
Genome Polymorphism Analysis and Selected Sweep Regions Detection via the Genome Resequencing of 91 Cabbage (Brassica oleracea) Accessions



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Material Culture and DNA Extraction
2.2. Database Building and Sequencing
2.3. Processing and Evaluation of Sequencing Data
2.4. Genomic SNP Detection, Filtering and Annotation
2.5. Population Evolution Analysis
2.6. Population Selection Analysis and Gene Function Enrichment Analysis
3. Results and Discussion
3.1. Sequencing Data Statistics and Quality Evaluation
3.2. Resequencing Data Were Mapped with Reference Genome
3.3. SNP Detection and Annotation
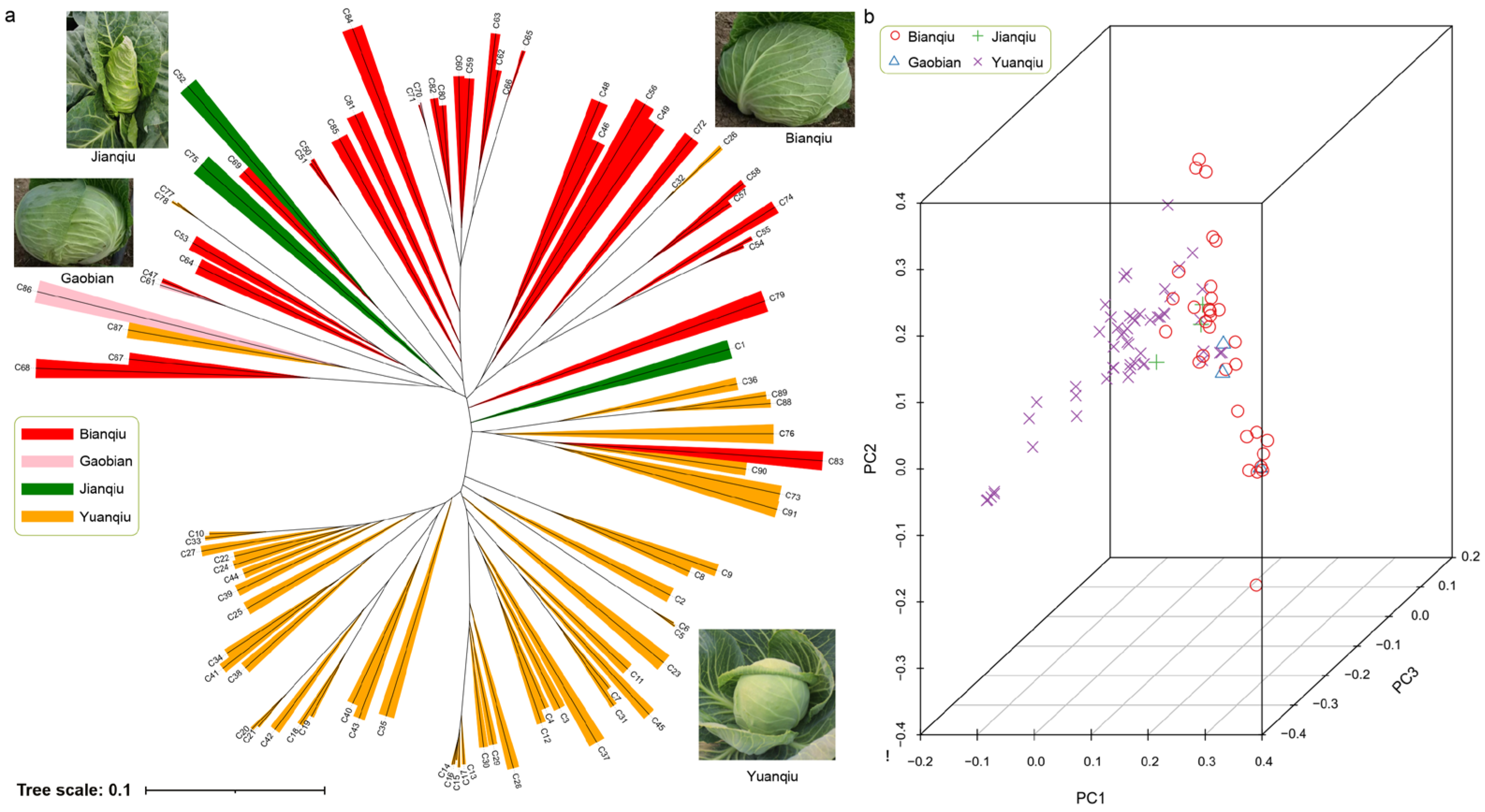
3.4. Population Evolution and Principal Component Analysis
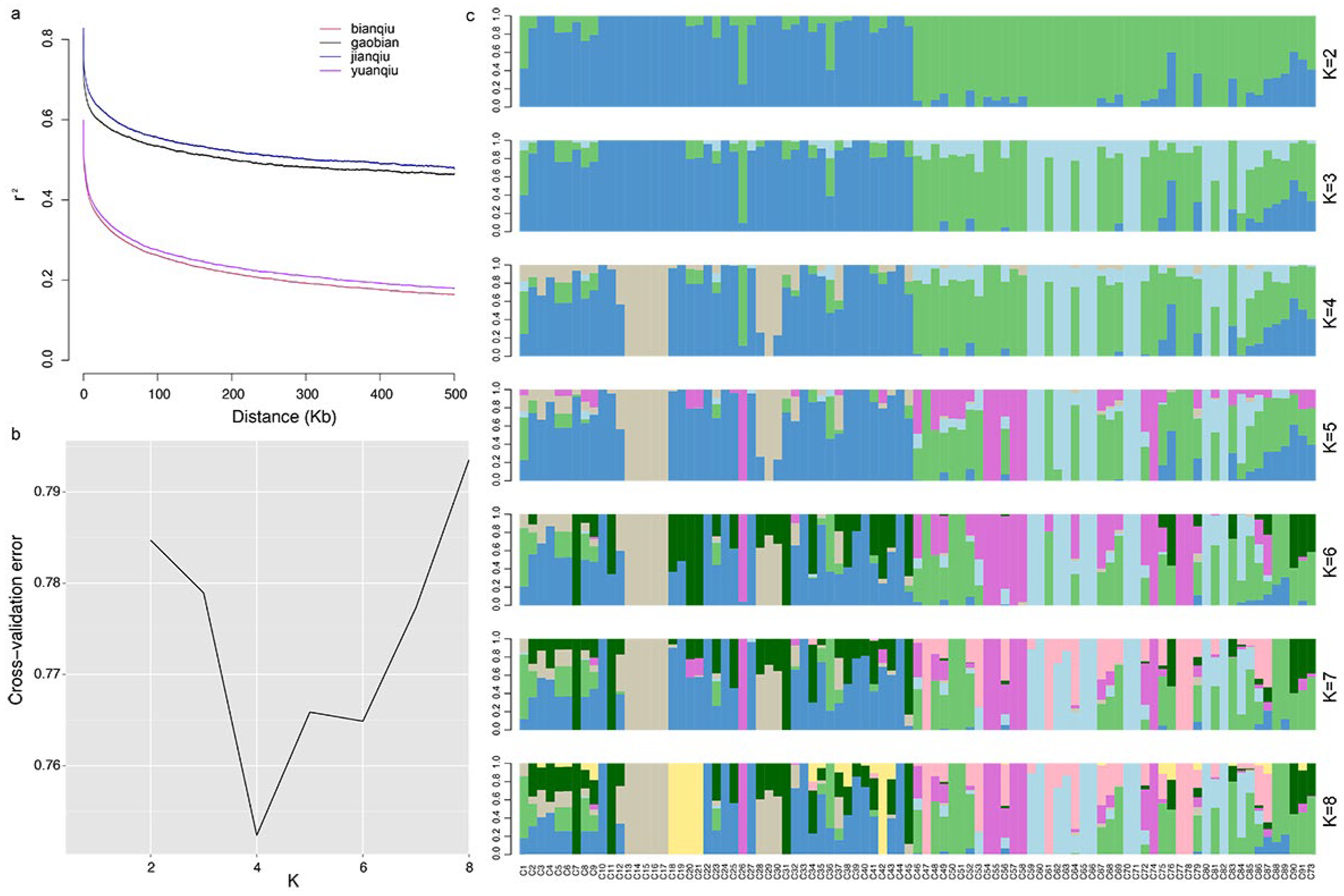
3.5. Linkage Disequilibrium Analysis
3.6. Analysis of Population Genetic Structure
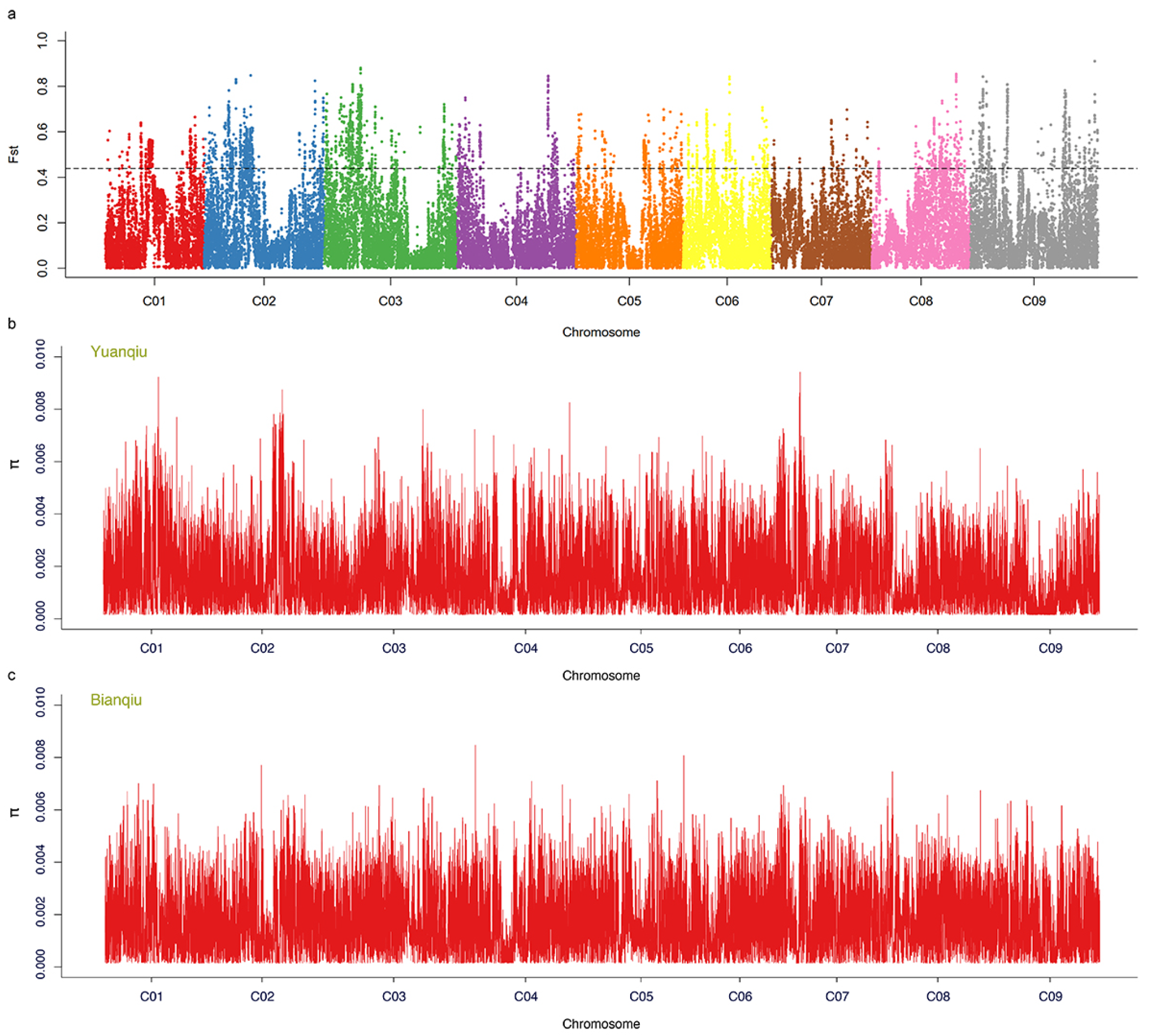
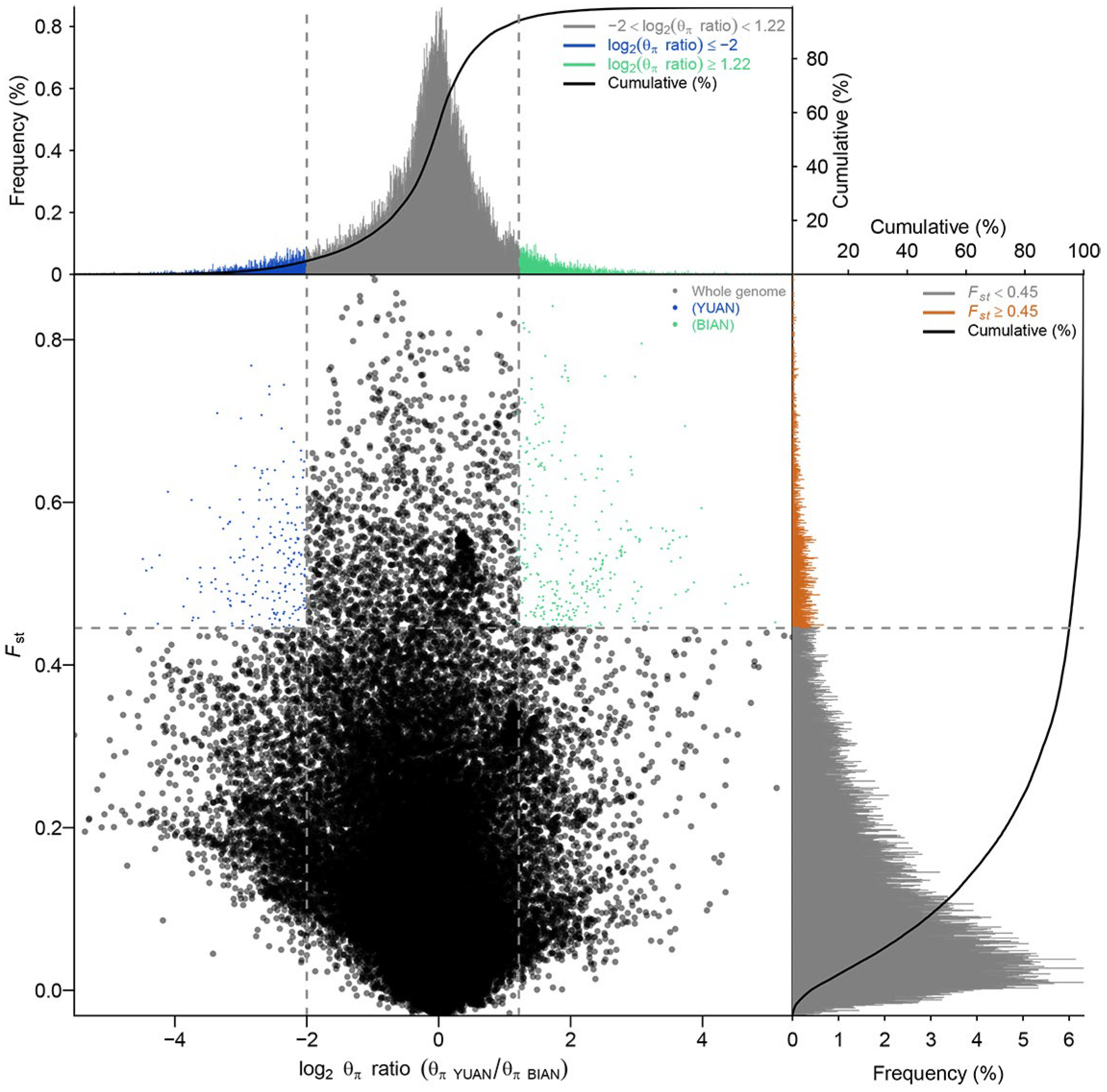
3.7. Selective Sweep Analysis
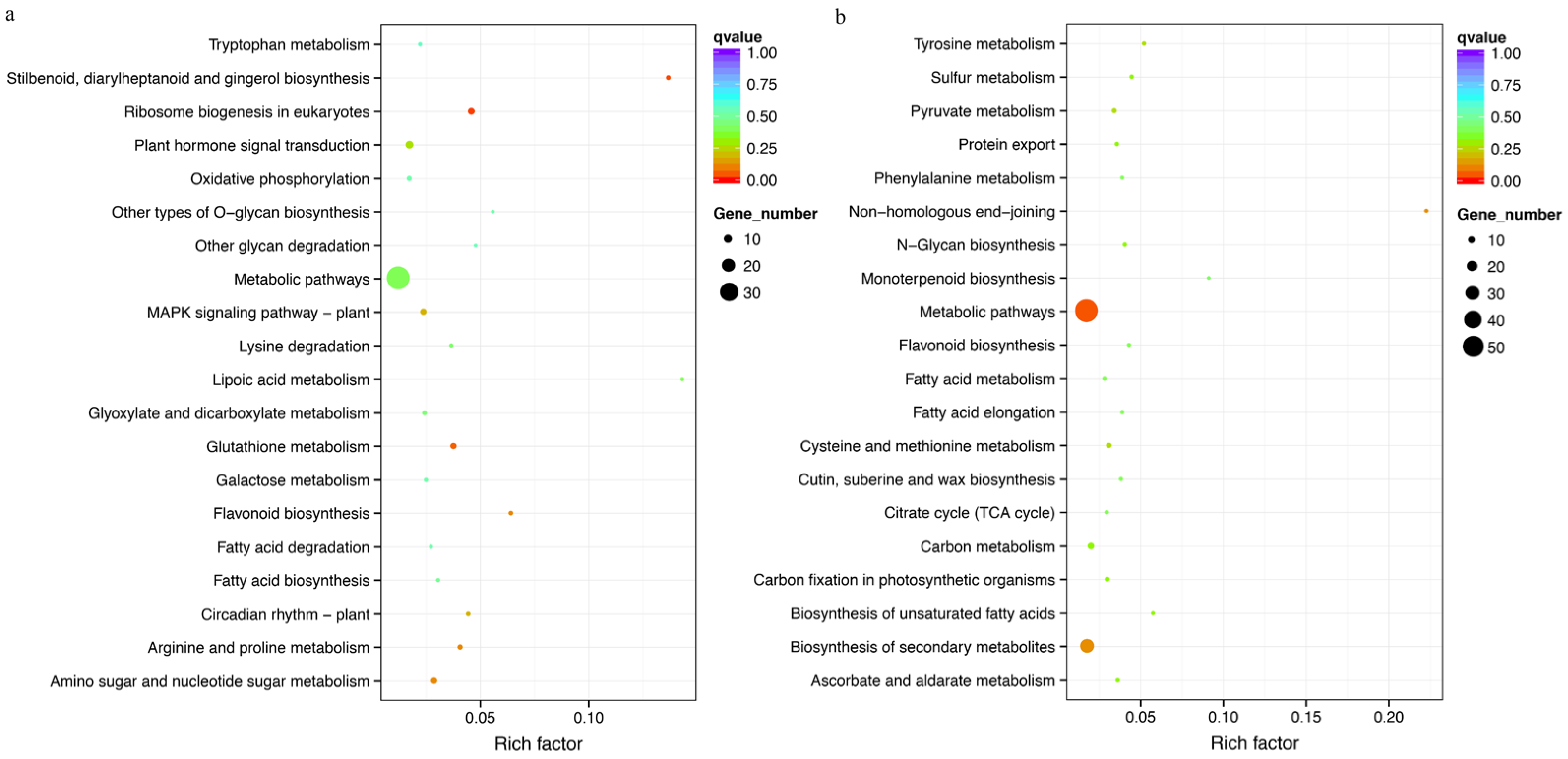
3.8. Gene Function Enrichment Analysis of Selection Sweep Regions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weng, Y. Inaugural Editorial: Vegetable Research. Veg. Res. 2021, 1, 1. [Google Scholar] [CrossRef]
- Yu, T.; Ma, X.; Liu, Z.; Feng, X.; Wang, Z.; Ren, J.; Cao, R.; Zhang, Y.; Nie, F.; Song, X. TVIR: A comprehensive vegetable information resource database for comparative and functional genomic studies. Hortic Res. 2022, 9, uhac213. [Google Scholar] [CrossRef]
- Liu, S.; Liu, Y.; Yang, X.; Tong, C.; Edwards, D.; Parkin, I.A.; Zhao, M.; Ma, J.; Yu, J.; Huang, S.; et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 2014, 5, 3930. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, M.; Li, S.; Chen, Q.; Teixeira da Silva, J.A.; Wang, A.; Yu, X.; Wang, L. Germplasm resources and genetic breeding of Paeonia: A systematic review. Hortic. Res. 2020, 7, 107. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, D.; Yu, K.; Ji, J.; Liu, N.; Zhang, Y.; Xu, M.; Zhang, Y.J.; Ma, X.; Liu, S.; et al. Frequent germplasm exchanges drive the high genetic diversity of Chinese-cultivated common apricot germplasm. Hortic. Res. 2021, 8, 215. [Google Scholar] [CrossRef]
- Shastry, B.S. SNPs: Impact on gene function and phenotype. Methods Mol. Biol. 2009, 578, 3–22. [Google Scholar]
- Aslam, M.L.; Bastiaansen, J.W.; Elferink, M.G.; Megens, H.J.; Crooijmans, R.P.; Blomberg, L.A.; Fleischer, R.C.; Van Tassell, C.P.; Sonstegard, T.S.; Schroeder, S.G.; et al. Whole genome SNP discovery and analysis of genetic diversity in Turkey (Meleagris gallopavo). BMC Genom. 2012, 13, 391. [Google Scholar] [CrossRef]
- Guo, Y.; Jamison, D.C. The distribution of SNPs in human gene regulatory regions. BMC Genom. 2005, 6, 140. [Google Scholar] [CrossRef]
- Lee, C.Y. A model for the clustered distribution of SNPs in the human genome. Comput. Biol. Chem. 2016, 64, 94–98. [Google Scholar] [CrossRef]
- Song, X.; Ge, T.; Li, Y.; Hou, X. Genome-wide identification of SSR and SNP markers from the non-heading Chinese cab-bage for comparative genomic analyses. BMC Genom. 2015, 16, 328. [Google Scholar] [CrossRef]
- Lehne, B.; Lewis, C.M.; Schlitt, T. From SNPs to genes: Disease association at the gene level. PLoS ONE 2011, 6, e20133. [Google Scholar] [CrossRef]
- Lu, L.; Chen, H.; Wang, X.; Zhao, Y.; Yao, X.; Xiong, B.; Deng, Y.; Zhao, D. Genome-level diversification of eight ancient tea populations in the Guizhou and Yunnan regions identifies candidate genes for core agronomic traits. Hortic. Res. 2021, 8, 388–406. [Google Scholar] [CrossRef]
- Song, X.; Wei, Y.; Xiao, D.; Gong, K.; Sun, P.; Ren, Y.; Yuan, J.; Wu, T.; Yang, Q.; Li, X.; et al. Brassica carinata genome characterization clarifies U’s triangle model of evolution and polyploidy in Brassica. Plant Physiol. 2021, 186, 388–406. [Google Scholar] [CrossRef]
- Parkin, I.A.; Koh, C.; Tang, H.; Robinson, S.J.; Kagale, S.; Clarke, W.E.; Town, C.D.; Nixon, J.; Krishnakumar, V.; Bidwell, S.L.; et al. Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biol. 2014, 15, R77. [Google Scholar] [CrossRef]
- Sun, D.; Wang, C.; Zhang, X.; Zhang, W.; Jiang, H.; Yao, X.; Liu, L.; Wen, Z.; Niu, G.; Shan, X. Draft genome sequence of cauliflower (Brassica oleracea L. var. botrytis) provides new insights into the C genome in Brassica species. Hortic. Res. 2019, 6, 82. [Google Scholar] [CrossRef]
- Lv, H.; Wang, Y.; Han, F.; Ji, J.; Fang, Z.; Zhuang, M.; Li, Z.; Zhang, Y.; Yang, L. A high-quality reference genome for cabbage obtained with SMRT reveals novel genomic features and evolutionary characteristics. Sci. Rep. 2020, 10, 12394. [Google Scholar] [CrossRef]
- Guo, N.; Wang, S.; Gao, L.; Liu, Y.; Wang, X.; Lai, E.; Duan, M.; Wang, G.; Li, J.; Yang, M.; et al. Genome sequencing sheds light on the contribution of structural variants to Brassica oleracea diversification. BMC Biol. 2021, 19, 93. [Google Scholar] [CrossRef]
- Cai, X.; Wu, J.; Liang, J.; Lin, R.; Zhang, K.; Cheng, F.; Wang, X. Improved Brassica oleracea JZS assembly reveals signif-icant changing of LTR-RT dynamics in different morphotypes. Theor. Appl. Genet. 2020, 133, 3187–3199. [Google Scholar] [CrossRef]
- Belser, C.; Istace, B.; Denis, E.; Dubarry, M.; Baurens, F.C.; Falentin, C.; Genete, M.; Berrabah, W.; Chevre, A.M.; Delourme, R.; et al. Chromosome-scale as-semblies of plant genomes using nanopore long reads and optical maps. Nat. Plants 2018, 4, 879–887. [Google Scholar] [CrossRef]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef]
- Shen, S.; Li, N.; Wang, Y.; Zhou, R.; Sun, P.; Lin, H.; Chen, W.; Yu, T.; Liu, Z.; Wang, Z.; et al. High-quality ice plant reference genome analysis provides insights into genome evolution and allows exploration of genes involved in the transition from C3 to CAM pathways. Plant Biotechnol. J. 2022, 20, 2107–2122. [Google Scholar] [CrossRef]
- Song, X.; Liu, H.; Shen, S.; Huang, Z.; Yu, T.; Liu, Z.; Yang, Q.; Wu, T.; Feng, S.; Zhang, Y.; et al. Chromo-some-level pepino genome provides insights into genome evolution and anthocyanin biosynthesis in Solanaceae. Plant J. 2022, 110, 1128–1143. [Google Scholar] [CrossRef]
- Liu, Z.; Li, N.; Yu, T.; Wang, Z.; Wang, J.; Ren, J.; He, J.; Huang, Y.; Shi, K.; Yang, Q.; et al. The Bras-sicaceae genome resource (TBGR): A comprehensive genome platform for Brassicaceae plants. Plant Physiol. 2022, 190, 226–237. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Sulovari, A.; Li, D. GACT: A Genome build and Allele definition Conversion Tool for SNP imputation and meta-analysis in genetic association studies. BMC Genom. 2014, 15, 610. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Vilella, A.J.; Severin, J.; Ureta-Vidal, A.; Heng, L.; Durbin, R.; Birney, E. EnsemblCompara GeneTrees: Complete, duplica-tion-aware phylogenetic trees in vertebrates. Genome Res. 2009, 19, 327–335. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, s13742-015. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Yu, N.; Jensen-Seaman, M.I.; Chemnick, L.; Ryder, O.; Li, W.H. Nucleotide diversity in gorillas. Genetics 2004, 166, 1375–1383. [Google Scholar] [CrossRef]
- Bimolata, W.; Kumar, A.; Reddy, M.S.K.; Sundaram, R.M.; Laha, G.S.; Qureshi, I.A.; Ghazi, I.A. Nucleotide diversity analysis of three major bacterial blight resistance genes in rice. PLoS ONE 2015, 10, e7276. [Google Scholar] [CrossRef]
- Hall, S.J.G. Genetic Differentiation among Livestock Breeds-Values for Fst. Animals 2022, 12, 1115. [Google Scholar] [CrossRef]
- Guo, T.; Zhao, H.; Yuan, C.; Huang, S.; Zhou, S.; Lu, Z.; Niu, C.; Liu, J.; Zhu, S.; Yue, Y.; et al. Selective Sweeps Uncovering the Genetic Basis of Horn and Adaptability Traits on Fine-Wool Sheep in China. Front. Genet. 2021, 12, 604235. [Google Scholar] [CrossRef]
- Feng, G.; Ai, X.; Yi, H.; Guo, W.; Wu, J. Genomic and transcriptomic analyses of Citrus sinensis varieties provide insights into Valencia orange fruit mastication trait formation. Hortic. Res. 2021, 8, 111785. [Google Scholar] [CrossRef]
- Li, C.; Li, Y.; Zheng, J.; Guo, Z.; Mei, X.; Lei, M.; Ren, Y.; Zhang, X.; Zhang, C.; Yang, C.; et al. Trait Analysis in Domestic Rabbits (Oryctolagus cuniculus f. domesticus) Using SNP Markers from Gen-otyping-by-Sequencing Data. Animals 2022, 12, 2052. [Google Scholar]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, 3787–3793. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, R14. [Google Scholar] [CrossRef]
- Song, X.; Hu, J.; Wu, T.; Yang, Q.; Feng, X.; Lin, H.; Feng, S.; Cui, C.; Yu, Y.; Zhou, R.; et al. Comparative analysis of long noncoding RNAs in angiosperms and characterization of long noncoding RNAs in response to heat stress in Chinese cabbage. Hortic. Res. 2021, 8, 48. [Google Scholar] [CrossRef]
- Armstrong, R.A. When to use the Bonferroni correction. Ophthalmic Physiol. Opt. 2014, 34, 502–508. [Google Scholar] [CrossRef]
- Purvis, A.; Bromham, L. Estimating the transition/transversion ratio from independent pairwise comparisons with an as-sumed phylogeny. J. Mol. Evol. 1997, 44, 112–119. [Google Scholar] [CrossRef]
- van der Merwe, N.A.; Gryzenhout, M.; Steenkamp, E.T.; Wingfield, B.D.; Wingfield, M.J. Multigene phylogenetic and pop-ulation differentiation data confirm the existence of a cryptic species within Chrysoporthe cubensis. Fungal Biol. 2010, 114, 966–979. [Google Scholar] [CrossRef]
- Bird, K.A.; An, H.; Gazave, E.; Gore, M.A.; Pires, J.C.; Robertson, L.D.; Labate, J.A. Population Structure and Phylogenetic Relationships in a Diverse Panel of Brassica rapa L. Front. Plant Sci. 2017, 8, 321. [Google Scholar] [CrossRef]
- Slatkin, M. Linkage disequilibrium—Understanding the evolutionary past and mapping the medical future. Nat. Rev. Genet. 2008, 9, 477–485. [Google Scholar] [CrossRef]
- Wang, M.; Jia, T.; Jiang, N.; Wang, L.; Hu, X.; Luo, Z. Inferring linkage disequilibrium from non-random samples. BMC Genom. 2010, 11, 328. [Google Scholar] [CrossRef]
- Kang, Y.; Guo, S.; Wang, X.; Cao, M.; Pei, J.; Li, R.; Bao, P.; Wang, J.; Lamao, J.; Gongbao, D.; et al. Whole-Genome Resequencing Highlights the Unique Characteristics of Kecai Yaks. Animals 2022, 12, 2682. [Google Scholar] [CrossRef]
- Flint-Garcia, S.A.; Thornsberry, J.M.; Buckler, E.S. Structure of Linkage Disequilibrium in Plants. Annu. Rev. Plant Biol. 2003, 54, 357–374. [Google Scholar] [CrossRef]
- Zhang, J.M.; Zhang, F. Population structure and genetic variation of the endangered species Elaeagnus mollis Diels (Elaeagnaceae). Genet. Mol. Res. 2015, 14, 5950–5957. [Google Scholar] [CrossRef]
- Nothnagel, M.; Lu, T.T.; Kayser, M.; Krawczak, M. Genomic and geographic distribution of SNP-defined runs of homozy-gosity in Europeans. Hum. Mol. Genet. 2010, 19, 2927–2935. [Google Scholar] [CrossRef]
- Nielsen, R. Population genetic analysis of ascertained SNP data. Hum. Genom. 2004, 1, 218–224. [Google Scholar] [CrossRef]
- Bandillo, N.; Jarquin, D.; Song, Q.; Nelson, R.; Cregan, P.; Specht, J.; Lorenz, A. A Population Structure and Genome-Wide Association Analysis on the USDA Soybean Germplasm Collection. Plant Genome 2015, 8, 1–13. [Google Scholar] [CrossRef]
- Flanagan, S.P.; Jones, A.G. Constraints on the FST-Heterozygosity Outlier Approach. J. Hered. 2017, 108, 561–573. [Google Scholar] [CrossRef]
- Holsinger, K.E.; Weir, B.S. Genetics in geographically structured populations: Defining, estimating and interpreting F(ST). Nat. Rev. Genet. 2009, 10, 639–650. [Google Scholar] [CrossRef]
- Feng, S.; Liu, Z.; Hu, Y.; Tian, J.; Yang, T.; Wei, A. Genomic analysis reveals the genetic diversity, population structure, evolutionary history and relationships of Chinese pepper. Hortic. Res. 2020, 7, 158. [Google Scholar] [CrossRef]
- Liu, Y.; Cheng, H.; Wang, S.; Luo, X.; Ma, X.; Sun, L.; Chen, N.; Zhang, J.; Qu, K.; Wang, M.; et al. Genomic Diversity and Selection Signatures for Weining Cattle on the Border of Yunnan-Guizhou. Front. Genet. 2022, 13, 848951. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Cai, Y.; Zhang, G.; Gu, L.; Wang, Y.; Zhao, Y.; Abdullah, S. Genome Polymorphism Analysis and Selected Sweep Regions Detection via the Genome Resequencing of 91 Cabbage (Brassica oleracea) Accessions. Horticulturae 2023, 9, 283. https://doi.org/10.3390/horticulturae9020283
Li Q, Cai Y, Zhang G, Gu L, Wang Y, Zhao Y, Abdullah S. Genome Polymorphism Analysis and Selected Sweep Regions Detection via the Genome Resequencing of 91 Cabbage (Brassica oleracea) Accessions. Horticulturae. 2023; 9(2):283. https://doi.org/10.3390/horticulturae9020283
Chicago/Turabian StyleLi, Qiang, Yumei Cai, Guoli Zhang, Liqiang Gu, Ying Wang, Yuqian Zhao, and Shamsiah Abdullah. 2023. "Genome Polymorphism Analysis and Selected Sweep Regions Detection via the Genome Resequencing of 91 Cabbage (Brassica oleracea) Accessions" Horticulturae 9, no. 2: 283. https://doi.org/10.3390/horticulturae9020283
APA StyleLi, Q., Cai, Y., Zhang, G., Gu, L., Wang, Y., Zhao, Y., & Abdullah, S. (2023). Genome Polymorphism Analysis and Selected Sweep Regions Detection via the Genome Resequencing of 91 Cabbage (Brassica oleracea) Accessions. Horticulturae, 9(2), 283. https://doi.org/10.3390/horticulturae9020283