
Field-Induced Single-Molecule Magnets of Dysprosium Involving Quinone Derivatives

 , , , ,
, , , ,  and
and 
Abstract
1. Introduction
2. Results and Discussion
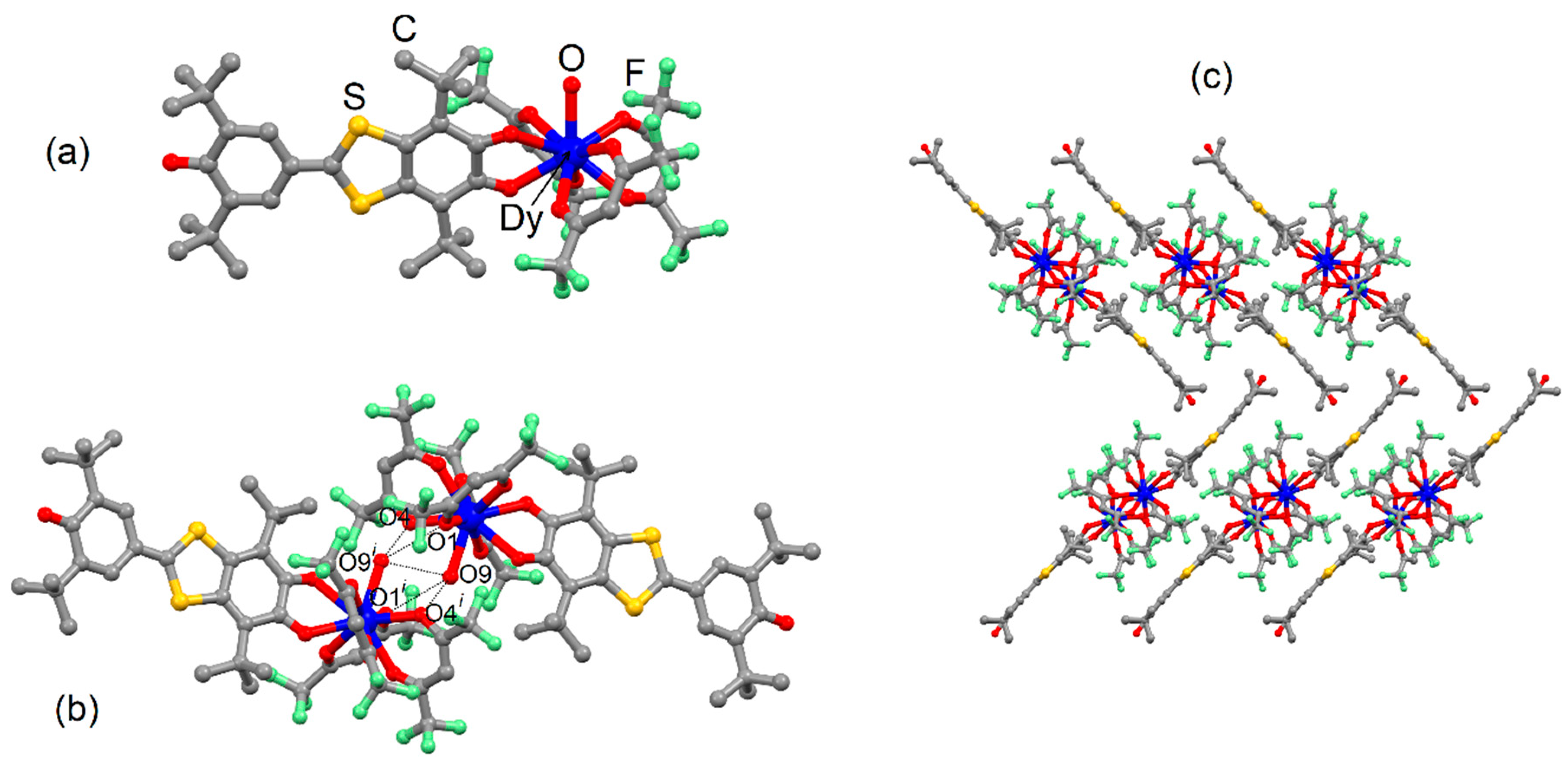
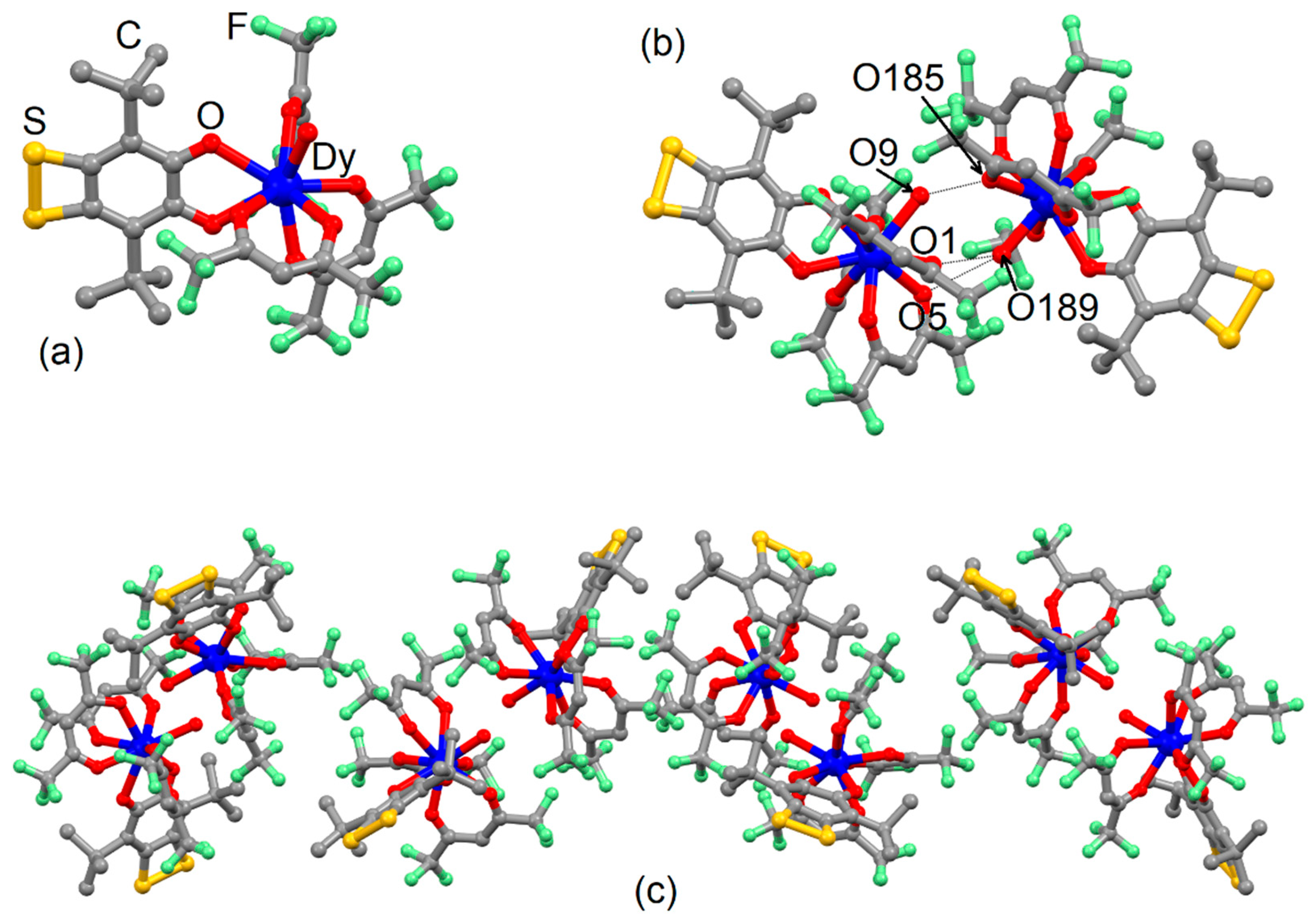
2.1. X-ray Structures
2.2. Magnetic Properties
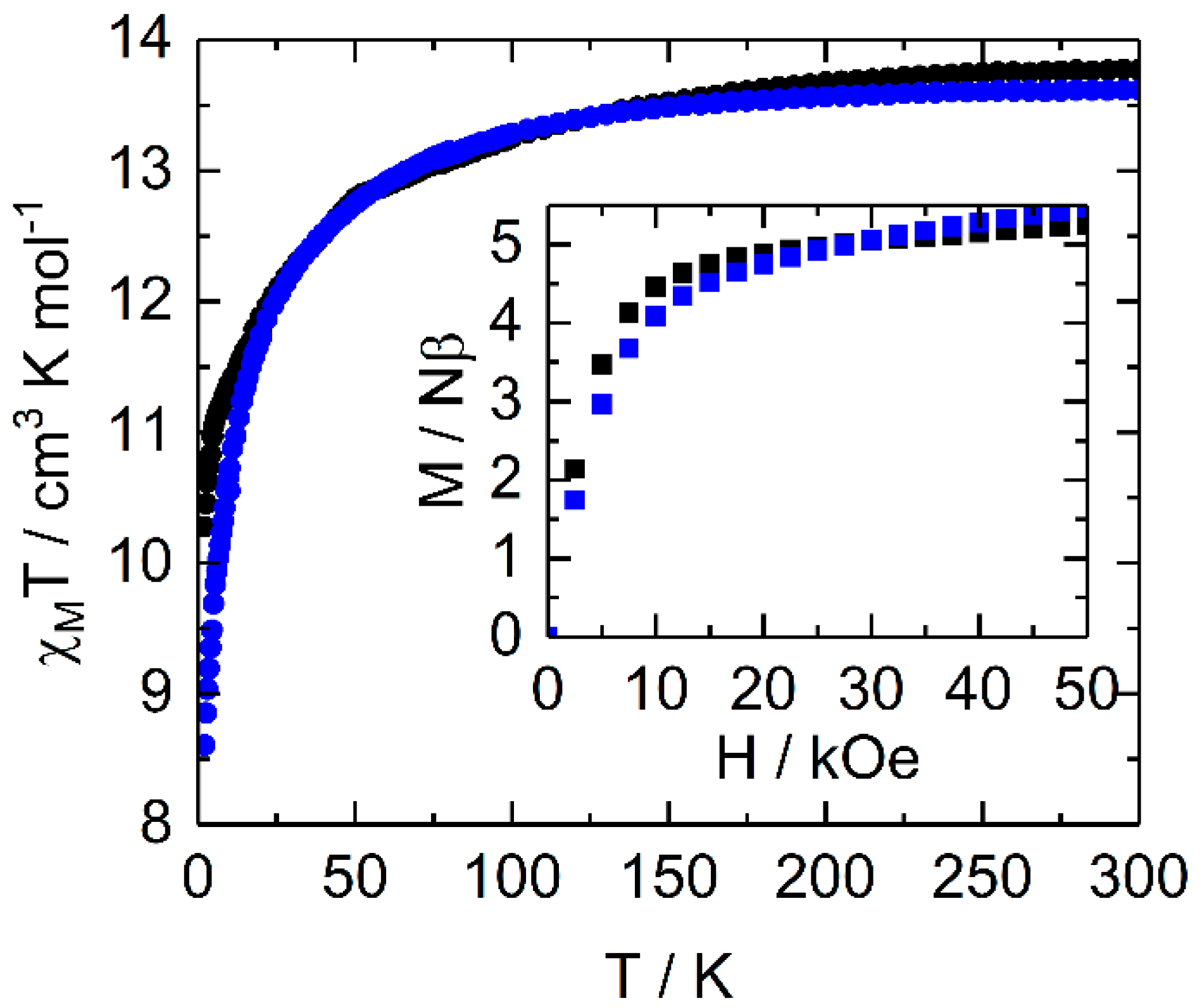
2.2.1. Static Magnetic Measurements
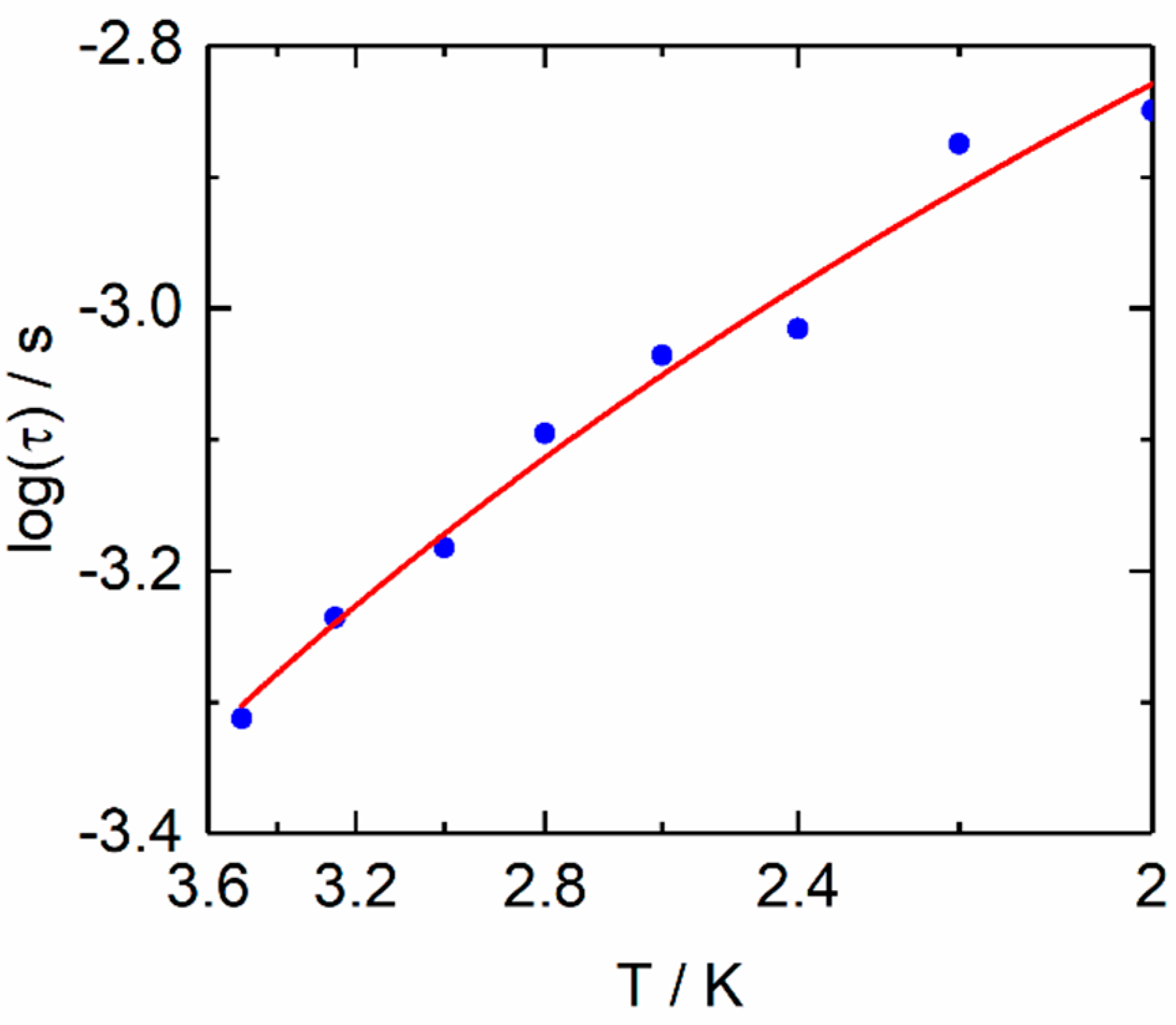
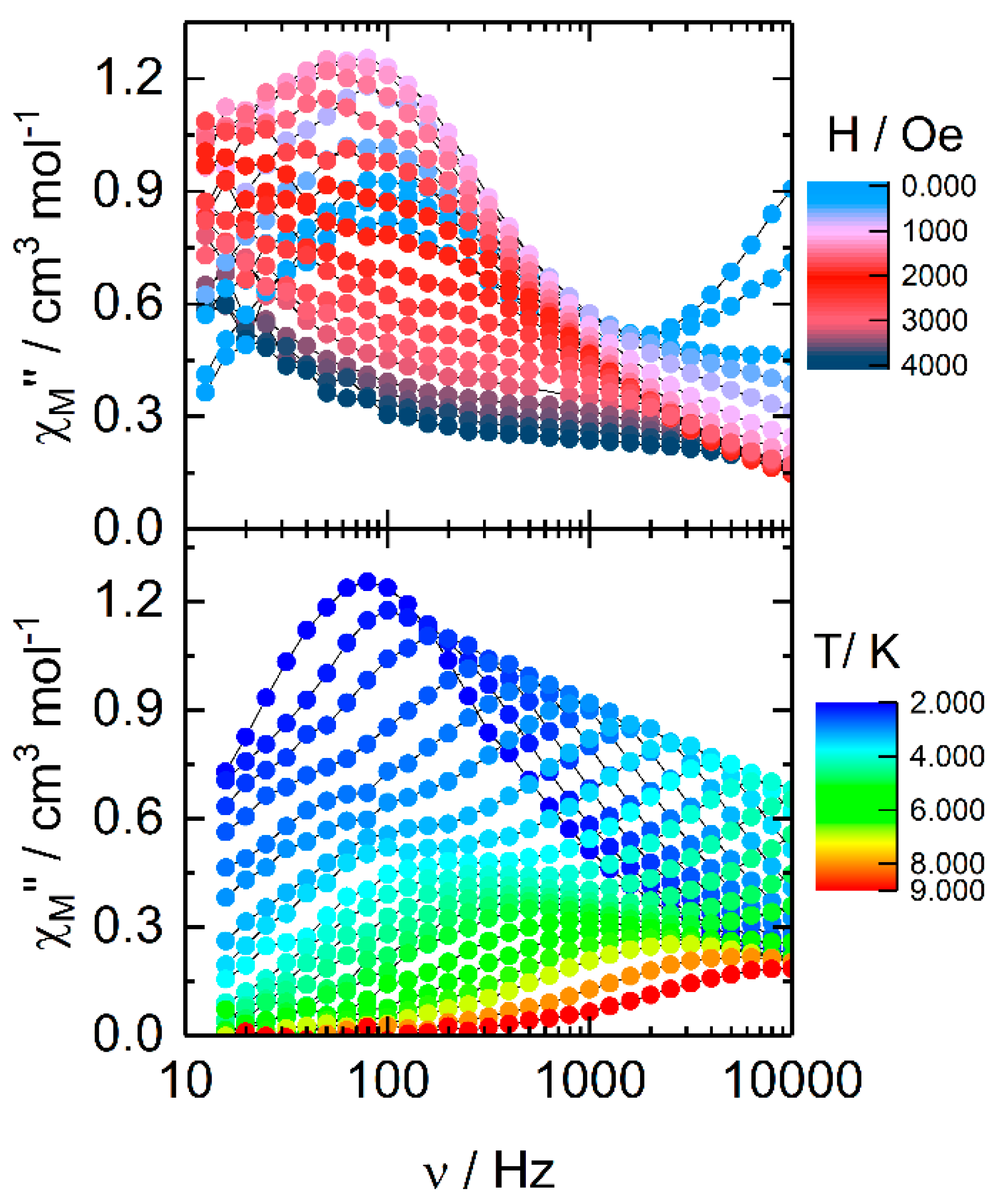
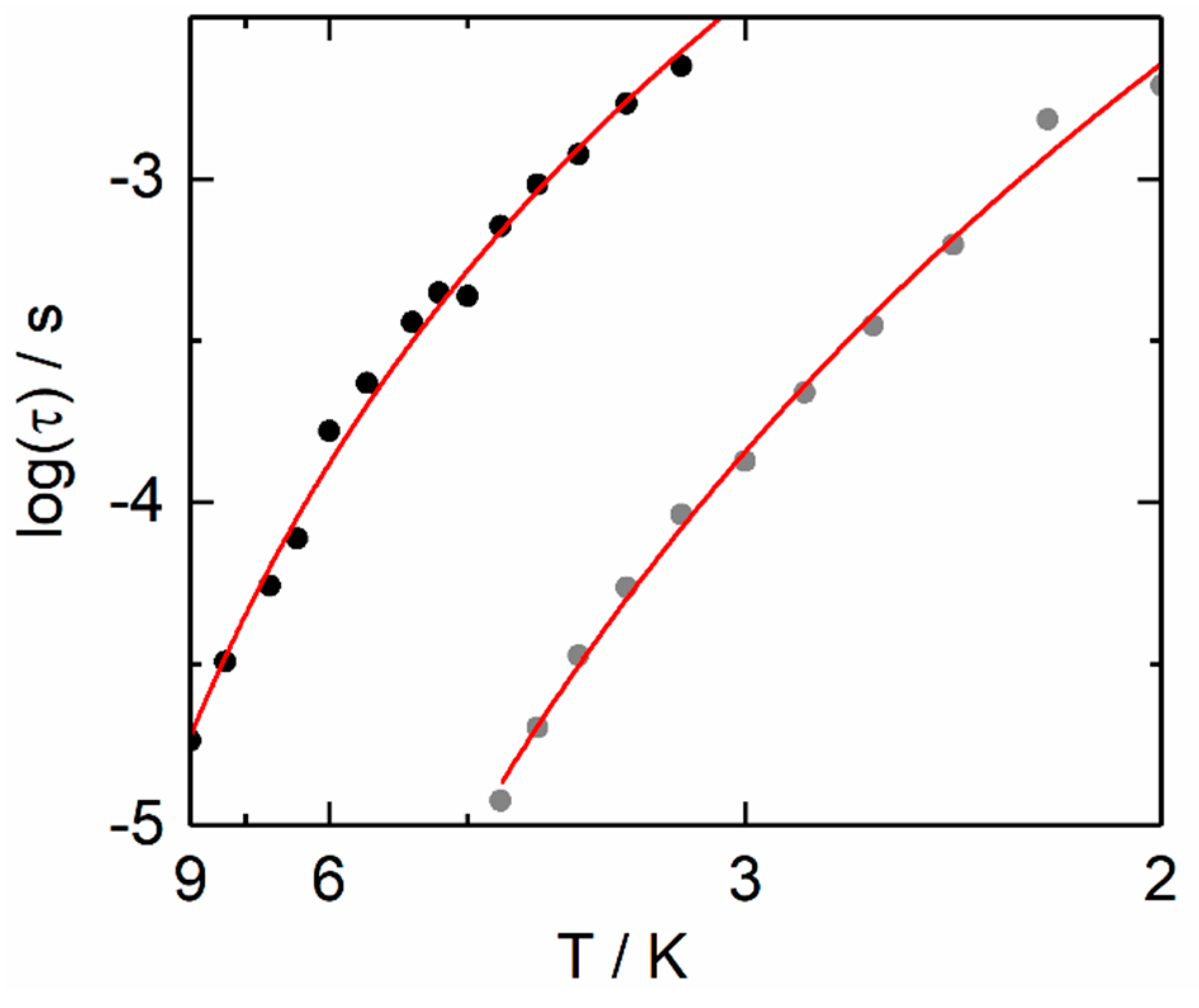
2.2.2. Dynamic Magnetic Measurements
3. Conclusions
4. Materials and Methods
4.1. Synthesis. General Procedures and Materials
4.2. Synthesis of Complexes [Dy(hfac)3(L1)] (1) and [Dy(hfac)3(L2)]⋅(C6H14)(CH2Cl2) (2)
4.3. Crystallography
4.4. Physical Measurements
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TTF | Tetrathiafulvalene |
| SMM | Single Molecule Magnet |
| QTM | Quantum Tunneling of the Magnetization |
| CH2Cl2 | dichloromethane |
| hfac- | 1,1,1,5,5,5-hexafluoroacetylacetonate |
References
- Patai, S. (Ed.) The Chemistry of the Quinoid Compounds, Part 1 and 2; Wiley: New York, NY, USA, 1974. [Google Scholar]
- Pierpont, C.G.; Buchanan, R.M. Transition metal complexes of o-benzoquinone, o-semiquinone, and catecholate ligands. Coord. Chem. Rev. 1981, 38, 45–87. [Google Scholar] [CrossRef]
- Pierpont, C.G.; Lange, C.W. The chemistry of transition metal complexes containing catechol and semiquinone ligands. Prog. Inorg. Chem. 1994, 41, 331–442. [Google Scholar]
- Pierpont, C.G. Studies on Charge Distribution and Valence Tautomerism in Transition Metal Complexes of Catecholate and Semiquinonate Ligands. Coord. Chem. Rev. 2001, 216-217, 99–125. [Google Scholar] [CrossRef]
- Bubnov, M.P.; Kozhanov, K.A.; Skorodumova, N.A.; Cherkasov, V.K. Photo- and Thermosensitive Molecular Crystals. Valence Tautomeric Interconversion as the Cause of the Photomechanical Effect in Crystals of Bis(dioxolene)cobalt Complex. Inorg. Chem. 2020, 59, 6679–6683. [Google Scholar] [CrossRef] [PubMed]
- Caneschi, A.; Dei, A.; Gatteschi, D.; Sorace, L.; Vostrikova, K. Antiferromagnetic coupling in a gadolinium(III) semiquinonato complex. Angew. Chem. Int. Ed. 2000, 39, 246–248. [Google Scholar] [CrossRef]
- Caneschi, A.; Dei, A.; Gatteschi, D.; Massa, C.A.; Pardi, L.A.; Poussereau, S.; Sorace, L. Evaluating the magnetic anisotropy in molecular rare earth compounds. Gadolinium derivatives with semiquinone radical and diamagnetic analogues. Chem. Phys. Lett. 2003, 371, 694–699. [Google Scholar] [CrossRef]
- Caneschi, A.; Dei, A.; Gatteschi, D.; Poussereau, S.; Sorace, L. Antiferromagnetic coupling between rare earth ions and semiquinones in a series of 1:1 complexes. Dalton Trans. 2004, 7, 1048–1055. [Google Scholar] [CrossRef]
- Claiser, N.; Souhassou, M.; Lecomte, C.; Gillon, B.; Carbonara, C.; Caneschi, A.; Dei, A.; Gatteschi, D.; Bencini, A.; Pontillon, Y.; et al. Combined charge and spin density experimental study of the yttrium(III) semiquinonato complex Y(HBPz(3))(2)(DTBSQ) and DFT calculations. J. Phys. Chem. B 2005, 109, 2723–2732. [Google Scholar] [CrossRef]
- Reynolds, R.A.; Coucouvanis, D. Mixed Carboxylate/Catecholate Polynuclear Complexes. Integral Association of Trivalent Lanthanide Ions with Mn4 Units in the Pentanuclear Mn4LaCl and Mn4Tb Clusters. J. Am. Chem. Soc. 1998, 120, 209–210. [Google Scholar] [CrossRef]
- Zhu, D.-H.; Kappel, M.J.; Raymond, K.N. Coordination chemistry of lanthanide catecholates. Inorg. Chim. Acta 1988, 147, 115–121. [Google Scholar] [CrossRef]
- Kuzyaev, D.M.; Vorozhtsov, D.L.; Druzhkov, N.O.; Lopatin, M.A.; Baranov, E.V.; Cherkasov, A.V.; Fukin, G.K.; Abakumov, G.A.; Bochkarev, M.N. 3,5-Di-tert-butyl-o-benzoquinone complexes of lanthanides. J. Organomet. Chem. 2012, 698, 35–41. [Google Scholar] [CrossRef]
- Pointillart, F.; Kuropatov, V.; Mitin, A.; Maury, O.; Le Gal, Y.; Golhen, S.; Cador, O.; Cherkasov, V.; Ouahab, L. Lanthanide-Based Dinuclear Complexes Involving an o-Quinone-Tetrathiafulvalene-o-Quinone Bridging Ligand: X-ray Structures, Magnetic and Photophysical properties. Eur. J. Inorg. Chem. 2012, 4708–4718. [Google Scholar] [CrossRef]
- Pointillart, F.; Klementieva, S.; Kuropatov, V.; Le Gal, Y.; Golhen, S.; Cador, O.; Cherkasov, V.; Ouahab, L. A single molecule magnet behavior in a D3h symmetry Dy(III) complex involving a quinone-tetrathiafulvalene-quinone bridge. Chem. Commun. 2012, 48, 714–716. [Google Scholar] [CrossRef]
- Dunstan, M.A.; Rousset, E.; Boulon, M.-E.; Gable, R.W.; Sorace, L.; Boskovic, C. Slow magnetisation relaxation in tetraoxolene-bridged rare earth complexes. Dalton Trans. 2017, 46, 13756–13767. [Google Scholar] [CrossRef]
- Gonzalez, J.F.; Cador, O.; Ouahab, O.; Norkov, S.; Kuropatov, V.; Pointillart, F. Field-Induced Dysprosium Single-Molecule Magnet Involving a Fused o-Semiquinone-Extended-Tetrathiafulvalene-o-Semiquinone Bridging Triad. Inorganics 2018, 6, 45. [Google Scholar] [CrossRef]
- Pointillart, F.; Gonzalez, J.F.; Montigaud, V.; Tesi, L.; Cherkasov, V.; Le Guennic, B.; Cador, O.; Ouahab, L.; Sessoli, R.; Kuropatov, V. Redox- and solvato-magnetic switching in a tetrathiafulvalene-based triad single-molecule magnet. Inorg. Chem. Front. 2020, 7, 2322–2334. [Google Scholar] [CrossRef]
- Tiaouinine, S.; Gonzalez, J.F.; Montigaud, V.; Mattei, C.A.; Dorect, V.; Kaboub, L.; Cherkasov, V.; Cador, O.; Le Guennic, B.; Ouahab, L.; et al. Redox Modulation of Field-Induced Tetrathiafulvalene-Based Single-Molecule Magnets of Dysprosium. Magnetochemistry 2020, 6, 34. [Google Scholar] [CrossRef]
- Zhang, P.; Perfetti, M.; Kern, M.; Hallmen, P.P.; Ungur, L.; Lenz, S.; Ringenberg, M.R.; Trey, W.; Stoll, H.; Rauhut, G.; et al. Exchange coupling and single molecule magnetism in redox-active tetraoxolene-bridged dilanthanide complexes. Chem. Sci. 2018, 9, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Lefeuvre, B.; Gonzalez, J.F.; Gendron, F.; Dorcet, V.; Riobé, F.; Cherkasov, V.; Maury, O.; Le Guennic, B.; Cador, O.; Kuropatov, V.; et al. Redox-Modulations of Photophysical and Single-Molecule Magnet Properties in Ytterbium Complexes Involving Extended-TTF Triads. Molecules 2020, 25, 492. [Google Scholar] [CrossRef]
- Guo, F.-S.; Day, B.-M.; Chen, Y.-C.; Tong, M.-L.; Mansikkamäki, A.; Layfield, R.A. A Dysprosium Metallocene Single-Molecule Magnet Functioning at the Axial Limit. Angew. Chem. Int. Ed. 2017, 56, 11445–11449. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, C.A.P.; Ortu, F.; Reta, D.; Chilton, N.F.; Mills, D.P. Molecular magnetic hysteresis at 60 kelvin in dysprosocenium. Nature 2017, 548, 439–442. [Google Scholar] [CrossRef]
- McClain, K.R.; Gould, C.A.; Chakarawet, K.; Teat, S.J.; Groshens, T.J.; Long, J.R.; Harvey, B.G. High temperature magnetic blocking and magneto-structural correlations in a series of dysprosium(III) metallocenium single-molecule magnets. Chem. Sci. 2018, 9, 8492–8503. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.-S.; Day, B.-M.; Chen, Y.-C.; Tong, M.-L.; Mansikkamäki, A.; Layfield, R.A. Magnetic hysteresis up to 80 kelvin in a dysprosium metallocene single-molecule magnet. Science 2018, 362, 1400–1403. [Google Scholar] [CrossRef] [PubMed]
- Martyanov, K.; Kuropatov, V. Functionalized o-Quinones: Concepts, Achievements and Prospects. Inorganics 2018, 6, 48. [Google Scholar] [CrossRef]
- Valade, L.; Tanaka, H. Molecular Materials; Bruce, D.W., O’Hare, D., Walton, R.I., Eds.; John & Sons, Inc.: Hoboken, NJ, USA, 2010. [Google Scholar]
- Zadrozny, J.M.; Niklas, J.; Poluektov, O.G.; Freedman, D.E. Millisecond Coherence Time in a Tunable Molecular Electronic Spin Qubit. ACS Cent. Sci. 2015, 1, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Atzori, M.; Morra, E.; Tesi, L.; Albino, A.; Chiesa, M.; Sorace, L.; Sessoli, R. Quantum Coherence Times Enhancement in Vanadium(IV)-based Potential Molecular Qubits: The key Role of the Vanadyl Moiety. J. Am. Chem. Soc. 2016, 138, 11234–11244. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.J.; Yu, C.-J.; Krzyaniak, M.D.; Wasielewski, M.R.; Freedman, D.E. Synthetic Approach To Determine the Effect of Nuclear Spin Distance on Electronic Spin Decoherence. J. Am. Chem. Soc. 2017, 139, 3196–3201. [Google Scholar] [CrossRef] [PubMed]
- Norkov, S.V.; Cherkasov, A.V.; Shavyrin, A.S.; Arsenyev, M.V.; Kuropatov, V.A.; Cherkasov, V.K. Annulation of 1,3-dithiole ring to sterically hindered o-quinone core. Novel ditopic redox-active ligands. Beilstein J. Org. Chem. 2021, 17, 273–282. [Google Scholar] [CrossRef]
- Cherkasov, V.K.; Abakumov, G.A.; Fukin, G.K.; Klementyeva, S.V.; Kuropatov, V.A. Sterically Hindered o-Quinone Annulated with Dithiete: A Molecule Comprising Diolate and Dithiolate Coordination Sites. Chem. Eur. J. 2012, 18, 13821–13827. [Google Scholar] [CrossRef]
- Martyanov, K.A.; Cherkasov, V.K.; Abakumov, G.A.; Samsonov, M.A.; Khrizanforova, V.V.; Budnikova, Y.H.; Kuropatov, V.A. New sterically-hindered o-quinones annelated with metal-dithiolates: Regiospecificity in oxidative addition reactions of a bifacial ligand to the Pd and Pt complexes. Dalton Trans. 2016, 45, 7400–7405. [Google Scholar] [CrossRef]
- Martyanov, K.A.; Cherkasov, V.K.; Abakumov, G.A.; Baranov, E.V.; Shavyrin, A.S.; Kuropatov, V.A. Regioisomerism in coordination chemistry: Oxidative addition of a bifunctional ligand to palladium, stabilized with 1,2-bis(diphenylphosphino)ethane. Dalton Trans. 2017, 46, 16783–16786. [Google Scholar] [CrossRef]
- Martyanov, K.A.; Abakumov, G.A.; Baranov, E.V.; Khrizanforova, V.V.; Khrizanforov, M.N.; Kholin, K.V.; Budnikova, Y.H.; Kuropatov, V.A.; Cherkasov, V.K. PdII(P-P) derivatives of o-quinone annulated with dithiete cycle: Electrochemical properties and coordination regioisomerism. Eur. J. Inorg. Chem. 2020, 2020, 4350–4357. [Google Scholar] [CrossRef]
- Llunell, M.; Casanova, D.; Cirera, J.; Alemany, P.; Alvarez, S. SHAPE Program for the Stereochemical Analysis of Molecular Fragments by Means of Continuous Shape Measures and Associated Tools; Departament de Quimica Fisica, Departament de Quimica Inorganica and Institut de Quimica Teorica i Computacional—Universitat de Barcelona: Barcelona, Spain, 2013. [Google Scholar]
- Kahn, O. Molecular Magnetism; VCH: Weinhem, Germany, 1993. [Google Scholar]
- Dekker, C.; Arts, A.F.M.; Wijn, H.W.; van Duyneveldt, A.J.; Mydosh, J.A. Activated dynamics in a two dimensional Ising spin glass: Rb2Cu1-xCoxF4. Phys. Rev. B 1989, 40, 11243–11251. [Google Scholar] [CrossRef] [PubMed]
- Cole, K.S.; Cole, R.H. Dipersion and Absorption in Dielectrics I. Alternating Current Characteristics. J. Chem. Phys. 1941, 9, 341–351. [Google Scholar] [CrossRef]
- Singh, A.; Shrivastava, K.N. Optical-acoustic two-phonon relaxation in spin systems. Phys. Status Solidi B 1979, 95, 273–277. [Google Scholar] [CrossRef]
- Shirivastava, K.N. Theory of Spin-Lattice Relaxation. Phys. Status Solidi B 1983, 177, 437–458. [Google Scholar] [CrossRef]
- Goodwin, C.A.P.; Reta, D.; Ortu, F.; Chilton, N.F.; Mills, D.P. Synthesis and Electronic Structures of Heavy Lanthanide Metallocenium Cations. J. Am. Chem. Soc. 2017, 139, 18714–18724. [Google Scholar] [CrossRef]
- Abragam, A.; Bleaney, B. Electron Paramagnetic Resonance of Transition Ions; Clarendon Press: Oxford, UK, 1970. [Google Scholar]
- Mattei, C.A.; Montigaud, V.; Gendron, F.; Denis-Quanquin, S.; Dorcet, V.; Giraud, N.; Riobé, F.; Argouarch, G.; Maury, O.; Le Guennic, B.; et al. Solid-state versus solution investigation of a luminescent chiral BINOL-derived bisphosphate single-molecule magnet. Inorg. Chem. Front. 2021. Advance Article. [Google Scholar] [CrossRef]
- Mattei, C.A.; Montigaud, V.; Dorect, V.; Riobé, F.; Argouarch, G.; Maury, O.; Le Guennic, B.; Cador, O.; Lalli, C.; Pointillart, F. Luminescent dysprosium single-molecule magnets made from designed chiral BINOL-derived bisphosphate ligands. Inorg. Chem. Front 2021. Advance Article. [Google Scholar] [CrossRef]
- Zhang, W.-Y.; Tian, Y.-M.; Li, H.-F.; Chen, P.; Sun, W.-B.; Zhang, Y.-Q.; Yan, P.-F. A series of dinuclear Dy(III) complexes bridged by 2-methyl-8-hydroxylquioline: Replacement on the periphery coordinated β-diketonate terminal leads to different single-molecule magnetic properties. Dalton Trans. 2016, 45, 3863–3873. [Google Scholar] [CrossRef]
- Diaz-Ortega, I.F.; Herrera, J.M.; Aravena, D.; Ruiz, E.; Gupta, T.; Rajaraman, G.; Nojiri, H.; Colacio, E. Designing a Dy2 Dingle-Molecule Magnet with Two Well-Differentiated Relaxation Processes by using a Nonsymmetric Bis-bidentate Bipyrimidine-N-oxide Ligand: A comparison with mononuclear Counterpart. Inorg. Chem. 2018, 57, 6362–6375. [Google Scholar] [CrossRef]
- Fondo, M.; Corredoira-Vazquez, J.; Garcia-Deibe, A.M.; Sanmartin-Matalobos, J.; Herrera, J.M.; Colacio, E. Field-Induced Single Molecule Magnets of Phosphine- and Asine-Oxides. Front. Chem. 2018, 6, 420. [Google Scholar] [CrossRef] [PubMed]
- Cen, P.; Liu, X.; Ferrando-Soria, J.; Zhang, Y.-Q.; Xie, G.; Chen, S.; Pardo, E. Capping N-Donor Ligands Modulate the Magnetic Dynamics of DyIII β-Diketonate Single-Ion Magnets with D4d Symmetry. Chem. Eur. J. 2019, 25, 3884–3892. [Google Scholar] [CrossRef] [PubMed]
- Errulat, D.; Gabidullin, B.; Mansikkamaki, A.; Murugesu, M. Two Heads are Better than One: Improving Magnetic Relaxation in the Dysprosium Metallocene upon Dimerization by Use of an Exceptionally Weakly-Coordinating Anion. Chem. Commun. 2020, 56, 5937–5940. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M.F.; Wagner, W.F.; Sands, D.E. Rare-earth trishexafluoroacetylacetonates and related compounds. J. Inorg. Nucl. Chem. 1968, 30, 1275–1289. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Local Geometries Around LnIII Centers | Magnetic Properties (Field Induced SMM) | Ref. |
|---|---|---|---|
| [Dy2(tta)6(Q)]∙2CH2Cl2 (tta = 2-thenoyltrifluoroacetylacetonate, Q = 2,2′-cyclohexa-2,5-diene-1,4-diylidenebis(4,7-di-tert-butyl-1,3-benzodithiole-5,6-dione) | Square antiprism geometry | H = 1200 Oe Ueff = 26.5(2) K τ0 = 1.9(7) × 10−7 s C = 289(93) s−1 K−n n = 1.88(39) | [17] |
| [(Dy(HB(pz)3)2)2(μ-tetraoxolene)]·2CH2Cl2 (HB(pz)3 = hydrotris(pyrazolyl)borate (μ-tetraoxolene = chloranilate) | Triangular dodecahedral geometry | H = 1600 Oe Ueff = 24 K τ0 = 3 × 10−5 s C = 2.7 × 10−3 s−1 K−n n = 7 (fixed) | [15] |
| [Dy(acac)2(CH3OH)]2(μ-Mq) (acac = acetylacetone) (μ-Mq = 2-methyl-8-hydroxyquinoline) | Hexagonal bipyramid geometry | H = 2000 Oe Ueff = 75.6 K τ0 = 2.1 × 10−8 s C = 0.76 s−1 K−n n = 3.4 | [45] |
| [Dy(tta)3(mbpymNO)] (mbpymNO = 4-methylbipyrimidine-2-N-oxide) | Square antiprism geometry | H = 1000 Oe Ueff = 99.6 K τ0 = 2.1 × 10−9 s C = 0.24 s−1 K−n n = 4.9 | [46] |
| [Dy(tta)3(phenNO)] (phenNO = 1,10-phenantroline-1-oxide) | Square antiprism geometry | H = 1000 Oe Ueff = 159.1 K τ0 = 1.9 × 10−10 s C = 5 × 10−3 s−1 K−n n = 5.2 | [46] |
| [Dy(tmhd)3(dppz)] (tmhd = 2,2,6,6-tetramethyl-3,5-heptanedione) (dppz = dipyrido[3,2-a:2′,3′-c]phenazine) | Distorted square-antiprism | H = 1000 Oe Ueff = 247.8 K τ0 = 2.22 × 10−11 s C = 4.28 × 10−4 s−1 K−n n = 5.43 | [48] |
| [Dy2Cp *4(μ-BPh4)][Al(OC(CF3)3)4] (Cp * = 1,2,3,4,5-pentamethylcyclopentadiene) (BPh4 = tetraphenylborate) | / | H = 1000 Oe Ueff = 490 K τ0 = 1.785 × 10−8 s C = 1.50 × 10−4 s−1 K−n n = 3.86 | [49] |
| [Dy(hfac)3(L1)]n | Square-antiprism geometry | H = 1000 Oe C = 0.12(9) s−1 K−n n = 6.10(48) | [43] |
| {[Dy(hfac)3(L2)]⋅0.5(CH2Cl2)⋅2(C6H14) | Square-antiprism geometry | H = 1000 Oe C = 0.71(6) s−1 K−n n = 3.75(25) | [44] |
| [Dy(hfac)3(L3)]n | Square-antiprism geometry | H = 1000 Oe C = 125(36) s−1 K−n n = 2.82(13) | [44] |
| [Dy(hfac)3(L4)]n | Square-antiprism geometry | H = 1000 Oe C = 48.8(41) s−1 K−n n = 2.73(6) | [44] |
| (1) | spherical tricapped trigonal prism geometry | H = 1000 Oe C = 175(19) s−1 K−n n = 1.95(11) | this work |
| (2) | spherical tricapped trigonal prism geometry and spherical capped square antiprism | H = 1000 Oe C = 3.93(69) and 1.44(27) s−1 K−n n = 6.81(16) and 7.78(11) | this work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martyanov, K.; Flores Gonzalez, J.; Norkov, S.; Lefeuvre, B.; Dorcet, V.; Cherkasov, V.; Cador, O.; Kuropatov, V.; Pointillart, F. Field-Induced Single-Molecule Magnets of Dysprosium Involving Quinone Derivatives. Magnetochemistry 2021, 7, 24. https://doi.org/10.3390/magnetochemistry7020024
Martyanov K, Flores Gonzalez J, Norkov S, Lefeuvre B, Dorcet V, Cherkasov V, Cador O, Kuropatov V, Pointillart F. Field-Induced Single-Molecule Magnets of Dysprosium Involving Quinone Derivatives. Magnetochemistry. 2021; 7(2):24. https://doi.org/10.3390/magnetochemistry7020024
Chicago/Turabian StyleMartyanov, Konstantin, Jessica Flores Gonzalez, Sergey Norkov, Bertrand Lefeuvre, Vincent Dorcet, Vladimir Cherkasov, Olivier Cador, Viacheslav Kuropatov, and Fabrice Pointillart. 2021. "Field-Induced Single-Molecule Magnets of Dysprosium Involving Quinone Derivatives" Magnetochemistry 7, no. 2: 24. https://doi.org/10.3390/magnetochemistry7020024
APA StyleMartyanov, K., Flores Gonzalez, J., Norkov, S., Lefeuvre, B., Dorcet, V., Cherkasov, V., Cador, O., Kuropatov, V., & Pointillart, F. (2021). Field-Induced Single-Molecule Magnets of Dysprosium Involving Quinone Derivatives. Magnetochemistry, 7(2), 24. https://doi.org/10.3390/magnetochemistry7020024