
Small Alcohols as Surfactants and Hydrate Promotors


Abstract
:
1. Introduction
2. The Use of Alcohols for Hydrate Prevention in Industrial Settings
3. Small Alcohols as Surfactants in Hydrate Production
4. Methodology
5. Non-Equilibrium Nature of Hydrates in Sediments
6. Heterogeneous Hydrate Formation in Systems Containing Alcohols
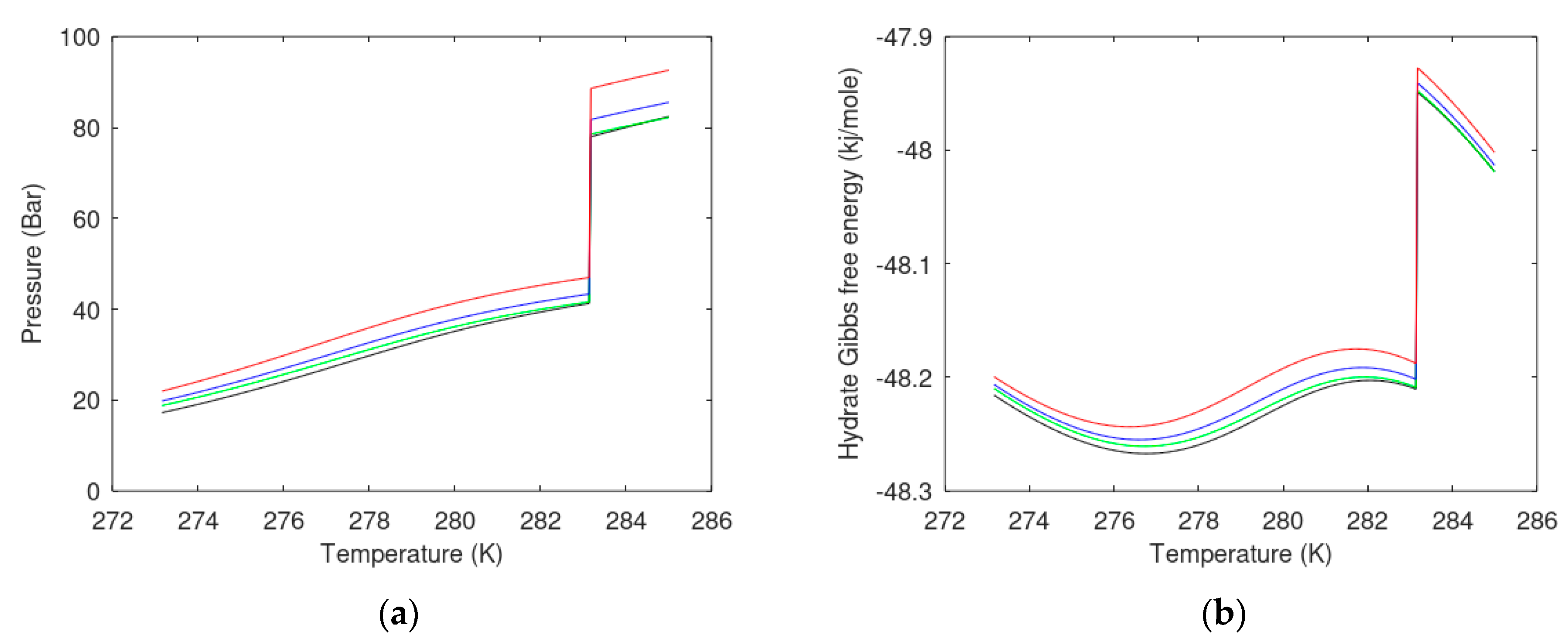
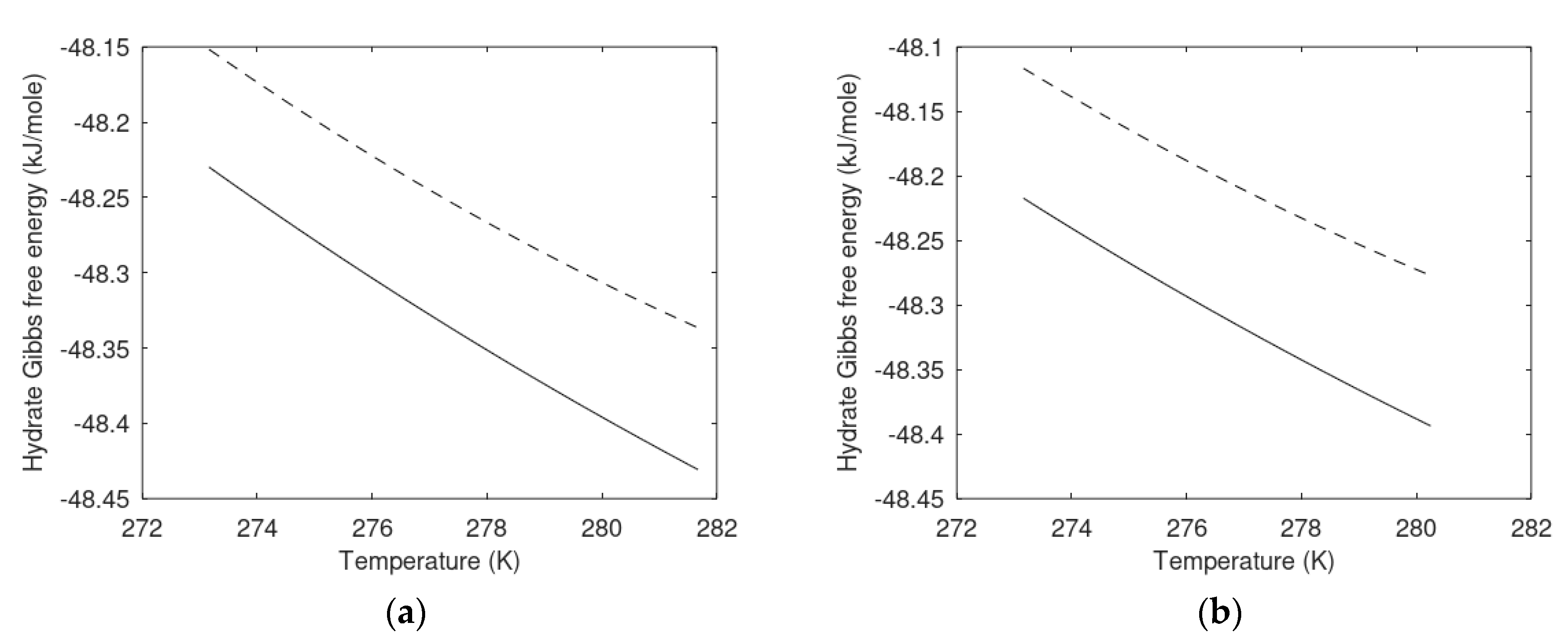
6.1. Hydrate Phase Transition and Free Energy Changes
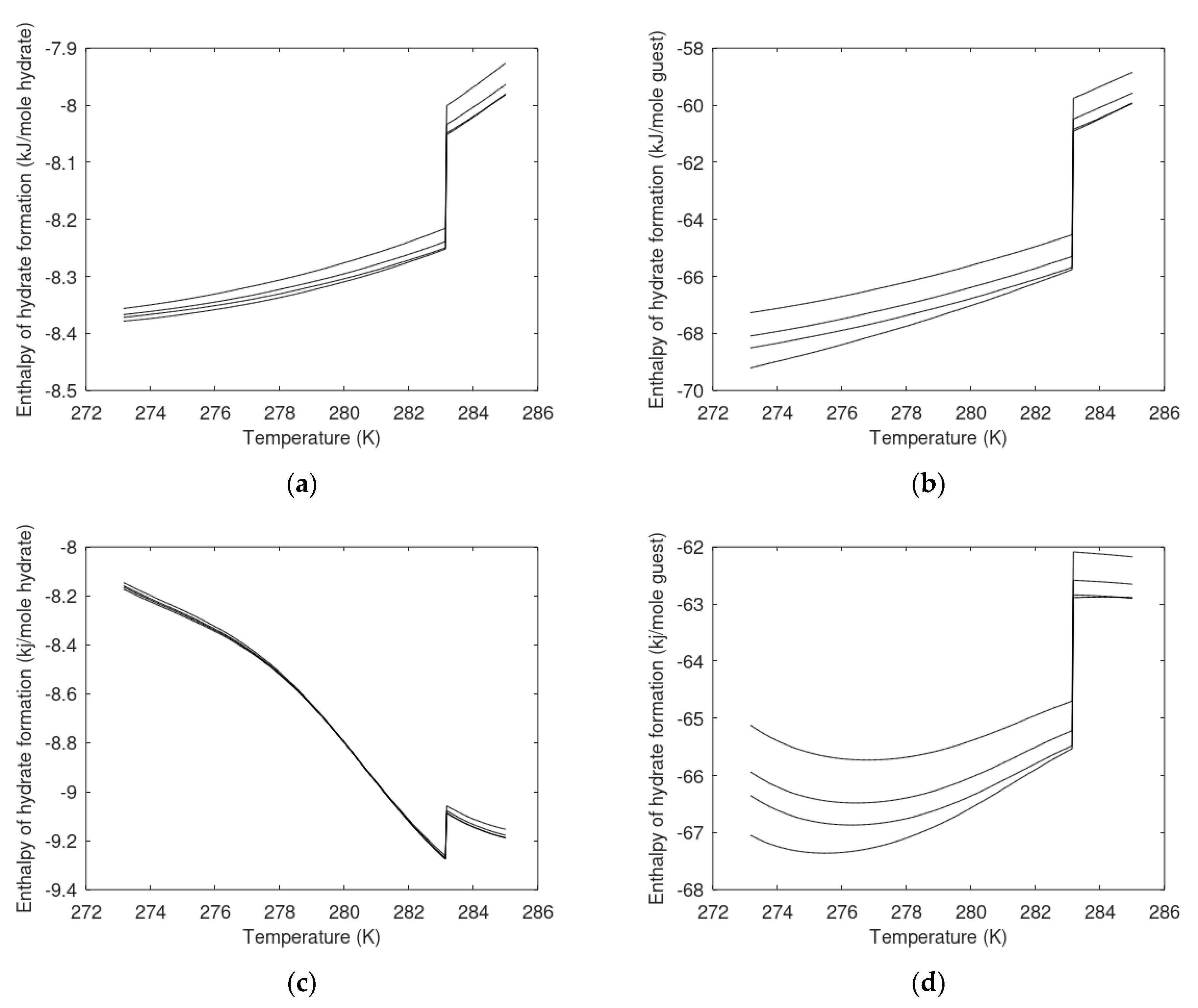
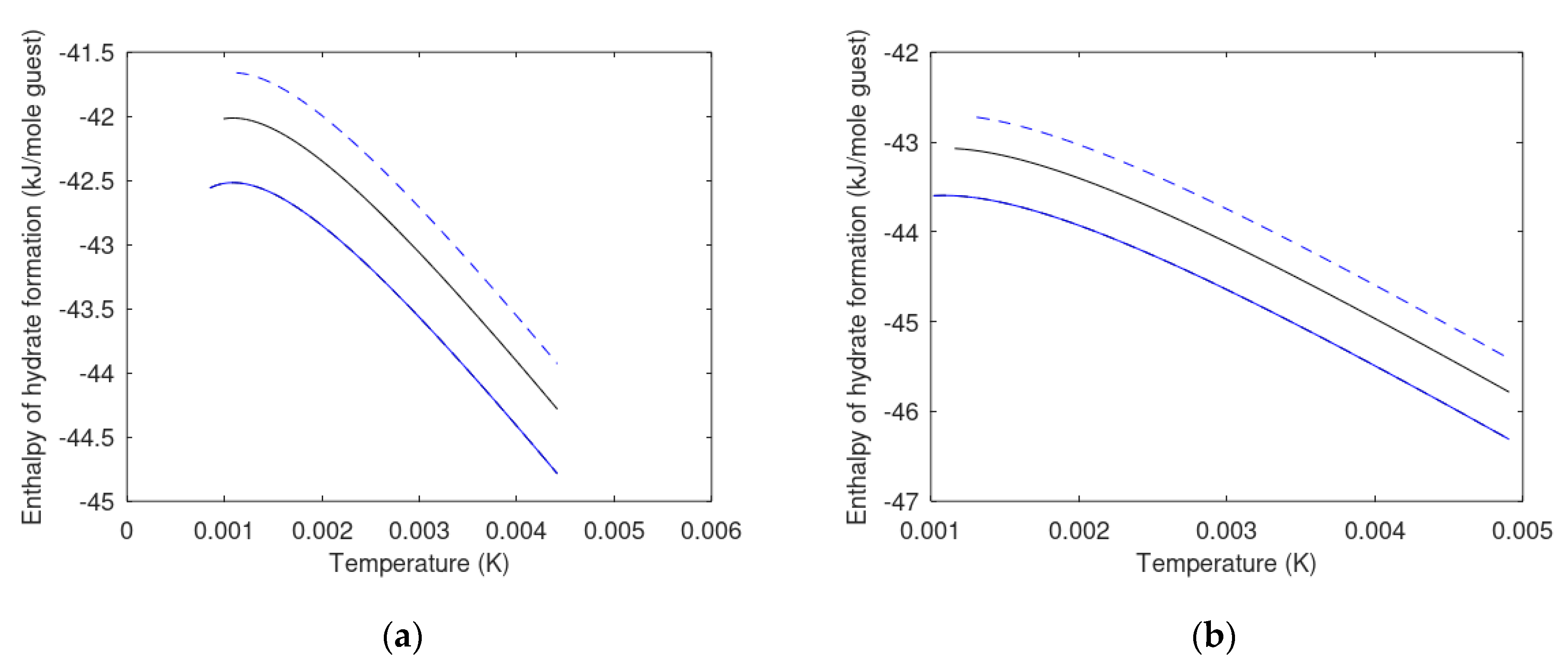
6.2. Enthalpies of Hydrate Formation
7. Homogeneous Hydrate Formation in Systems Containing Alcohols
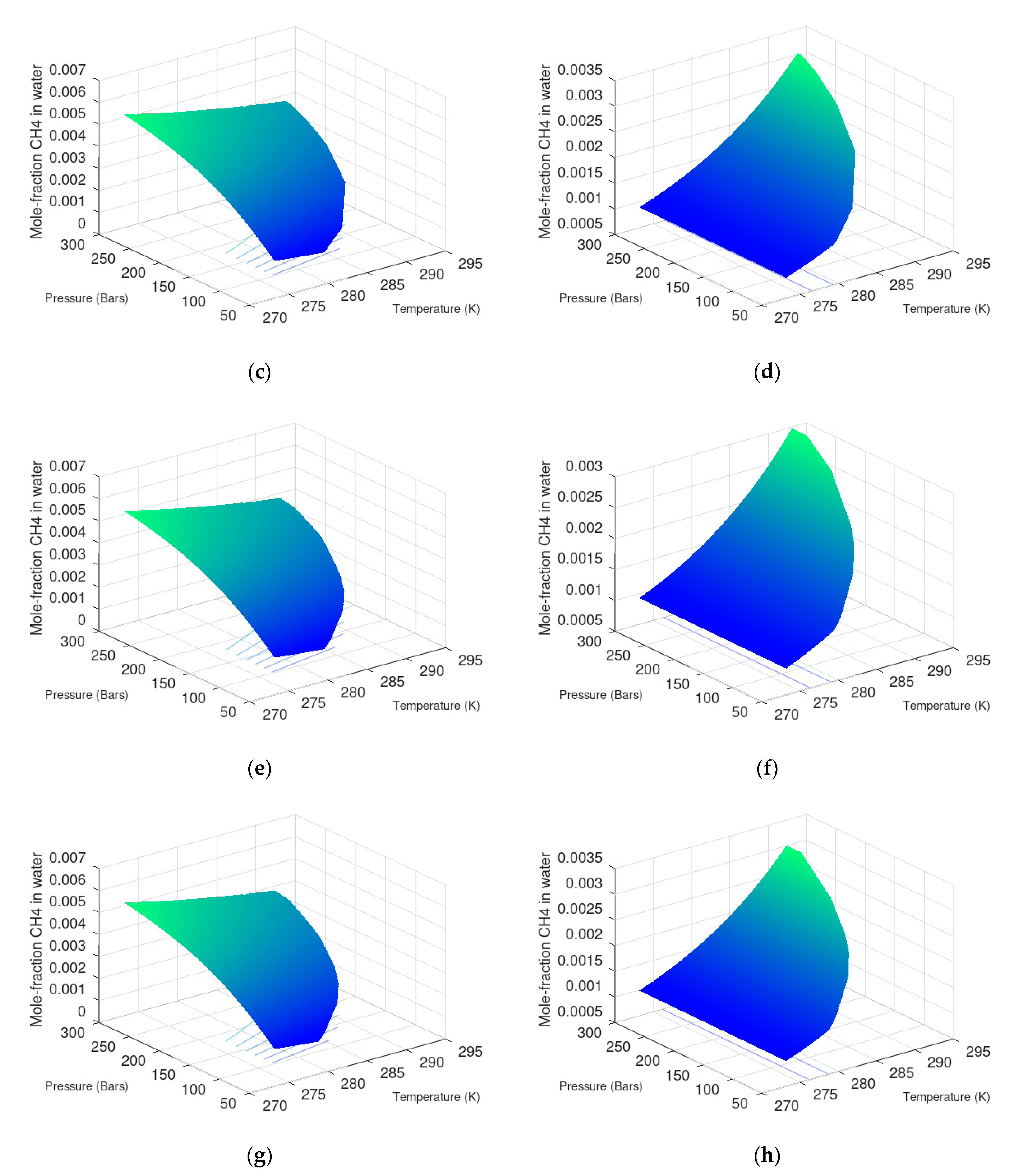
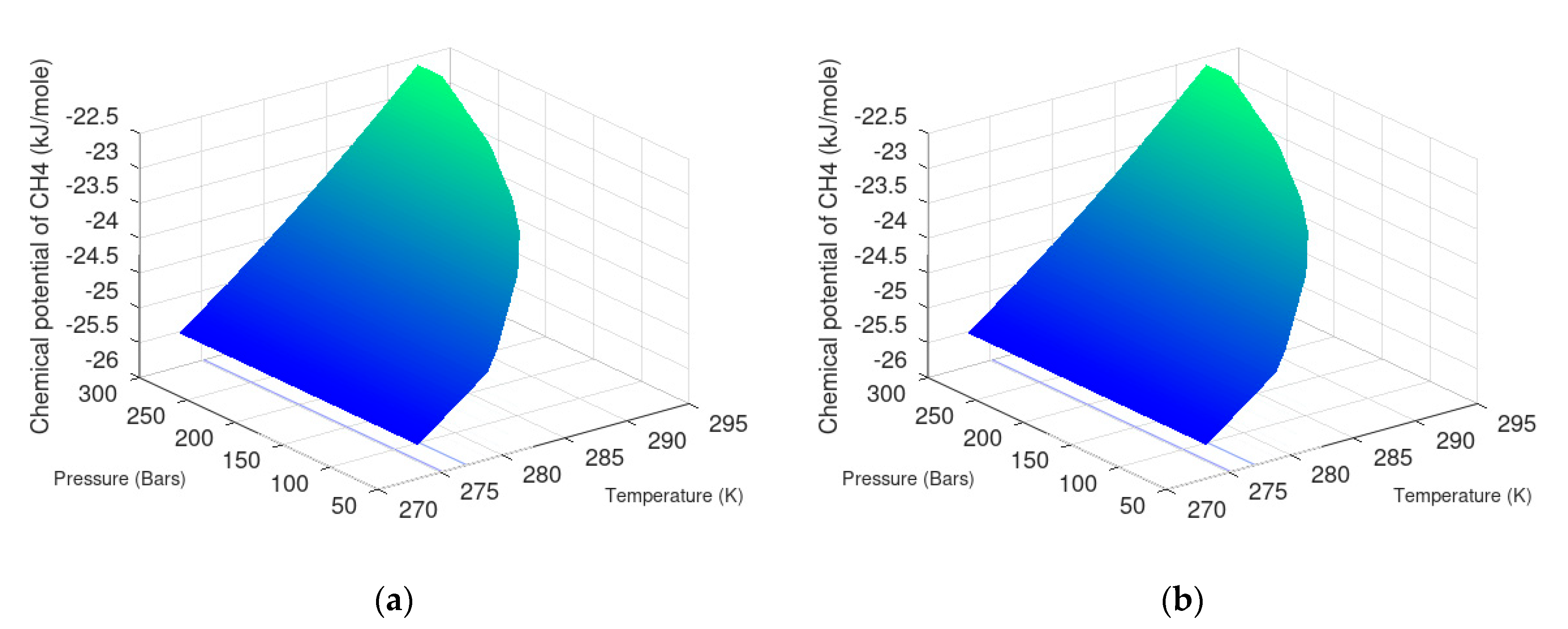
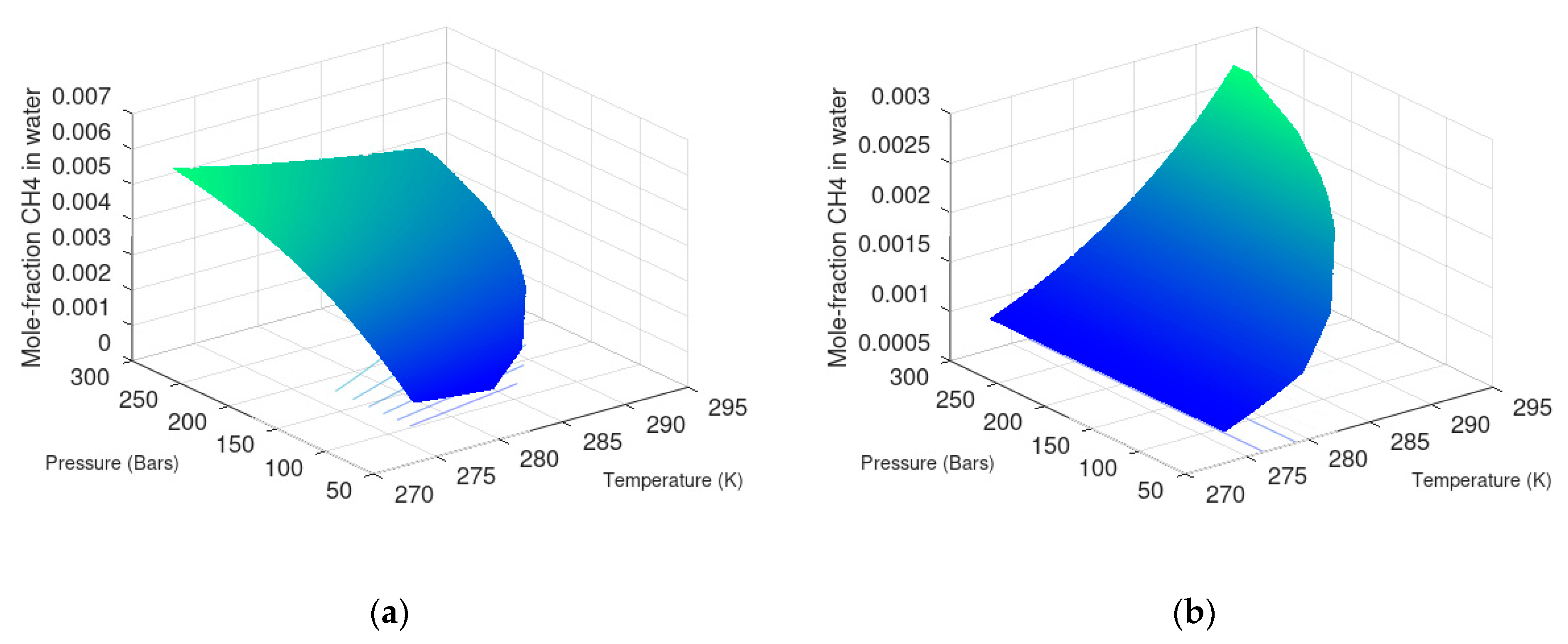
7.1. Methane Hydrate Formation from Water Solution
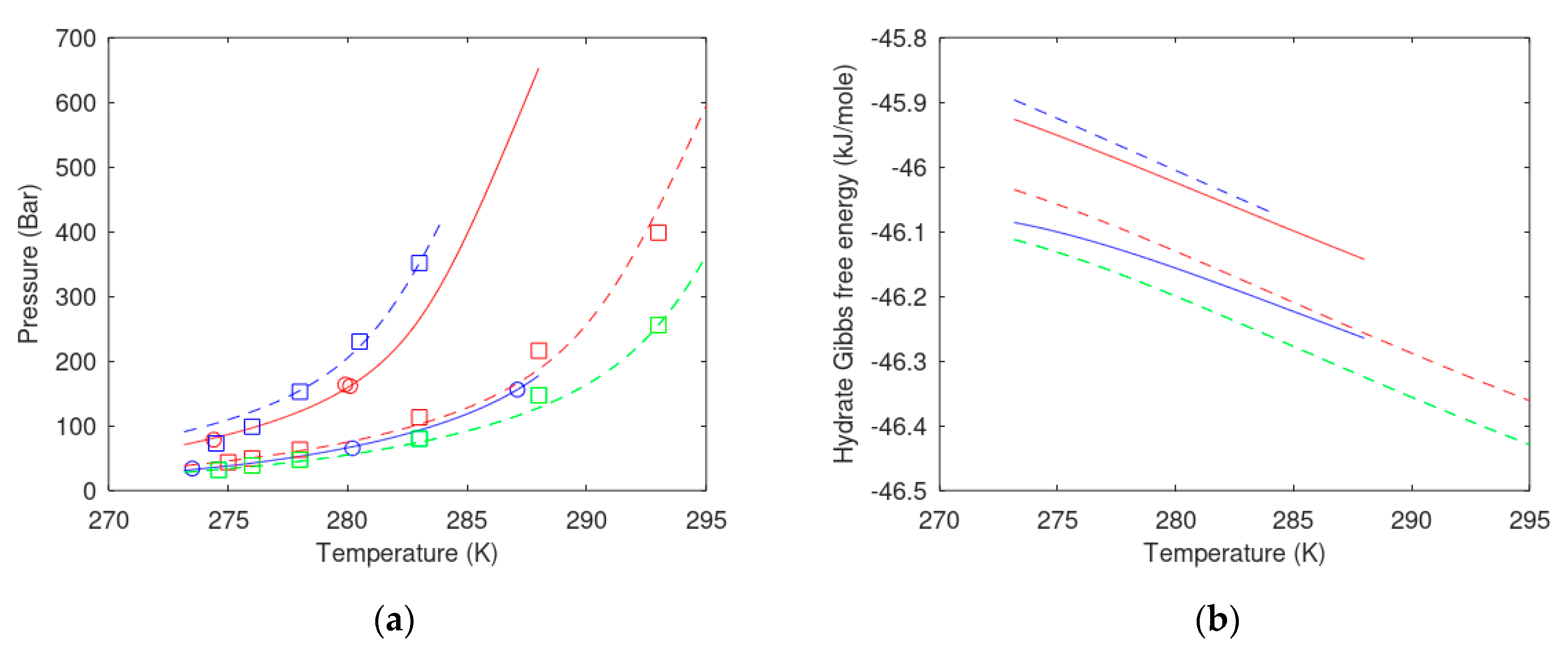
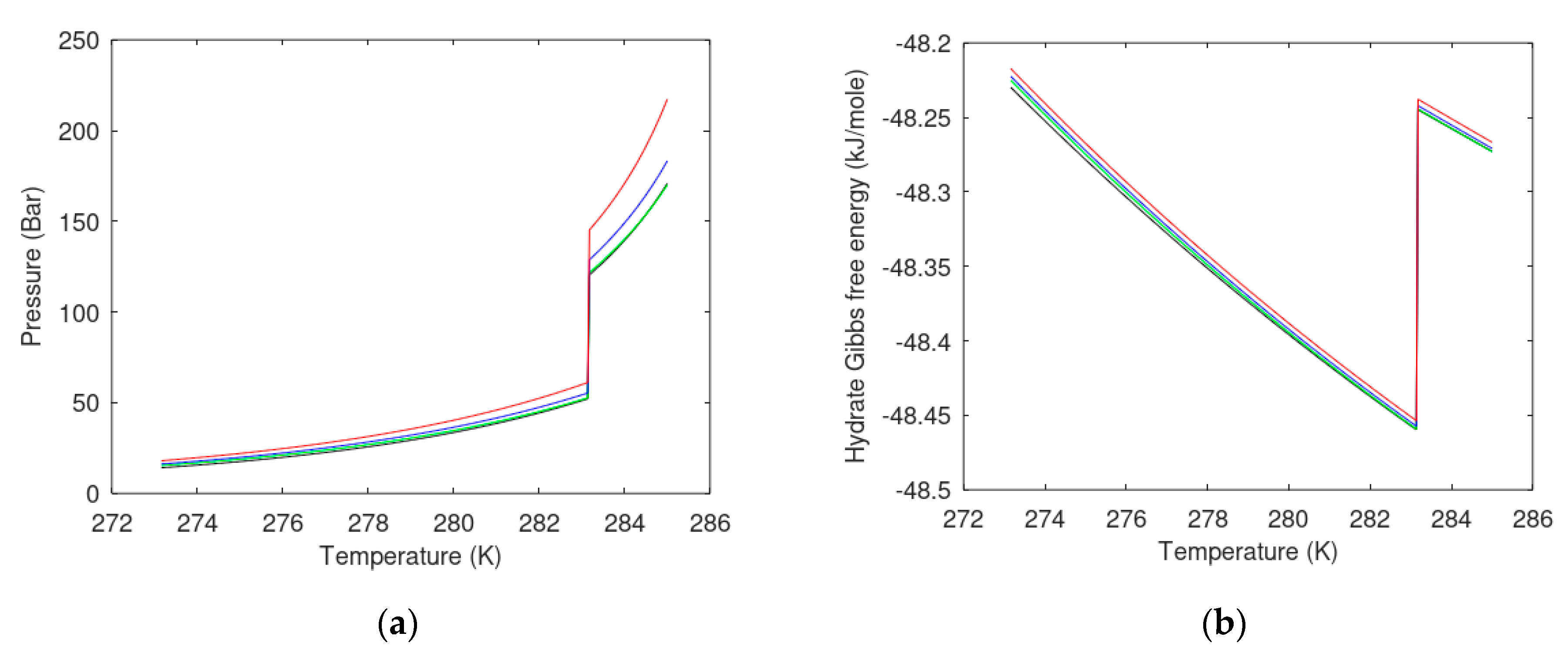
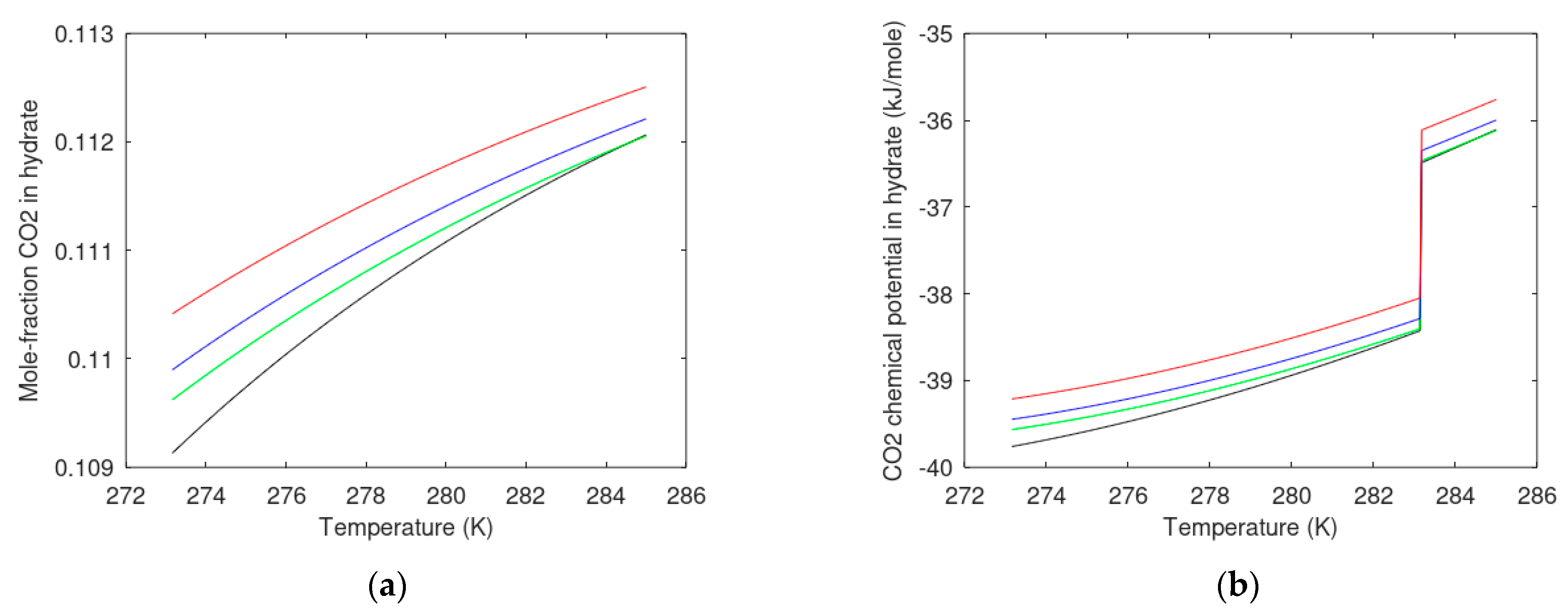
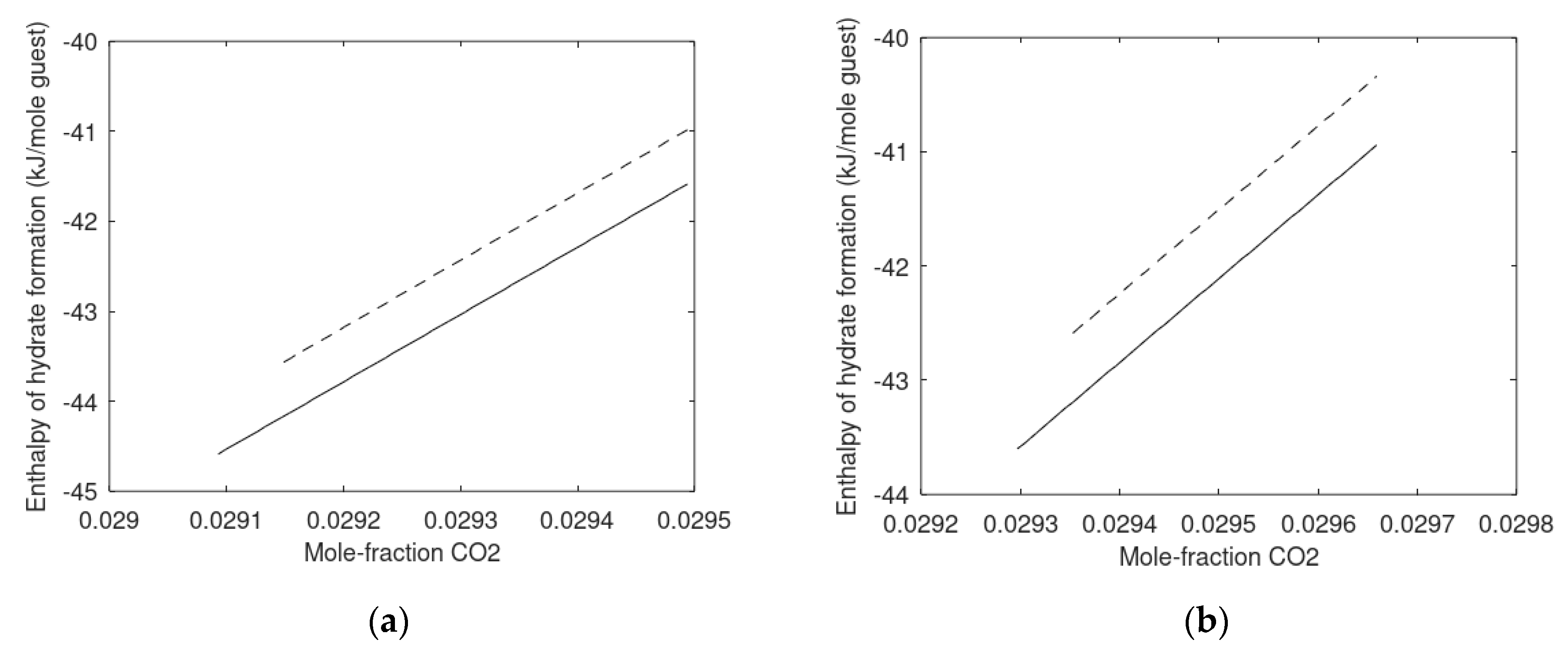
7.2. Carbon Dioxide Hydrate Formation from Water Solution
8. Discussion
9. Conclusions
Funding
Conflicts of Interest
References
- Sloan, E.D.; Koh, C.A. Clathrate Hydrates of Natural Gases, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Mokogan, Y.F. Hydrates of Hydrocarbons, 1st ed.; PennWell Books: Tulsa, OK, USA, 1997; ISBN 978-0878147182. [Google Scholar]
- Kvamme, B.; Selvåg, J.; Aromada, S.A.; Saeidi, N.; Kuznetsova, T. Methanol as hydrate inhibitor and hydrate activator. Phys. Chem. Chem. Phys. 2018, 20, 21968–21987. [Google Scholar] [CrossRef]
- Austvik, T.; Hustvedt, E.; Gjertsen, L.H. Formation and Removal of Hydrate Plugs—Field Trial at Tommeliten. In Proceedings of the 76 Annual Meeting of the Gas Processors Association (GPA), San Antonio, TX, USA, 10–12 March 1997; p. 249. [Google Scholar]
- Kvamme, B. Enthalpies of Hydrate Formation from Hydrate Formers Dissolved in Water. Energies 2019, 12, 1039. [Google Scholar] [CrossRef] [Green Version]
- Kvamme, B.; Aromada, S.A.; Gjerstad, P.B. Consistent Enthalpies of the Hydrate Formation and Dissociation Using Residual Thermodynamics. J. Chem. Eng. Data 2019, 64, 3493–3504. [Google Scholar] [CrossRef]
- Aromada, S.A.; Kvamme, B.; Wei, N.; Saeidi, N. Enthalpies of Hydrate Formation and Dissociation from Residual Thermodynamics. Energies 2019, 12, 4726. [Google Scholar] [CrossRef] [Green Version]
- Kvamme, B. Consistent Thermodynamic Calculations for Hydrate Properties and Hydrate Phase Transitions. J. Chem. Eng. Data 2020, 65, 2872–2893. [Google Scholar] [CrossRef]
- Kvamme, B.; Zhao, J.; Wei, N.; Sun, W.; Zarifi, M.; Saeidi, N.; Zhou, S.; Kuznetsova, T.; Li, Q. Why Should We Use Residual Thermodynamics for Calculation of Hydrate Phase Transitions? Energies 2020, 13, 4135. [Google Scholar] [CrossRef]
- Kvamme, B. Thermodynamic properties and dielectric constants in water/methanol mixtures by integral equation theory and molecular dynamics simulations. Phys. Chem. Chem. Phys. 2002, 4, 942–948. [Google Scholar] [CrossRef]
- Kvamme, B.; Graue, A.; Buanes, T.; Kuznetsova, T.; Ersland, G. Storage of CO2 in natural gas hydrate reservoirs and the effect of hydrate as an extra sealing in cold aquifers. Int. J. Greenh. Gas Control 2007, 1, 236–246. [Google Scholar] [CrossRef]
- Tegze, G.; Pusztai, T.; Tóth, G.; Gránásy, L.; Svandal, A.; Buanes, T.; Kuznetsova, T.; Kvamme, B. Multiscale approach to CO2 hydrate formation in aqueous solution: Phase field theory and molecular dynamics. Nucleation and growth. J. Chem. Phys. 2006, 124, 234710. [Google Scholar] [CrossRef]
- Kvamme, B.; Coffin, R.B.; Zhao, J.; Wei, N.; Zhou, S.; Li, Q.; Saeidi, N.; Chien, Y.-C.; Dunn-Rankin, D.; Sun, W.; et al. Stages in the Dynamics of Hydrate Formation and Consequences for Design of Experiments for Hydrate Formation in Sediments. Energies 2019, 12, 3399. [Google Scholar] [CrossRef] [Green Version]
- Bybee, K. Natural Gas Technology/Monetization: Overview of the Mallik Gas-Hydrate Production Research Well. J. Pet. Technol. 2004, 56, 53–54. [Google Scholar] [CrossRef]
- Moridis, G.J.; Collett, T.S.; Dallimore, S.R.; Satoh, T.; Hancock, S.; Weatherill, B. Numerical studies of gas production from several CH4 hydrate zones at the Mallik site, Mackenzie Delta, Canada. J. Pet. Sci. Eng. 2004, 43, 219–238. [Google Scholar] [CrossRef] [Green Version]
- Konno, Y.; Fujii, T.; Sato, A.; Akamine, K.; Naiki, M.; Masuda, Y.; Yamamoto, K.; Nagao, J. Key Findings of the World’s First Offshore Methane Hydrate Production Test off the Coast of Japan: Toward Future Commercial Production. Energy Fuels 2017, 31, 2607–2616. [Google Scholar] [CrossRef]
- Yamamoto, K.; Wang, X.-X.; Tamaki, M.; Suzuki, K. The second offshore production of methane hydrate in the Nankai Trough and gas production behavior from a heterogeneous methane hydrate reservoir. RSC Adv. 2019, 9, 25987–26013. [Google Scholar] [CrossRef] [Green Version]
- Oyama, A.; Masutani, S.M. A Review of the Methane Hydrate Program in Japan. Energies 2017, 10, 1447. [Google Scholar] [CrossRef] [Green Version]
- Tenma, N. Recent Status of Methane Hydrate R&D Program in Japan. Presented at the 11th IMHRD, Corpus Christie, TX, USA, 7 December 2017. [Google Scholar]
- Svandal, A.; Kvamme, B.; Granasy, L.; Pusztai, T. The influence of diffusion on hydrate growth. In Proceedings of the 1st International Conference on Diffusion in Solids and Liquids DSL-2005, Aveiro, Portugal, 6–8 July 2005. [Google Scholar]
- Kvamme, B.; Kivelæ, P.-H.; Kuznetsova, T. Adsorption of water and carbon dioxide on hematite and consequences for possible hydrate formation. Phys. Chem. Chem. Phys. 2012, 14, 4410–4424. [Google Scholar] [CrossRef]
- Olsen, R.; Leirvik, K.; Kvamme, B.; Kuznetsova, T. A molecular dynamics study of triethylene glycol on a hydrated calcite surface. Langmuir 2015, 31, 8606–8617. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.; Kvamme, B. Effects of glycol on adsorption dynamics of idealized water droplets on LTA-3A zeolite surfaces. AIChE J. 2019, 65, e16567. [Google Scholar] [CrossRef]
- Olsen, R.; Leirvik, K.N.; Kvamme, B.; Kuznetsova, T. Effects of Sodium Chloride on Acidic Nanoscale Pores Between Steel and Cement. J. Phys. Chem. C 2016, 120, 29264–29271. [Google Scholar] [CrossRef]
- Olsen, R.; Leirvik, K.N.; Kvamme, B. Adsorption characteristics of glycols on calcite and hematite. AIChE J. 2019, 65, e16728. [Google Scholar] [CrossRef]
- Kvamme, B.; Zhao, J.; Wei, N.; Sun, W.; Saeidi, N.; Pei, J.; Kuznetsova, T. Hydrate Production Philosophy and Thermodynamic Calculations. Energies 2020, 13, 672. [Google Scholar] [CrossRef] [Green Version]
- Geissbühler, P.; Fenter, P.; DiMasi, E.; Srajer, G.; Sorensen, L.; Sturchio, N. Three-dimensional structure of the calcite–water interface by surface X-ray scattering. Surf. Sci. 2004, 573, 191–203. [Google Scholar] [CrossRef]
- Phan Van, C. Transport and Adsorption of CO2 and H2O on Calcite and Clathrate Hydrate. Ph.D. Thesis, University of Bergen, Bergen, Norway, 2012. [Google Scholar]
- Van Cuong, P.; Kvamme, B.; Kuznetsova, T.; Jensen, B. Adsorption of water and CO2 on calcite and clathrate hydrate: The effect of short-range forces and temperature. Int. J. Energy Env. 2012, 6, 301. [Google Scholar]
- Van Cuong, P.; Kvamme, B.; Kuznetsova, T.; Jensen, B. Molecular dynamics study of calcite, hydrate and the temperature effect on CO2 transport and adsorption stability in geological formations. Mol. Phys. 2012, 110, 1097–1106. [Google Scholar] [CrossRef]
- Van Cuong, P.; Kvamme, B.; Kuznetsova, T.; Jensen, B. Adsorption energy and stability of H2O and CO2 on calcite effect by short-range force field parameters and temperature. In Recent Researches in Applied Mathematics and Economics; WSEAS Press: Athens, Greece, 2012; pp. 66–72. ISBN 978-1-61804-076-3. [Google Scholar]
- Kuznetsova, T.; Jensen, B.; Kvamme, B.; Sjøblom, S. Water-wetting surfaces as hydrate promoters during transport of carbon dioxide with impurities. Phys. Chem. Chem. Phys. 2015, 17, 12683–12697. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B. Investigations into the Impact of Solid Surfaces in Aqueous Systems. Ph.D. Thesis, University of Bergen, Bergen, Norway, 2016. [Google Scholar]
- Mohammad, N. Heterogeneous Hydrate Nucleation on Calcite {1014} and Kaolinite {001} Surfaces: A Molecular Dynamics Simulation Study. Master’s Thesis, University of Bergen, Bergen, Norway, 2016. [Google Scholar]
- Nesse Knarvik, A.B. Examination of Water and Methane Structuring at a Hematite Surface in the Presence of MEG. Master’s Thesis, Department of Physics and Technology, University of Bergen, Bergen, Norway, 2017. [Google Scholar]
- Austrheim, M.H. Evaluation of Methane and Water Structure at a Hematite Surface—A Hydrate Prevention Perspective. Master’s Thesis, Department of Physics and Technology, University of Bergen, Bergen, Norway, 2017. [Google Scholar]
- Kvamme, B. Thermodynamic Limitations of the CO2/N2 Mixture Injected into CH4 Hydrate in the Ignik Sikumi Field Trial. J. Chem. Eng. Data 2016, 61, 1280–1295. [Google Scholar] [CrossRef]
- Robinson, D.B.; Ng, H.-J. Hydrate Formation and Inhibition in Gas or Gas Condensate Streams. J. Can. Pet. Tech. 1986, 25, 26–30. [Google Scholar] [CrossRef]
- Ross, M.J.; Toczylkin, L.S. Hydrate Dissociation Pressures for Methane or Ethane in the Presence of Aqueous Solutions of Triethylene Glycol. J. Chem. Eng. Data 1992, 37, 488–491. [Google Scholar] [CrossRef]
- Kobayashi, R.; Withrow, H.J.; Williams, G.B.; Katz, D.L. Gas hydrate formation with brine and ethanol solutions. Proc. NGAA 1951, 27, 57–66. [Google Scholar]
- Ng, H.-J.; Robinson, D.B. Hydrate formation in systems containing methane, ethane, propane, carbon dioxide or hydrogen sulfide in the presence of methanol. Fluid Phase Equilibria 1985, 21, 145. [Google Scholar] [CrossRef]
- Sabil, K.M.; Nashed, O.; Lal, B.; Ismail, L.; Japper-Jaafar, A. Experimental investigation on the dissociation conditions of methane hydrate in the presence of imidazolium-based ionic liquids. Thermodyn. J. Chem. 2015, 84, 7–13. [Google Scholar]
- Tumba, K.; Tumba, P.; Reddy, P.; Naidoo, D.; Ramjugernath, A.; Eslamimanesh, A.; Mohammadi, H.; Richon, D. Phase equilibria of methane and carbon dioxide clathrate hydrates in the presence of aqueous solutions of tributylmethyl-phosphonium methylsulfate ionic liquid. J. Chem. Eng. Data 2011, 56, 3620–3629. [Google Scholar] [CrossRef]
- Kvamme, B.; Zhao, J.; Wei, N.; Saeidi, N. Hydrate—A Mysterious Phase or Just Misunderstood? Energies 2020, 13, 880. [Google Scholar] [CrossRef] [Green Version]
- Kvamme, B.; Zhao, J.; Wei, N.; Sun, W.; Zarifi, M.; Saeidi, N.; Zhou, S.; Kuznetsova, T.; Li, Q. Thermodynamics of hydrate systems using a uniform reference state. Asia-Pac. J. Chem. Eng. 2021, e2706. [Google Scholar] [CrossRef]
- Kvamme, B.; Tanaka, H. Thermodynamic Stability of Hydrates for Ethane, Ethylene, and Carbon Dioxide. J. Phys. Chem. 1995, 99, 7114–7119. [Google Scholar] [CrossRef]
- Van der Waals, J.H.; Platteeuw, J.C. Clathrate solutions. In Advances in Chemical Physics, 1st ed.; Prigogine, I., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1958; pp. 1–57. [Google Scholar]
- Falenty, A.; Salamatin, A.N.; Kuhs, W.F. Kinetics of CO2-Hydrate Formation from Ice Powders: Data Summary and Modeling Extended to Low Temperatures. J. Phys. Chem. C 2013, 117, 8443–8457. [Google Scholar] [CrossRef]
- Salamatin, A.N.; Falenty, A.; Hansen, T.C.; Kuhs, W.F. Guest Migration Revealed in CO2 Clathrate Hydrates. Energy Fuels 2015, 29, 5681–5691. [Google Scholar] [CrossRef]
- Falenty, A.; Genov, G.; Hansen, T.C.; Kuhs, W.F.; Salamatin, A.N. Kinetics of CO2 Hydrate Formation from Water Frost at Low Temperatures: Experimental Results and Theoretical Model. J. Phys. Chem. C 2011, 115, 4022–4032. [Google Scholar] [CrossRef]
- Lee, H.; Seo, Y.; Seo, Y.-T.; Moudrakovski, I.L.; Ripmeester, J.A. Recovering Methane from Solid Methane Hydrate with Carbon Dioxide. Angew. Chem. 2003, 115, 5202–5205. [Google Scholar] [CrossRef]
- Soave, G. Equilibrium constants from a modified Redlich-Kwong equation of state. Chem. Eng. Sci. 1972, 27, 1197–1203. [Google Scholar] [CrossRef]
- Kvamme, B.; Lund, A.; Hertzberg, T. The influence of gas-gas interactions on the Langmuir constants for some natural gas hydrates. Fluid Phase Equilibria 1993, 90, 15–44. [Google Scholar] [CrossRef]
- Kvamme, B.; Førrisdahl, O.K. Polar guest-molecules in natural gas hydrates. Fluid Phase Equilibria 1993, 83, 427–435. [Google Scholar] [CrossRef]
- Selvåg, J.; Kuznetsova, T.; Kvamme, B. Molecular dynamics study of surfactant-modified water–carbon dioxide systems. Mol. Simul. 2017, 44, 128–136. [Google Scholar] [CrossRef]
- Selvåg, J. Molecular simulations for surfactants at aqueous-nonpolar liquid interfaces. Ph.D. Thesis, University of Bergen, Bergen, Norway, May 2021. [Google Scholar]
- Baig, K. Nano to Micro Scale Modeling of Hydrate Phase Transition Kinetics. Ph.D. Thesis, University of Bergen, Bergen, Norway, 2017. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ak | Methanol | Ethanol | ||||
|---|---|---|---|---|---|---|
| c0 | c1 | c2 | c0 | c1 | c2 | |
| a0 | 0.74821 | 0.52077 | −0.59936 | 0.73743 | 0.51369 | −0.58176 |
| a1 | 0.54174 | −0.47388 | 0.54721 | 0.53390 | −0.45996 | 0.53981 |
| a2 | −0.53859 | 0.56767 | −0.52552 | −0.52277 | 0.55995 | −0.51009 |
| a3 | 0.35068 | −0.53117 | 0.37324 | 0.34566 | −0.51558 | 0.36817 |
| ak | Ethylene Glycol (MEG) | Triethylene Glycol (TEG) | ||||
| c0 | c1 | c2 | c0 | c1 | c2 | |
| a0 | 0.59006 | 0.44750 | −0.49897 | 0.10473 | 0.19307 | −0.12389 |
| a1 | 0.47286 | −0.39499 | 0.47783 | 0.15496 | 0.17274 | −0.15457 |
| a2 | −0.44892 | 0.49855 | −0.43804 | 0.08770 | 0.08731 | 0.11456 |
| a3 | 0.37487 | −0.44859 | 0.36800 | 0.37947 | 0.80576 | −0.82038 |
| ak | Glycerol | NaCl | ||||
| c0 | c1 | c2 | c0 | c1 | c2 | |
| a0 | 0.10453 | 0.08003 | 0.35094 | 0.14486 | 0.15079 | 0.26256 |
| a1 | −0.00151 | 0.32200 | 0.21219 | 0.09071 | −0. 09709 | −0.63019 |
| a2 | 0.04988 | 0.04965 | 0.00515 | 0.13827 | −0.28171 | 0.57169 |
| a3 | 0.44152 | 0.56533 | −1.25419 | 0.13053 | 1.11504 | −0.55605 |
| i | a0 | a1 |
|---|---|---|
| 1 | 1.360608 | 3.796962 |
| 3 | 0.033630 | −0.703216 |
| 5 | 0.656974 | −12.441339 |
| 7 | 1.763890 | −21.119318 |
| 9 | 5.337858 | −33.298760 |
| 11 | −0.024750 | 12.387276 |
| 13 | 48.353808 | 17.261174 |
| 15 | −11.580192 | 16.384626 |
| 17 | −0.087295 | 13.171333 |
| 19 | −0.558793 | 13.556732 |
| 21 | −23.753020 | 16.573197 |
| 23 | −10.128675 | 13.591099 |
| 25 | −41.212178 | 5.060082 |
| 27 | −31.279868 | 31.289978 |
| 29 | −23.855418 | 31.720767 |
| 31 | −35.125907 | 37.064849 |
| 33 | −33.675110 | 41.544360 |
| 35 | −27.027285 | 57.609882 |
| 37 | −19.026786 | 54.961702 |
| 39 | −37.872252 | 57.204781 |
| i | a0 | a1 |
|---|---|---|
| 1 | −139.137483 | −138.899061 |
| 3 | −76.549658 | −72.397006 |
| 5 | −20.868725 | −14.715982 |
| 7 | 18.030987 | 24.548835 |
| 9 | 44.210433 | 52.904238 |
| 11 | 63.353037 | 71.596515 |
| 13 | 74.713278 | 82.605791 |
| 15 | 80.411175 | 88.536302 |
| 17 | 82.710575 | 90.262518 |
| 19 | 82.017332 | 89.094887 |
| 21 | 79.373137 | 85.956670 |
| 23 | 75.429910 | 81.519167 |
| 25 | 70.680932 | 76.270320 |
| 27 | 65.490785 | 70.551406 |
| 29 | 60.126698 | 64.683147 |
| 31 | 54.782421 | 58.865478 |
| 33 | 49.592998 | 53.235844 |
| 35 | 44.500001 | 47.728622 |
| 37 | 39.869990 | 42.730831 |
| 39 | 35.597488 | 38.125674 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kvamme, B. Small Alcohols as Surfactants and Hydrate Promotors. Fluids 2021, 6, 345. https://doi.org/10.3390/fluids6100345
Kvamme B. Small Alcohols as Surfactants and Hydrate Promotors. Fluids. 2021; 6(10):345. https://doi.org/10.3390/fluids6100345
Chicago/Turabian StyleKvamme, Bjørn. 2021. "Small Alcohols as Surfactants and Hydrate Promotors" Fluids 6, no. 10: 345. https://doi.org/10.3390/fluids6100345
APA StyleKvamme, B. (2021). Small Alcohols as Surfactants and Hydrate Promotors. Fluids, 6(10), 345. https://doi.org/10.3390/fluids6100345