorithm identified unique poly(A) sites by calculating the depth of reads within a 5-nucleotide window. Sites with a minimum depth of two and at least 15 nucleotides from existing sites were added to the list, and the process continued until no further candidate sites remained. Transcription factors (TFs) were identified using the Animal Transcription Factor Database (AnimalTFDB) []. Four computational methods (CPC, CNCI, CPAT, and Pfam) were employed to differentiate between non-protein-coding and putative protein-coding RNAs [,,]. Putative protein-coding RNAs were filtered based on minimum length (>200 nt) and exon number (>2) and further assessed using the aforementioned methods for differentiation between coding and non-coding genes.
2.6. Protein–Protein Interaction Analysis and Molecular Characterization of Immune Genes
Protein–protein interactions (PPIs) among immune-related gene products were predicted using STRING 12.0 (https://STRING-db.org/, accessed on 20 November 2024). A confidence threshold of 0.40 was applied to retrieve PPI information from text mining, experimental data, databases, co-expression patterns, and neighborhood sources. The results were then visualized using Cytoscape (v3.10.0), with a degree cutoff value of 2, a node score cutoff of 0.2, a k-core of 2, and a maximum depth of 100. The transcripts of immune-related genes were analyzed using NCBI’s CD-Search tool to predict conserved domains, and the results were visualized using TBtools (v2.357) []. The gene structures associated with AS events were visualized through the GSDS online platform (https://gsds.gao-lab.org/, accessed on 25 November 2024).
3. Results
3.1. Quality Control of the Full-Length Transcriptomes
Full-length transcriptome sequencing of Platax teira was performed using the PacBio Sequel platform based on pooled RNA extracted from ten major tissues, including black skin, light skin, fin, black scale, light scale, liver, muscle, intestine, gill, and heart samples. The results revealed 461,095 raw polymerase reads (58.7 Gb nucleotides). Raw sequencing reads were filtered to eliminate low-quality sequences, obtaining 25,608,838 clean subreads. The average subread length was 2222 bp, and the N50 length was 2488 bp (Table 1). Following error correction among subreads, a total of 362,543 circular consensus sequencing (CCS) reads were generated, with an average read length of 2333 bp and an N50 length of 2496 bp (Figure 1A). Among these, 304,085 reads were identified as full-length non-chimeric (FLNC), exhibiting an average length of 2250 bp and an N50 of 2421 bp (Figure 1B). The FLNC ratio (FLNC/CCS) was 83.88%. Following clustering and removal of redundant sequences, the FLNC reads underwent a final round of error correction, yielding 62,807 polished consensus reads with an N50 of 2881 bp (Figure 1C).

Table 1.
The description of full-length sequencing in Platax teira.

Figure 1.
CCS, FLNC, and consensus reads’ length distribution. (A) Length distribution of CCS reads. The abscissa represents the length of CCS reads, and the ordinate represents the number of CCS reads. (B) Length distribution of FLNC reads. The abscissa represents the length of FLNC reads, and the ordinate represents the number of FLNC reads. (C) Length distribution of consensus reads. The abscissa represents the length of consensus reads, and the ordinate represents the number of consensus reads.
To evaluate the accuracy and genomic representation of the full-length transcriptome generated by the PacBio SMRT platform (v2.3), the polished consensus reads were mapped to the reference genome. The mapped sequences exhibited excellent concordance with the genome (Figure 2A). In total, 61,095 reads (97.23%) were successfully aligned, whereas 1712 reads (2.73%) remained unmapped (Figure 2B). These results highlight the high fidelity and accuracy of the sequencing data. On one hand, the splice donor–acceptor results revealed that the GT-AG dinucleotide combination was predominant, accounting for 97.52% of splicing events. In comparison, GC-AG and AT-AC motifs were observed at lower frequencies, representing 0.01% and 0.13%, respectively (Figure 2C). On the other hand, examining the distribution of FLNC reads across increasing sequencing depths showed that the gene discovery curve reached a plateau (Figure 2D). Furthermore, mapped reads exhibited a higher isoform density than typically observed in second-generation sequencing data (Figure 2E,F).
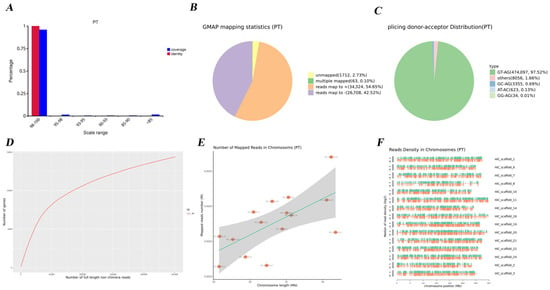
Figure 2.
Quality detection of Iso-Seq reads and isoforms. (A) Distributions of coverage (blue) and identity (red) for every transcript. The abscissa is the range of coverage and identity, and the ordinate is the percentage of transcripts. (B) GMAP mapping statistics of full-length transcripts, including those unmapped, multi-mapped, mapped to +, and mapped to −. (C) The distribution of splicing donors/acceptors among all transcripts. Values are shown to two decimal places. (D) The depth of sequencing. The abscissa is the number of full-length non-chimeric reads, and the ordinate is the number of genes in the genome. Values are shown to two decimal places. (E) The number of mapped reads in chromosomes. The abscissa is the chromosome length, and the ordinate is the number of mapped reads. (F) Read density in chromosomes of full-length transcripts. The abscissa is the chromosome position, and the ordinate is the median read density.
3.2. Transcript Identification and Gene Functional Annotation
A total of 39,770 non-redundant isoforms were aligned to the reference genome, with an N50 length of 2976 bp. It comprised 2775 previously reported isoforms (6.98%), 32,265 novel isoforms of known genes (81.12%), and 4730 isoforms derived from novel genes (11.89%) (Figure 3A). Among these non-redundant isoforms, 38,714 (97.34%) were successfully annotated at least once in the NR, SwissProt, KEGG, KOG, or NT databases. These 4730 transcripts corresponded to 3455 previously unannotated genes (Table S1). To investigate the potential functions of these novel genes, we compared their protein sequences against seven public databases. In total, 1871, 1366, 1767, 945, 1449, 2905, and 1449 genes were successfully annotated in the NR, Swiss-Prot, KEGG, KOG, GO, NT, and Pfam databases, respectively (Figure 3B). Within the NR database, the closest homologs were most frequently assigned to Larimichthys crocea (607 genes), followed by Lates calcarifer (473 genes) and Stegastes partitus (126 genes) (Figure 3C).
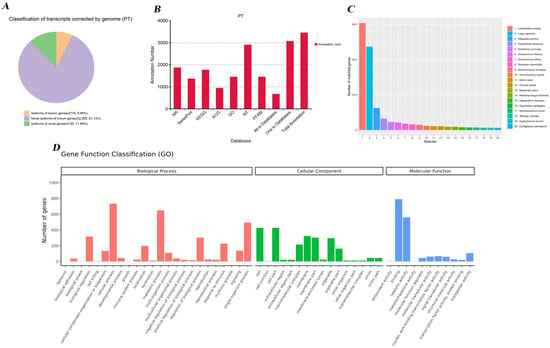
Figure 3.
Characteristics of transcripts and novel gene annotation. (A) Classification statistics of full-length transcripts, including isoforms of known genes, novel isoforms of known genes, and isoforms of novel genes. (B) Statistic results of NR, SwissProt protein, KEGG, KOG, GO, NR, and PFAM databases. The abscissa represents different databases, and the ordinate represents the number of annotations. (C) NR database annotation statistics results. The abscissa represents the species ID, and the ordinate represents the number of genes on the annotation. (D) GO database annotation statistics. The abscissa represents the lower-level GO terms of the three major categories of GO, while the ordinate represents the number of genes annotated to that term (including the sub-terms of that term). The three different classifications represent the three basic classifications of GO terms (from left to right: biological process, cellular component, and molecular function).
GO annotation revealed that most genes were associated with binding activity (790 genes) and catalytic activity (559 genes), while substantial numbers were linked to “cell” and “cell part terms” (427 genes each) (p < 0.05). Additional prominent categories included metabolic processes (647 genes), cellular processes (733 genes), and responses to the stimulus (227 genes) (Figure 3D, Table S2). KEGG pathway analysis indicated that the most highly represented pathway was signaling molecules and their interaction with 183 genes (p < 0.05), followed by the global and overview metabolic pathways (187 genes) and aging (120 genes). A substantial number of genes were also involved in cancers: specific types (120 genes) and the immune system (84 genes). Additionally, cellular processes like transport and catabolism (86 genes) and signal transduction (88 genes) also accounted for a large portion of gene annotations (Figure 4, Table S3).
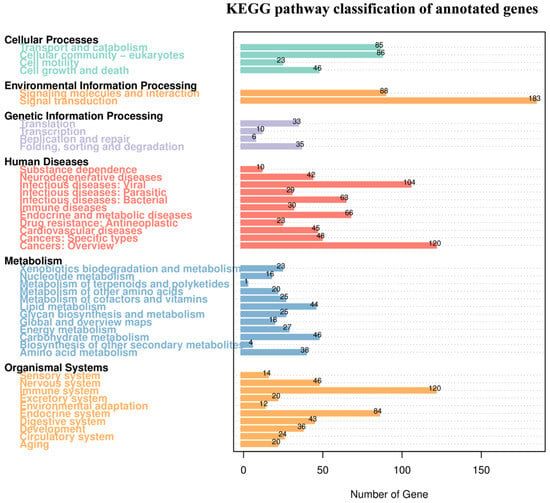
Figure 4.
KEGG pathway classification of annotated genes. The numbers on the bar chart represent the number of genes annotated; the other axis shows the codes of level 1 functional categories in the database, and the explanations of the codes can be found in the corresponding legend.
3.3. SSR Detection, Alternative Splicing (AS) Analysis, and Alternative Polyadenylation (APA) Events
A total of 39,421 sequences longer than 500 bp were analyzed for simple sequence repeats (SSRs), resulting in the identification of 42,250 SSRs, with 21,321 sequences containing at least one SSRs in P. teira (Table S4). Among these, 10,968 sequences contained more than one SSR, and 4770 SSRs were found in compound formations (Figure 5A). Regarding the repeat number, SSRs in P. teira were predominantly found with repeat lengths of fewer than 15 (Figure 5B).
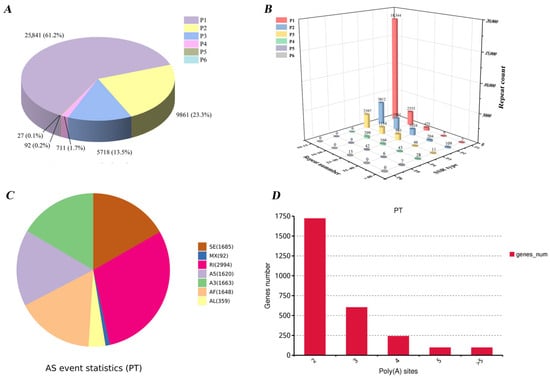
Figure 5.
Gene structure analysis based on the full-length transcripts. (A) Pie chart shows the proportion of different SSR types. Values are shown to one decimal places. (B) The 3D bar chart displays the repeat counts of SSRs (Z-axis) plotted against the SSR type (X-axis) and repeat number (Y-axis). (C) Statistic results of AS events. SE, skipped exon; MX, mutually exclusive exon; RI, retained intron; A5, alternative 5′ splice site; A3, alternative 3′ splice site; AF, alternative first exon; AL, alternative last exon. (D) Polyadenylation profile. The abscissa is the variable number of polyadenylate sites, and the ordinate is the number of genes.
In this study, AS analysis based on full-length transcripts was performed using SUPPA, resulting in the identification of 14,398 AS events. Among these, 10,061 events were classified into seven distinct AS types (Figure 5C, Table S5). Among them, intron retention (IR) was the most prevalent, accounting for 20.79% of the total events, followed by skipped exon (SE) at 11.7%.
APA analysis identified 1720 transcripts that contained at least two supported poly(A) sites, while 602 and 240 transcripts possessed three and four or more poly(A) sites, respectively (Figure 5D). Among the 2754 sites exhibiting APA events, genes with two polyadenylation sites accounted for the largest proportion, reaching 62.4%.
3.4. Transcription Factors (TFs) and Long Non-Coding RNAs
In this study, we visualized the top 30 transcription factor (TF) families predicted from 1568 transcripts using a bar chart (Figure 6A). The results showed that the most abundant TF family was zf-C2H2 (zinc finger C2H2), comprising 450 members, followed by ZBTB (zinc finger- and BTB domain-containing family) and TF_bZIP (basic leucine zipper transcription factor), with 158 and 121 members, respectively.
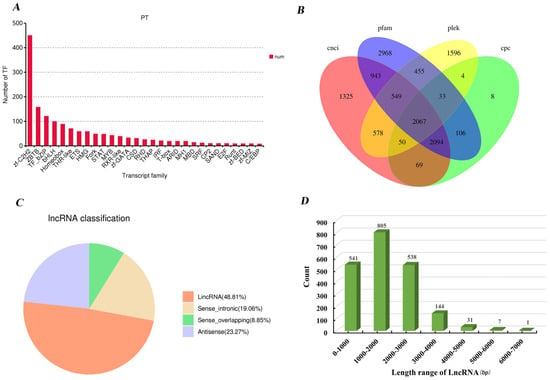
Figure 6.
(A) Transcription factor analysis results. The abscissa represents different families of transcription factors, and the ordinate represents the number of transcript factor. (B) Venn diagram of lncRNA prediction results. (C) Statistical graph of lncRNA classification results. Values are shown to two decimal places. (D) The length distribution of lncRNA.
Because lncRNAs lack the capacity to encode proteins, assessing transcript coding potential is a crucial step in their discovery. In the present study, agreement among all four algorithms yielded 2067 candidate lncRNA transcripts (Figure 6B). Genomic classification revealed 1009 intergenic lncRNAs (lincRNAs), 394 sense lncRNAs, 481 antisense lncRNAs, and 183 sense-overlapping lncRNAs among the full-length transcripts (Figure 6C). The length distribution analysis of lncRNAs revealed that transcripts of around 2000 nucleotides were the most prevalent, accounting for approximately 38.94%. In addition, a large proportion of lncRNAs fell within the 1000-nucleotide range and between 3000 and 4000 nucleotides, each group representing around 26% of the total. The longest lncRNA identified was 6375 nucleotides in length (Figure 6D).
3.5. Screening Key Genes by PPI and Molecular Analysis of Immune Genes
The “Immune system” and “Immune disease” pathways were screened from KEGG annotations of novel genes, and 128 non-redundant immune-related genes enriched in these two pathways were used for further analysis. Protein–protein interactions (PPIs) among these novel genes were predicted using the STRING 12.0 database. The results showed that genes such as cxcr3.1, cxcr5, and ccl19a.2 occupied central positions in the network, while most chemokines, including ccl39.10, ccl34a.3, and ccl25a, exhibited distinct clustering patterns (Figure 7). We analyzed a subset of immune genes from SE and RI with the largest number of transcripts, including NLRC3 (evm.TU.ctg000770_np12.11), tyrosine kinase 2 (evm.TU.ctg000280_np12.83), interleukin 17 receptor A (evm.TU.ctg000850_np12.563), and alpha-2-macroglobulin (evm.TU.ctg000220_np12.61) (Figure 8A–D, Table S6). The results revealed that NLR3, tyrosine kinase 2, and interleukin 17 receptor A had only two, three, and three transcripts, respectively, containing protein domain families. In contrast, alpha-2-macroglobulin, which was involved in more SE events, had 25 transcripts containing protein domains. These findings suggest that transcripts with protein domains, particularly those associated with SE events, may play a more significant role in immune functions. Additionally, the structural analysis of some alpha-2-macroglobulin transcripts (e.g., evm.TU.ctg000220_np12.61_novel07, evm.TU.ctg000220_np12.61_novel08, evm.TU.ctg000220_np12.61_novel010, evm.TU.ctg000220_np12.61_novel23, and evm.TU.ctg000220_np12.61_novel24) revealed different splicing types, which may be closely related to the formation of their domains (Figure 8E).
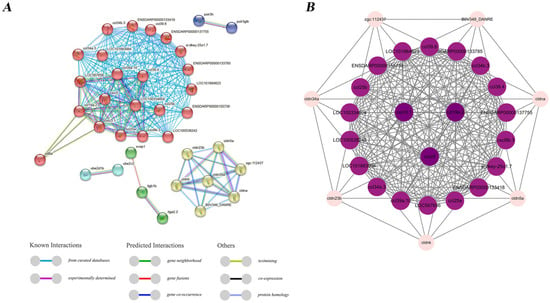
Figure 7.
Interaction networks in immune-related genes among novel genes. (A) The nodes in the network represent the proteins corresponding to the immune-related genes, and the lines connecting the different nodes indicate the interactions between the immune-related genes. (B) The interaction network of the immune-related genes was constructed in Cytoscape based on DC values, with node colors ranging from dark to light to represent a gradient from high to low DC values.
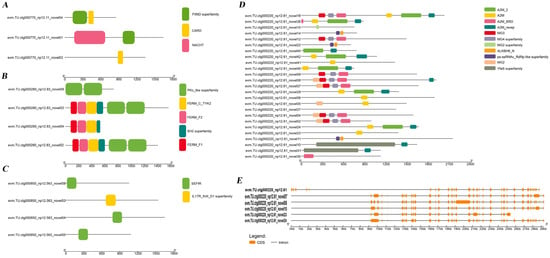
Figure 8.
Immune gene domain prediction and alternative splicing event structure analysis. (A) NLRC3, (B) tyrosine kinase 2, (C) IL17RA, (D) A2M. (E) Partial alternative splicing events of alpha-2-macroglobulin.
4. Discussion
4.1. Transcriptome Completeness and Annotation Quality
We performed full-length transcriptome sequencing of P. teira using the PacBio Iso-Seq platform, which effectively addressed the challenges of short-read assembly commonly encountered in species lacking high-quality reference genomes. The Iso-Seq enables the direct capture of complete transcripts, thereby avoiding assembly errors caused by repetitive sequences and substantially improving the accuracy and completeness of transcriptome annotation []. To further refine the genomic information, we employed GMAP to align the full-length transcripts to the reference genome, facilitating the identification of novel genes and transcripts and improving gene structure annotations []. As a powerful tool for capturing transcriptomic information from complex genomes, full-length transcriptome sequencing has been increasingly applied in studies on growth, development, stress responses, and adaptive evolution in aquatic organisms [,]. This study represents the first comprehensive characterization of the full-length transcripts in P. teira, providing valuable genetic resources and a foundation for future investigations into functional genes and molecular regulatory mechanisms.
Furthermore, 39,770 non-redundant full-length transcripts were acquired, with an N50 length of 2976 bp. This is substantially longer than transcript assemblies that were previously produced in our work utilizing NGS technologies []. The number of full-length transcripts in our analysis was slightly lower than the results reported for Sebastes schlegelii, but it was greater than those reported in studies on Scophthalmus maximus and Acipenser sinensis [,,]. With 38,714 transcripts (97.34%) effectively annotated in public databases, the excellent quality and completeness of these full-length transcripts significantly increased annotation efficiency. Our annotation rate was higher than those reported in previous studies of Ictiobus cyprinellus (96.54%) and Misgurnus anguillicaudatus (95.21%) [,], further highlighting the significant advantages of full-length transcriptome sequencing in improving annotation accuracy and completeness. However, a small number of transcripts remained unannotated, which may be due to the limited availability of genomic information for P. teira and its closely related species, or they may represent previously uncharacterized or species-specific genes. Notably, a total of 4730 transcripts were identified as novel genes in this study, of which 3070 were successfully annotated in multiple databases, including NR, Swiss-Prot, KEGG, KOG, GO, NT, and Pfam. In the NR annotation results, L. crocea and L. calcarifer exhibited the highest numbers of homologous sequence matches. Both species are Perciformes with well-assembled and extensively annotated reference genomes [,], suggesting that the novel transcripts in P. teira have relatively complete structures and a high potential for protein translation and functional execution.
4.2. Functional Implications of Novel Genes
Gene Ontology (GO) annotation provided a structured framework for understanding the functional roles of genes at the molecular, cellular, and biological process levels. Some tyrosine-related enzymes (Novelgene0253; novelgene0384; novelgene0573; novelgene0938; novelgene1184, etc.; see red marker in Table S2) were found in the GO annotation of the new transcript in this study. Early research has shown that the tyrosinase family, which includes the rate-limiting enzymes in melanin biosynthesis, plays a pivotal role in regulating pigment production in vertebrates. The expression level and enzymatic activity of tyr directly influence the balance between eumelanin and pheomelanin synthesis, thereby determining pigmentation patterns such as hair or skin color in animals []. In our study, different alternative splicing types of tyrosinase genes were analyzed, and the body color changes caused by immune stress may be related to these splicing types. KEGG annotation provided a pathway-level understanding of gene function and offered valuable insights into how genes coordinated in complex biological processes such as metabolism, signal transduction, and immune responses. In teleost fish, appropriate levels of melanin not only protected the skin from ultraviolet radiation but also helped defend against harmful environmental factors such as pollution []. Studies showed that immune responses and pigmentation pathways were often interconnected, commonly regulated through shared signaling cascades such as the MAPK pathway and cytokine signaling networks []. The KEGG pathway analysis in this study further supported this observation, showing that these novel genes were significantly enriched in multiple signaling pathways, including PI3K-Akt, MAPK, Wnt, Jak-STAT, and Ras signaling pathways. These results suggest that the annotated genes play crucial roles in signaling, metabolic regulation, immune response, and disease-related pathways.
Chemokines are a group of structurally conserved yet functionally diverse cytokines that orchestrate immune cell migration and exert pleiotropic roles under both physiological and pathological conditions []. Studies have demonstrated that rSsCXCL12a and rSsCXCL12b exhibit potent antibacterial activity and that the CXCL12/CXCR4 axis plays a critical role in mediating the immune response of S. schlegelii against bacterial infection []. Protein–protein interaction network analysis of immune-related pathway genes in P. teira showed that factors such as CXCR5, CCL25a, and CCL25b, which scored highly, form a flexible regulatory network. It has been reported that the CXCL13/CXCR5 axis is involved in the immune response of Oreochromis niloticus by mediating the chemotaxis of IgM+ B cells []. Furthermore, Yang et al. identified EcCCL25 and EcCCR9 sequences from the transcriptome database of Epinephelus coioides and reported that the CCL25/CCR9a complex may be involved in the host’s defense against Cryptocaryon irritans infection []. Based on these findings, we hypothesize that chemokines and their receptors may play an important role in the immune regulation of P. teira, although their specific functions still need to be further validated experimentally.
4.3. Simple Sequence Repeats, Transcription Factors, and lncRNAs
Simple sequence repeats (SSRs), also called short tandem repeats or microsatellites, are 1–6 bp motifs widely distributed throughout eukaryotic genomes [,]. Due to the variability in the nucleotide sequences of repeat units and the number of repeats, SSR lengths can vary considerably. Among the six types, mononucleotides were the most abundant, followed by dinucleotides and trinucleotides, which is consistent with the findings of a study on Schizothorax prenanti []. Regarding the repeat number, SSRs in P. teira were predominantly found with repeat lengths of fewer than 15, a result that aligns with studies on Heliocidaris crassispina, Platypharodon extremus, and Hucho bleekeri and is in accordance with the typical characteristics of microsatellite markers [,,]. These findings lay the foundation for genetic diversity and phylogeny studies and will support the development and application of SSRs in P. teira.
Transcription factors (TFs) play a central role in regulating gene expression, cell differentiation, immune responses, and environmental adaptation by binding to specific sequences in genomic regulatory regions []. In this study, the most abundant TF family predicted from the transcriptome was zf-C2H2, followed by ZBTB and bZIP, which is consistent with findings from most teleost species []. Zf-C2H2 proteins are known to be widely involved in transcriptional regulation, particularly under environmental stress conditions. Specifically, certain members of the zf-C2H2 family, such as the KLF subfamily, have been shown to participate in immune regulation and play roles in disease resistance [,]. This study found that a novel Kruppel-like factor (MrKLF) from Macrobrachium rosenbergii is expressed in various tissues and its expression is modulated by Vibrio parahaemolyticus and Aeromonas hydrophila challenges. RNA interference analysis revealed that MrKLF regulates antimicrobial peptides, playing a role in innate immunity []. In addition, several members of the bZIP family, including C/EBP proteins, are closely associated with lipid metabolism and are known to be involved in the differentiation and activation of immune cells [].
LncRNAs, typically more than 200 nucleotides in length, represent non-coding transcripts with essential roles in immune regulation, cell cycle progression, alternative splicing, differentiation, and epigenetic control []. The researchers identified multiple lncRNAs in Penaeus vannamei hemocytes that were highly co-expressed with immune-related genes, suggesting that lncRNAs may be involved in regulating the immune defense in shrimp hemocytes []. In addition, lncRNAs in large yellow croaker have been studied through Vibrio parahaemolyticus challenge. Tissue expression profiling revealed that most lncRNAs were tissue-specific, with 163 lncRNAs specifically expressed in the spleen and likely involved in immune responses []. However, no lncRNAs had previously been reported in P. teira. In conclusion, our analysis identified multiple SSRs, TFs, and lncRNAs in P. teira, and their specific biological functions remain to be elucidated in future studies.
4.4. Alternative Splicing and Immune Regulation
Alternative splicing (AS) and alternative polyadenylation (APA) are key post-transcriptional regulatory mechanisms in both plants and animals []. In P. teira, we identified seven major AS types, with intron retention (IR) being the most prevalent (20.79%), followed by exon skipping (SE) (11.7%). This pattern is consistent with other marine fish species (S. schlegelii and S. maximus), suggesting that IR and SE are common AS events across various animal taxa [,]. Although IR is dominant in plants and SE is more common in animals [], certain IR events also play a significant role in animals. These events can act as precursor RNAs stored within the cell, which can be rapidly spliced and translated upon specific stimuli, thereby enhancing protein synthesis efficiency []. Our findings indicate that IR events are more frequent than SE in P. teira, suggesting that gene structural variations are more often driven by IR. Immune-related genes such as CD3G, Nacht, LRR, and PYD domain-containing proteins and MAFB generate multiple transcripts through IR splicing, highlighting IR’s significant role in regulating immune functions and cellular processes []. Genome-wide studies also show that APA, which generates alternative 3′ ends, is widespread in animals, with over 70% of human genes containing multiple APA sites []. In P. teira, tandem APA events in the 3′ untranslated region (3′UTR) were most common, influencing mRNA stability, localization, and translation efficiency by altering the length and sequence of 3′UTRs []. These APA patterns are consistent with those found in other teleosts and are also implicated in responses to infection, as seen with Grass Carp Reovirus (GCRV), where APA plays a role in processes like cytoskeleton organization and microtubule regulation [].
Previous studies have shown that alternative splicing of immune-related genes regulates immune responses by enhancing functional diversity and the ability to respond to external stimuli []. Specifically, the novel1 transcript of the NLRC3 gene in Platax teira contains a NACHT domain, while the novel3 and novel4 transcripts contain CARD domains. The NACHT domain is typically involved in intracellular signal transduction and the regulation of immune responses, while the CARD domain is closely associated with inflammatory responses, particularly in programmed cell death and immune responses [,]. This study found that the overexpression of zebrafish NLRC3-like 1, which contains FISNA, NACHT, and LRR domains, led to decreased expression of the proinflammatory cytokines TNF-α, IFN-γ, and CCL4 in zebrafish larvae infected with E. piscicida []. This suggests that NLRC3-like 1 may play a role in regulating and suppressing inflammatory responses. Tyrosine kinase 2 transcripts primarily contain the FERM domain family, which is involved in the transmission of extracellular signals and plays a crucial role in connecting the cytoskeleton and cell signaling. This suggests that tyrosine kinase 2 may play an important role in cytokine signaling pathways []. The comparative transcriptomic study of the skin in snakehead fish (GCAS) and Channa argus (CAS) revealed that the expression level of the TYR gene was downregulated in GCAS. These results indicated that the TYR gene, which encodes tyrosinase, was expressed at lower levels or suppressed in GCAS, thereby inhibiting melanin production and inducing the occurrence of leucism []. Furthermore, the alpha-2-macroglobulin (A2M) transcripts predominantly contain the A2M and MG superfamily domains. These domains are involved in immune regulation, pathogen clearance, and immune evasion mechanisms. The A2M domain is known for its role in inhibiting protease activity and clearing pathogens, while the MG superfamily domain contributes to protein binding and detoxification functions []. Researchers have studied two distinct A2M genes in Portunus trituberculatus and found that PtA2Ms are maternally transferred and can be induced by bacteria and fungi. They play a role in regulating the proPO system, likely limiting PO activity by inhibiting the activation of relevant serine proteases. Silencing of PtA2M-1 or PtA2M-2 genes resulted in reduced lysozyme activity and lower expression of phagocytosis-related genes. Additionally, PtA2Ms may have been involved in the transcriptional regulation of AMP genes through the Toll and NF-κB pathways []. Interestingly, different transcripts of alpha-2-macroglobulin exhibit various splicing types, which may be closely linked to the formation and function of their domains. Previous research has demonstrated that alternative splicing enhances the functional diversity of immune genes in Tenualosa ilisha, thus strengthening the host’s ability to respond to pathogens []. Our analysis indicates that SE events could significantly impact the function of alpha-2-macroglobulin, and further investigation of these splice variants may provide new insights into the regulation of immune mechanisms. To summarize, transcripts containing protein domains, particularly those associated with SE events, may play a more significant role in immune functions.
5. Conclusions
In summary, our study conducted a comprehensive full-length transcriptome analysis of Platax teira using the PacBio Iso-Seq platform, resulting in a high-quality transcript dataset. Long-read sequencing improved gene structure annotation and made it easier to identify AS, SSRs, and lncRNAs by accurately reconstructing full-length transcripts. Functional annotation and enrichment analysis of novel transcripts and genes revealed potential involvement in pigmentation and immune-related pathways, suggesting their roles in immune responses. Therefore, the full-length transcriptome generated in this research will be invaluable for future investigations into functional gene discovery, molecular marker development, key regulatory events, and signaling pathways in P. teira.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/fishes10110575/s1, Table S1: Statistics of the non-redundant isoform annotation results; Table S2: Annotation of novel gene transcripts in GO database; Table S3: Annotation of novel gene transcripts in KEGG database; Table S4: Predicted SSRs; Table S5: Predicted alternative splicing events; Table S6: Transcripts sequences of immune-related genes.
Author Contributions
D.Z. and J.S.: Study concept and design. L.L.: Writing—original draft, writing—review and editing. B.L.: Data curation, writing—review and editing. H.G.: Formal analysis, visualization. K.Z.: Validation, investigation. N.Z.: Supervision and resources. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Hainan Province Science and Technology Special Fund (ZDYF2024XDNY231), the Guangxi Key Research and Development Program (Guinongke AB241484019), the Central Public-Interest Scientific Institution Basal Research Fund, CAFS (NO. 2023TD33), and the Seed Industry Revitalization Project of Special Fund for Rural Revitalization Strategy in Guangdong Province (2023SBH22002).
Institutional Review Board Statement
All animal experiments were conducted in compliance with the guidelines and regulations of the Animal Care and Use Committee of the South China Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, and were approved under protocol number SCSFRI96-253; approval date: 8 October 2018.
Data Availability Statement
The raw full-length transcriptome sequencing data have been stored in NCBI under BioProject accession PRJNA1305163 and SRA accession SRR34970248. The transcriptome was deposited in the figshare database (https://doi.org/10.6084/m9.figshare.30415042, accessed on 22 October 2025).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Priyashadi, M.S.V.H.; Deepananda, K.H.M.A.; Jayasinghe, A. Socio-Economic Development of Marine Ornamental Reef Fish Fishers in Eastern Sri Lanka through the Lenses of Human Development Index. Mar. Policy 2022, 143, 105136. [Google Scholar] [CrossRef]
- Liu, B.; Guo, H.-Y.; Zhu, K.-C.; Liu, B.-S.; Guo, L.; Zhang, N.; Jiang, S.-G.; Zhang, D.-C. Nutritional Compositions in Different Parts of Muscle in the Longfin Batfish, Platax Teira (Forsskål, 1775). J. Appl. Anim. Res. 2019, 47, 403–407. [Google Scholar] [CrossRef]
- Leu, M.-Y.; Tai, K.-Y.; Meng, P.-J.; Tang, C.-H.; Wang, P.-H.; Tew, K.S. Embryonic, Larval and Juvenile Development of the Longfin Batfish, Platax teira (Forsskål, 1775) under Controlled Conditions with Special Regard to Mitigate Cannibalism for Larviculture. Aquaculture 2018, 493, 204–213. [Google Scholar] [CrossRef]
- Li, S.; Xie, Z.; Chen, P.; Tang, J.; Tang, L.; Chen, H.; Wang, D.; Zhang, Y.; Lin, H. The Complete Mitochondrial Genome of the Platax teira (Osteichthyes: Ephippidae). Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 796–797. [Google Scholar] [CrossRef]
- Parker, J.; Roth, O. Comparative Assessment of Immunological Tolerance in Fish with Natural Immunodeficiency. Dev. Comp. Immunol. 2022, 132, 104393. [Google Scholar] [CrossRef]
- Zhang, F.; Wan, W.; Li, Y.; Wang, B.; Shao, Y.; Di, X.; Zhang, H.; Cai, W.; Wei, Y.; Ma, X. Construction of a Full-Length Transcriptome Resource for the African Sharptooth Catfish (Clarias Gariepinus), a Prototypical Air-Breathing Fish, Based on Isoform Sequencing (Iso-Seq). Gene 2024, 930, 148802. [Google Scholar] [CrossRef]
- Chen, X.; Tang, Y.Y.; Yin, H.; Sun, X.; Zhang, X.; Xu, N. A Survey of the Full-Length Transcriptome of Gracilariopsis lemaneiformis Using Single-Molecule Long-Read Sequencing. BMC Plant Biol. 2022, 22, 597. [Google Scholar] [CrossRef]
- Liao, X.; Zhang, L.; Tian, H.; Yang, B.; Wang, E.; Zhu, B. Transcript Annotation of Chinese Sturgeon (Acipenser sinensis) Using Iso-Seq and RNA-Seq Data. Sci. Data 2023, 10, 105. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Zhang, F.; Smith, J.; Kuo, R.; Hou, Z.-C. Full-Length Transcriptome Sequencing from Multiple Tissues of Duck, Anas Platyrhynchos. Sci. Data 2019, 6, 275. [Google Scholar] [CrossRef]
- Tian, K.; Zhang, C.; Gao, C.; Shi, J.; Xu, C.; Xie, W.; Yan, S.; Xiao, C.; Jia, X.; Tian, Y.; et al. Full-Length Transcriptome Sequencing of Seven Tissues of GuShi Chickens. Poult. Sci. 2024, 104, 104697. [Google Scholar] [CrossRef]
- Liu, M.-J.; Gao, J.; Guo, H.-Y.; Zhu, K.-C.; Liu, B.-S.; Zhang, N.; Sun, J.-H.; Zhang, D.-C. Transcriptomics Reveal the Effects of Breeding Temperature on Growth and Metabolism in the Early Developmental Stage of Platax Teira. Biology 2023, 12, 1161. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.D.; Watanabe, C.K. GMAP: A Genomic Mapping and Alignment Program for mRNA and EST Sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef]
- Yan, O.; Pan, J.; Lin, X.; Liu, B.; Guo, H.; Zhu, T.; Zhang, N.; Zhu, K.; Zhang, D. Chromosome-Level Genome and Characteristic Analysis of Platax Teira. South China Fish. Sci. 2024, 20, 31–42. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Rogers, M.F.; Thomas, J.; Reddy, A.S.; Ben-Hur, A. SpliceGrapher: Detecting Patterns of Alternative Splicing from RNA-Seq Data in the Context of Gene Models and EST Data. Genome Biol. 2012, 13, R4. [Google Scholar] [CrossRef]
- Trincado, J.L.; Entizne, J.C.; Hysenaj, G.; Singh, B.; Skalic, M.; Elliott, D.J.; Eyras, E. SUPPA2: Fast, Accurate, and Uncertainty-Aware Differential Splicing Analysis across Multiple Conditions. Genome Biol. 2018, 19, 40. [Google Scholar] [CrossRef]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S.N. A Survey of the Sorghum Transcriptome Using Single-Molecule Long Reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef]
- Zhang, H.-M.; Liu, T.; Liu, C.-J.; Song, S.; Zhang, X.; Liu, W.; Jia, H.; Xue, Y.; Guo, A.-Y. AnimalTFDB 2.0: A Resource for Expression, Prediction and Functional Study of Animal Transcription Factors. Nucleic Acids Res. 2015, 43, D76–D81. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam Protein Families Database: Towards a More Sustainable Future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: Assess the Protein-Coding Potential of Transcripts Using Sequence Features and Support Vector Machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A Tool for Predicting Long Non-Coding RNAs and Messenger RNAs Based on an Improved k-Mer Scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, S.; Shi, Y.; Yan, Y.; Liu, Q. Full-Length Transcriptome of Anadromous Coilia Nasus Using Single Molecule Real-Time (SMRT) Sequencing. Aquac. Fish. 2022, 7, 420–426. [Google Scholar] [CrossRef]
- Cheng, J.; Zhang, L.; Hui, M.; Li, Y.; Sha, Z. Insights into Adaptive Divergence of Japanese Mantis Shrimp Oratosquilla Oratoria Inferred from Comparative Analysis of Full-Length Transcriptomes. Front. Mar. Sci. 2022, 9, 975686. [Google Scholar] [CrossRef]
- Wang, A.; Sha, Z.; Hui, M. Full-Length Transcriptome Comparison Provides Novel Insights into the Molecular Basis of Adaptation to Different Ecological Niches of the Deep-Sea Hydrothermal Vent in Alvinocaridid Shrimps. Diversity 2022, 14, 371. [Google Scholar] [CrossRef]
- Fu, Q.; Zhang, P.; Zhao, S.; Li, Y.; Li, X.; Cao, M.; Yang, N.; Li, C. A Novel Full-Length Transcriptome Resource from Multiple Immune-Related Tissues in Turbot (Scophthalmus maximus) Using Pacbio SMART Sequencing. Fish Shellfish. Immunol. 2022, 129, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Zhang, M.; Yang, N.; Fu, Q.; Su, B.; Zhang, X.; Li, Q.; Yan, X.; Thongda, W.; Li, C. Full Length Transcriptome Profiling Reveals Novel Immune-Related Genes in Black Rockfish (Sebastes schlegelii). Fish Shellfish. Immunol. 2020, 106, 1078–1086. [Google Scholar] [CrossRef]
- Ge, H.; Zhang, H.; Yang, L.; Wang, H.; Tu, L.; Jiang, Z.; Zheng, J.; Chen, B.; Chen, J.; Li, Y.; et al. Full-Length Transcriptome Sequencing from the Longest-Lived Freshwater Bony Fish of the World: Bigmouth Buffalo (Ictiobus cyprinellus). Front. Mar. Sci. 2021, 8, 736188. [Google Scholar] [CrossRef]
- Luo, W.; Wu, Q.; Wang, T.; Xu, Z.; Wang, D.; Wang, Y.; Yang, S.; Long, Y.; Du, Z. Full-Length Transcriptome Analysis of Misgurnus anguillicaudatus. Mar. Genom. 2020, 54, 100785. [Google Scholar] [CrossRef]
- Ao, J.; Li, J.; You, X.; Mu, Y.; Ding, Y.; Mao, K.; Bian, C.; Mu, P.; Shi, Q.; Chen, X. Construction of the High-Density Genetic Linkage Map and Chromosome Map of Large Yellow Croaker (Larimichthys crocea). Int. J. Mol. Sci. 2015, 16, 26237–26248. [Google Scholar] [CrossRef]
- Vij, S.; Kuhl, H.; Kuznetsova, I.S.; Komissarov, A.; Yurchenko, A.A.; Van Heusden, P.; Singh, S.; Thevasagayam, N.M.; Prakki, S.R.S.; Purushothaman, K.; et al. Chromosomal-Level Assembly of the Asian Seabass Genome Using Long Sequence Reads and Multi-Layered Scaffolding. PLoS Genet. 2016, 12, e1005954. [Google Scholar] [CrossRef]
- Gratten, J.; Beraldi, D.; Lowder, B.V.; McRae, A.F.; Visscher, P.M.; Pemberton, J.M.; Slate, J. Compelling Evidence That a Single Nucleotide Substitution in TYRP1 Is Responsible for Coat-Colour Polymorphism in a Free-Living Population of Soay Sheep. Proc. Biol. Sci. 2007, 274, 619–626. [Google Scholar] [CrossRef]
- Fujii, R. The Regulation of Motile Activity in Fish Chromatophores. Pigment. Cell Res. 2000, 13, 300–319. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, S.; Xu, J.; Feng, J.; Mahboob, S.; Al-Ghanim, K.A.; Sun, X.; Xu, P. Comparative Transcriptome Analysis Reveals the Genetic Basis of Skin Color Variation in Common Carp. PLoS ONE 2014, 9, e108200. [Google Scholar] [CrossRef]
- Bhatt, P.; Kumaresan, V.; Palanisamy, R.; Ravichandran, G.; Mala, K.; Amin, S.M.N.; Arshad, A.; Yusoff, F.M.; Arockiaraj, J. A Mini Review on Immune Role of Chemokines and Its Receptors in Snakehead Murrel Channa striatus. Fish Shellfish. Immunol. 2018, 72, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Chen, C.; Lin, F.; Xu, X.; Zhang, X.; Liu, Y.; Li, C.; Cui, P.; Fu, Q. Characterization, Immune Response and Antibacterial Mechanism of CXCL12 in Black Rockfish (Sebastes schlegelii). Int. J. Biol. Macromol. 2025, 309, 143153. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Chai, Y.; Gao, A.; Han, J.; Guo, Y.; Lin, Y.; Wu, L.; Ye, J. CXCL13/CXCR5 Axis Mediates IgM+ B Cell Migration through AKT and STAT3 Signaling Pathways in Nile Tilapia (Oreochromis niloticus). Aquaculture 2024, 591, 741109. [Google Scholar] [CrossRef]
- Yang, M.; Zhou, L.; Wang, H.-Q.; Luo, X.-C.; Dan, X.-M.; Li, Y.-W. Molecular Cloning and Expression Analysis of CCL25 and Its Receptor CCR9s from Epinephelus coioides Post Cryptocaryon irritans Infection. Fish Shellfish. Immunol. 2017, 67, 402–410. [Google Scholar] [CrossRef]
- Han, Z.; Xiao, S.; Li, W.; Ye, K.; Wang, Z.Y. The Identification of Growth, Immune Related Genes and Marker Discovery through Transcriptome in the Yellow Drum (Nibea albiflora). Genes Genom. 2018, 40, 881–891. [Google Scholar] [CrossRef]
- Li, C.; Teng, T.; Shen, F.; Guo, J.; Chen, Y.; Zhu, C.; Ling, Q. Transcriptome Characterization and SSR Discovery in Squaliobarbus Curriculus. J. Ocean. Limnol. 2019, 37, 235–244. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, P.; Mo, Q.; Luo, W.; Du, Z.; Jiang, J.; Yang, S.; Zhao, L.; Gong, Q.; Wang, Y. Comprehensive Analysis of Full-Length Transcriptomes of Schizothorax prenanti by Single-Molecule Long-Read Sequencing. Genomics 2022, 114, 456–464. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, H.; Chen, Y.; Song, M.; Liu, B.; Song, J.; Liu, X.; Li, H. Full-Length Transcriptome Sequencing and Identification of Immune-Related Genes in the Critically Endangered Hucho bleekeri. Dev. Comp. Immunol. 2021, 116, 103934. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, L.; Huang, S.; Wang, G. Full-Length Transcriptome Sequencing of Heliocidaris crassispina Using PacBio Single-Molecule Real-Time Sequencing. Fish Shellfish. Immunol. 2022, 120, 507–514. [Google Scholar] [CrossRef]
- Wu, X.; Gong, Q.; Chen, Y.; Liu, Y.; Song, M.; Li, F.; Li, P.; Lai, J. Full-Length Transcriptome and Analysis of Bmp-Related Genes in Platypharodon extremus. Heliyon 2022, 8, e10783. [Google Scholar] [CrossRef] [PubMed]
- Weidemüller, P.; Kholmatov, M.; Petsalaki, E.; Zaugg, J.B. Transcription Factors: Bridge between Cell Signaling and Gene Regulation. Proteomics 2021, 21, e2000034. [Google Scholar] [CrossRef] [PubMed]
- Rosewell Shaw, A.; Bennett, A.; Fan, D.; Ai, W. Alternative Splicing of KLF4 in Myeloid Cells: Implications for Cellular Plasticity and Trained Immunity in Cancer and Inflammatory Disease. Front. Immunol. 2025, 16, 1585528. [Google Scholar] [CrossRef] [PubMed]
- Salmon, J.M.; Adams, H.; Magor, G.W.; Perkins, A.C. KLF Feedback Loops in Innate Immunity. Front. Immunol. 2025, 16, 1606277. [Google Scholar] [CrossRef]
- Huang, Y.; Ren, Q. A Kruppel-like Factor from Macrobrachium rosenbergii (MrKLF) Involved in Innate Immunity against Pathogen Infection. Fish Shellfish. Immunol. 2019, 95, 519–527. [Google Scholar] [CrossRef]
- Ren, Q.; Liu, Z.; Wu, L.; Yin, G.; Xie, X.; Kong, W.; Zhou, J.; Liu, S. C/EBPβ: The Structure, Regulation, and Its Roles in Inflammation-Related Diseases. Biomed. Pharmacother. 2023, 169, 115938. [Google Scholar] [CrossRef]
- Özdemir, S.; Aydın, Ş.; Laçin, B.B.; Arslan, H. Identification and Characterization of Long Non-Coding RNA (lncRNA) in Cypermethrin and Chlorpyrifos Exposed Zebrafish (Danio rerio) Brain. Chemosphere 2023, 344, 140324. [Google Scholar] [CrossRef]
- Ren, Y.; Li, J.; Guo, L.; Liu, J.N.; Wan, H.; Meng, Q.; Wang, H.; Wang, Z.; Lv, L.; Dong, X.; et al. Full-Length Transcriptome and Long Non-Coding RNA Profiling of Whiteleg Shrimp Penaeus vannamei Hemocytes in Response to Spiroplasma eriocheiris Infection. Fish Shellfish. Immunol. 2020, 106, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, W.; Jiang, L.; Lü, Z.; Liu, M.; Gong, L.; Liu, B.; Liu, L.; Yin, X. Immunity-Associated Long Non-Coding RNA and Expression in Response to Bacterial Infection in Large Yellow Croaker (Larimichthys crocea). Fish Shellfish. Immunol. 2019, 94, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, X.; Zhang, A.; Yang, S.; Li, H. Role of Alternative Splicing in Fish Immunity. Fish Shellfish. Immunol. 2024, 149, 109601. [Google Scholar] [CrossRef]
- Zhang, H.; Jia, J.; Zhai, J. Plant Intron-Splicing Efficiency Database (PISE): Exploring Splicing of ∼1,650,000 Introns in Arabidopsis, Maize, Rice, and Soybean from ~57,000 Public RNA-Seq Libraries. Sci. China Life Sci. 2023, 66, 602–611. [Google Scholar] [CrossRef]
- Schmitz, U.; Pinello, N.; Jia, F.; Alasmari, S.; Ritchie, W.; Keightley, M.-C.; Shini, S.; Lieschke, G.J.; Wong, J.J.-L.; Rasko, J.E.J. Intron Retention Enhances Gene Regulatory Complexity in Vertebrates. Genome Biol. 2017, 18, 216. [Google Scholar] [CrossRef]
- Chaudhary, S.; Khokhar, W.; Jabre, I.; Reddy, A.S.N.; Byrne, L.J.; Wilson, C.M.; Syed, N.H. Alternative Splicing and Protein Diversity: Plants Versus Animals. Front. Plant Sci. 2019, 10, 708. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative Isoform Regulation in Human Tissue Transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef]
- Ren, F.; Zhang, N.; Zhang, L.; Miller, E.; Pu, J.J. Alternative Polyadenylation: A New Frontier in Post Transcriptional Regulation. Biomark. Res. 2020, 8, 67. [Google Scholar] [CrossRef]
- Tan, S.; Zhang, J.; Peng, Y.; Du, W.; Yan, J.; Fang, Q. Integrative Transcriptome Analysis Reveals Alternative Polyadenylation Potentially Contributes to GCRV Early Infection. Front. Microbiol. 2023, 14, 1269164. [Google Scholar] [CrossRef]
- Liu, D.; Yu, H.; Xue, N.; Bao, H.; Gao, Q.; Tian, Y. Alternative Splicing Patterns of Hnrnp Genes in Gill Tissues of Rainbow Trout (Oncorhynchus mykiss) during Salinity Changes. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2024, 271, 110948. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.X.; Xiong, F.; Wu, X.M.; Hu, Y.W. The Expanding and Function of NLRC3 or NLRC3-like in Teleost Fish: Recent Advances and Novel Insights. Dev. Comp. Immunol. 2021, 114, 103859. [Google Scholar] [CrossRef]
- Wang, T.; Yan, B.; Lou, L.; Lin, X.; Yu, T.; Wu, S.; Lu, Q.; Liu, W.; Huang, Z.; Zhang, M.; et al. Nlrc3-like Is Required for Microglia Maintenance in Zebrafish. J. Genet. Genom. 2019, 46, 291–299. [Google Scholar] [CrossRef]
- Fang, H.; Wu, X.M.; Hu, Y.W.; Song, Y.J.; Zhang, J.; Chang, M.X. NLRC3-like 1 Inhibits NOD1-RIPK2 Pathway via Targeting RIPK2. Dev. Comp. Immunol. 2020, 112, 103769. [Google Scholar] [CrossRef]
- Li, F.-L.; Wu, X.-J.; Feng, H.-Y.; Liu, X.-S.; Pan, X.-Y.; Chen, Q.-L.; Li, R.; Wang, C. Harnessing Tyrosine Kinase 2: Breakthroughs in Autoimmune Disease and Cancer Therapy. Bioorganic Chem. 2025, 164, 108802. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Zhu, Y.; Yan, J.; Zhang, L.; Zhu, S.; An, L.; Meng, Q.; Zhang, Z.; Wang, X. Full-Length Transcriptome Sequencing Analysis Reveals Differential Skin Color Regulation in Snakeheads Fish Channa argus. Aquac. Fish. 2024, 9, 590–596. [Google Scholar] [CrossRef]
- Vandooren, J.; Itoh, Y. Alpha-2-Macroglobulin in Inflammation, Immunity and Infections. Front. Immunol. 2021, 12, 803244. [Google Scholar] [CrossRef]
- Ning, J.; Liu, Y.; Gao, F.; Song, C.; Cui, Z. Two Alpha-2 Macroglobulin from Portunus trituberculatus Involved in the Prophenoloxidase System, Phagocytosis and Regulation of Antimicrobial Peptides. Fish Shellfish. Immunol. 2019, 89, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Mohindra, V.; Dangi, T.; Chowdhury, L.M.; Jena, J.K. Tissue Specific Alpha-2-Macroglobulin (A2M) Splice Isoform Diversity in Hilsa Shad, Tenualosa Ilisha (Hamilton, 1822). PLoS ONE 2019, 14, e0216144. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).