
High Strength and Strong Thixotropic Gel Suitable for Oil and Gas Drilling in Fractured Formation

Abstract
1. Introduction
2. Results and Discussion
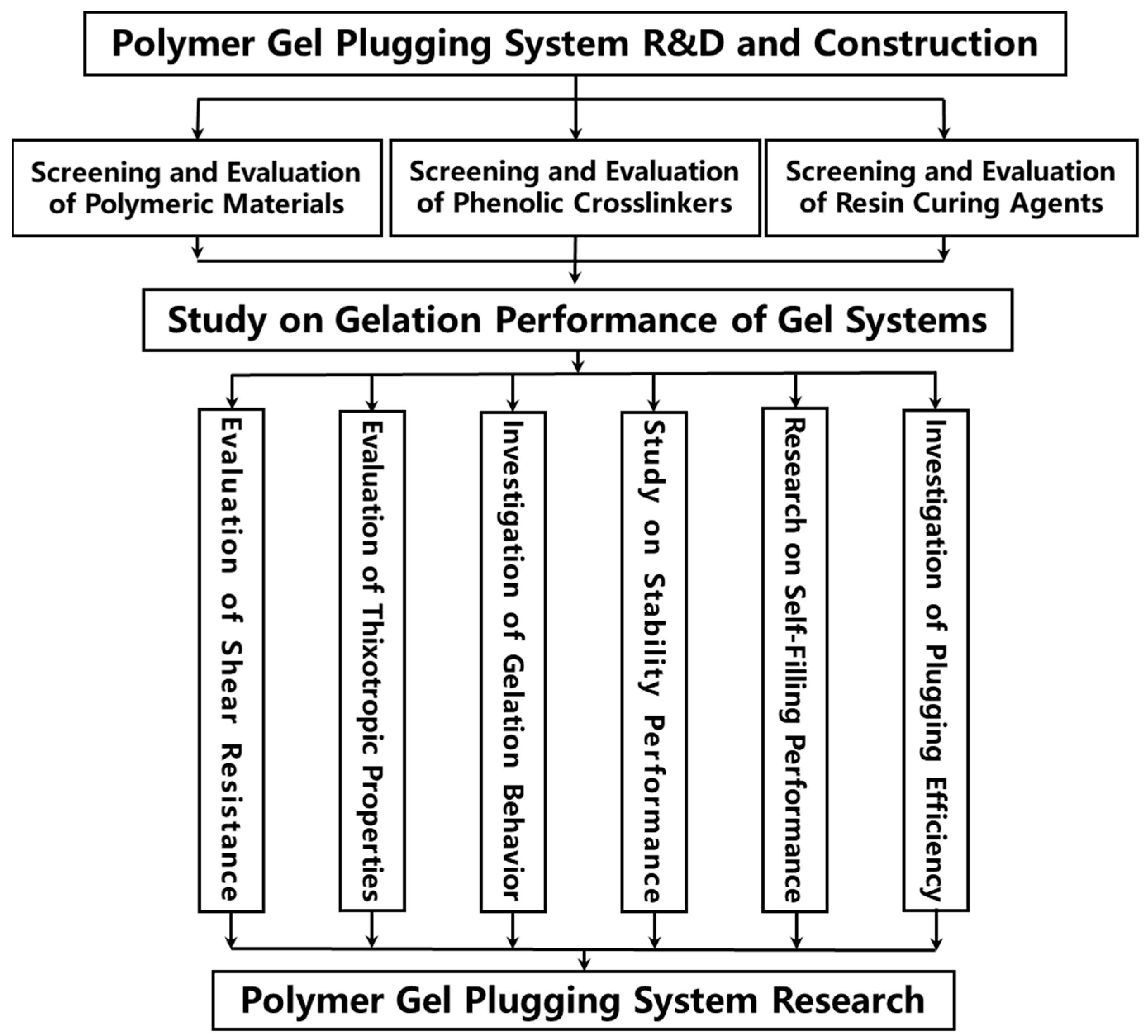
2.1. Construction of the Polymer Gel System
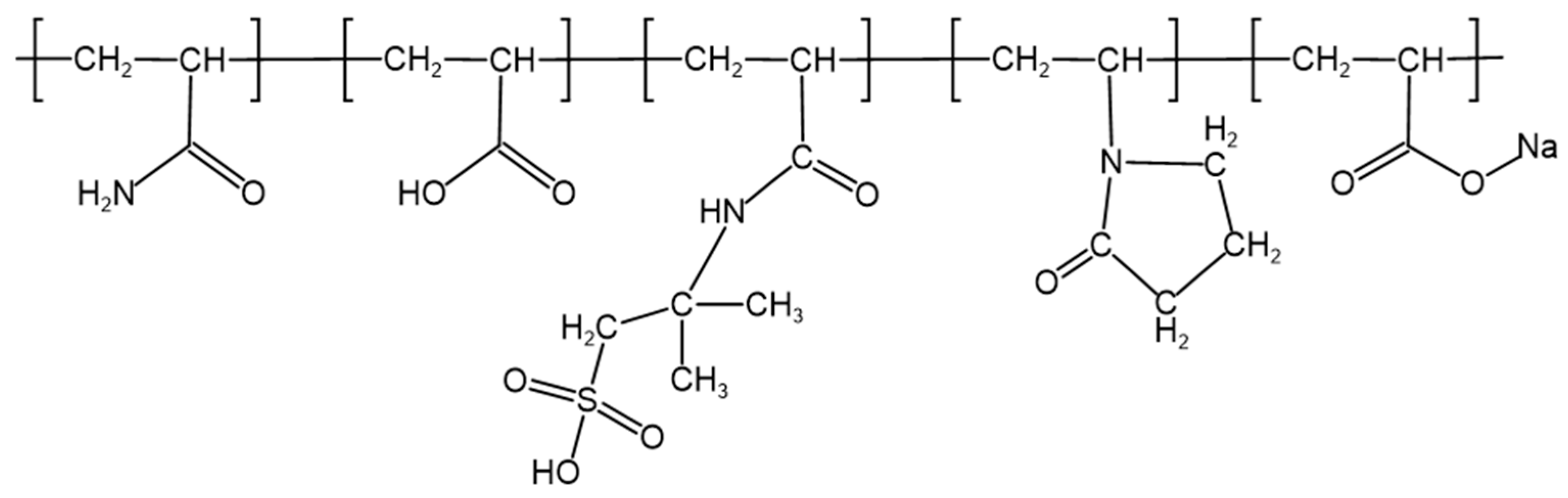
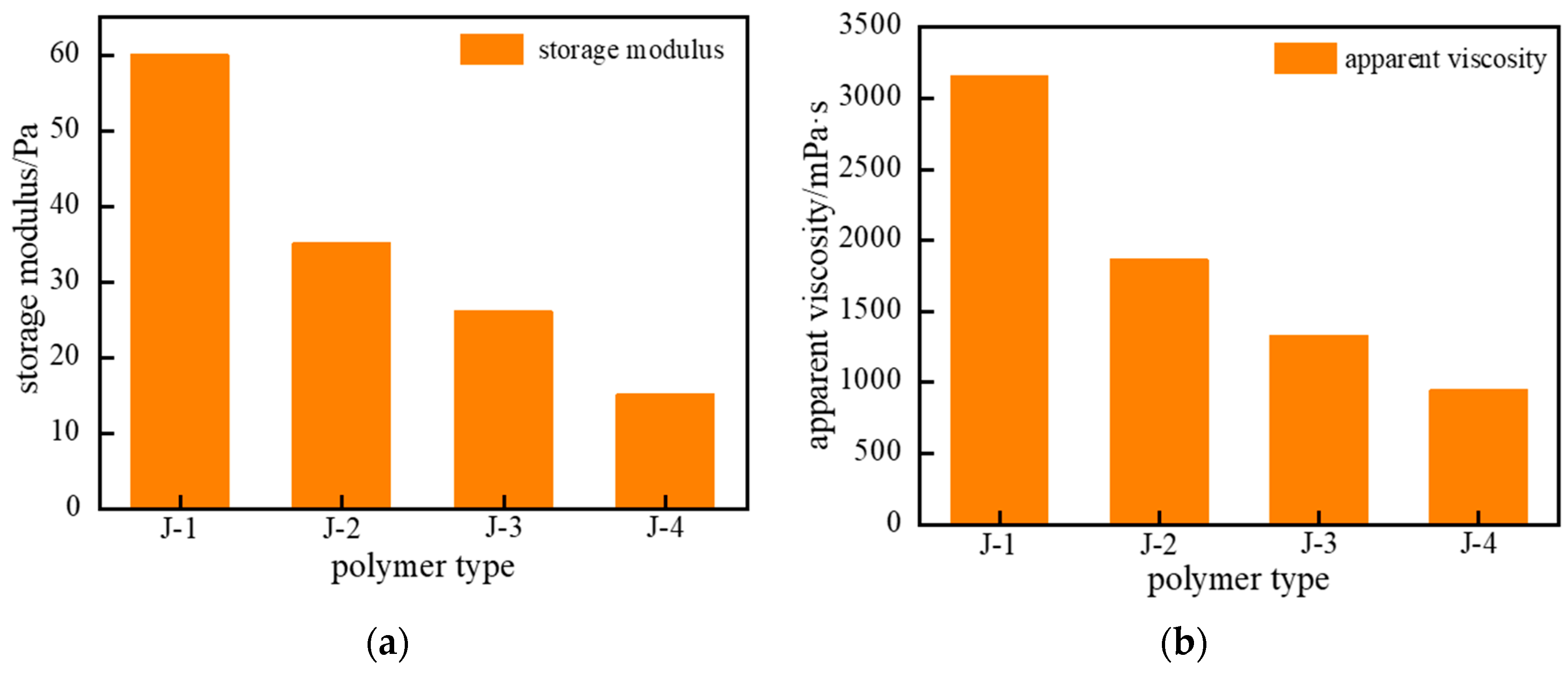
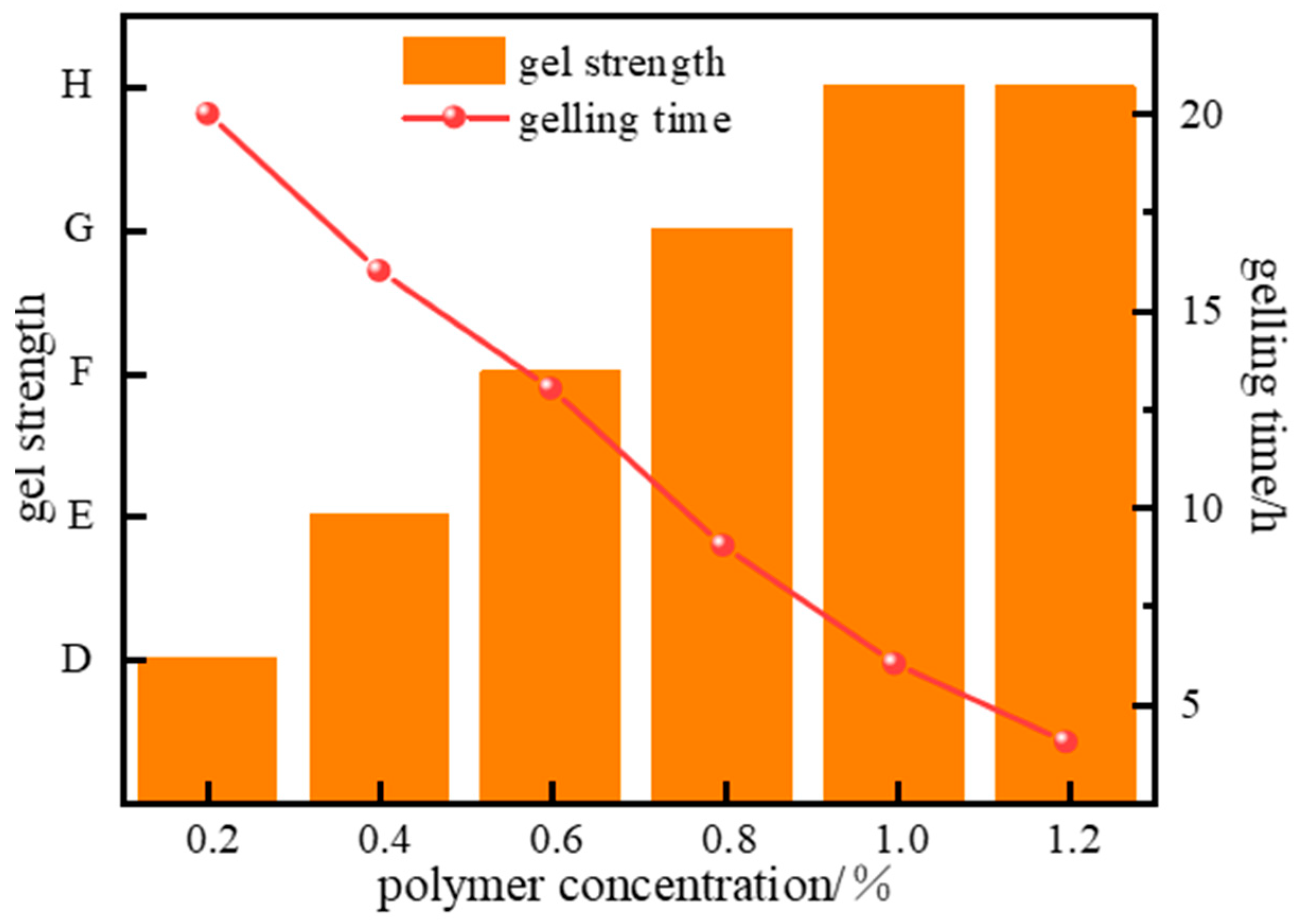
2.1.1. Optimization of Polymer Type and Concentration
2.1.2. Optimization of Crosslinker Type and Concentration
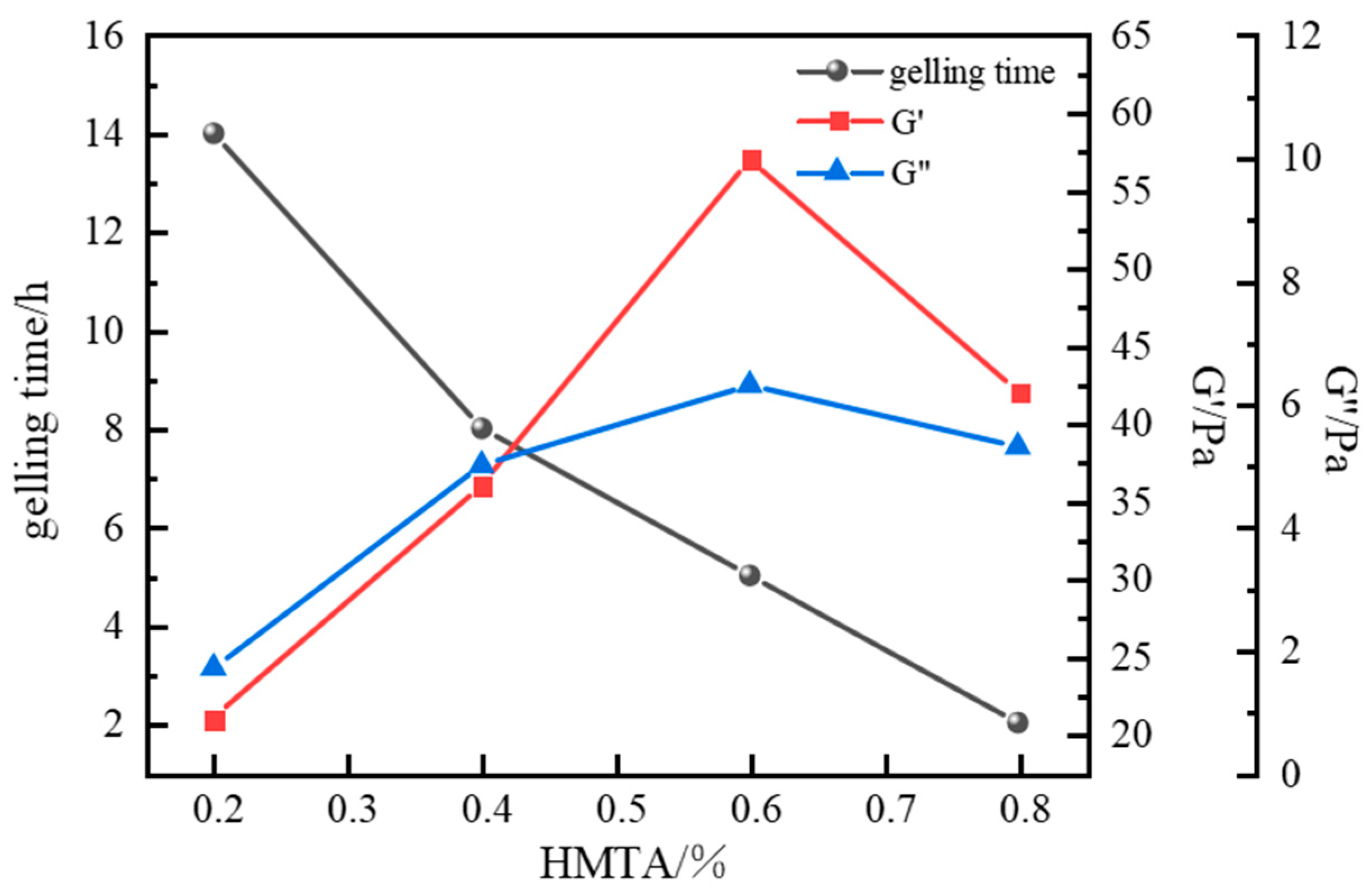
Optimization of Aldehyde Crosslinker
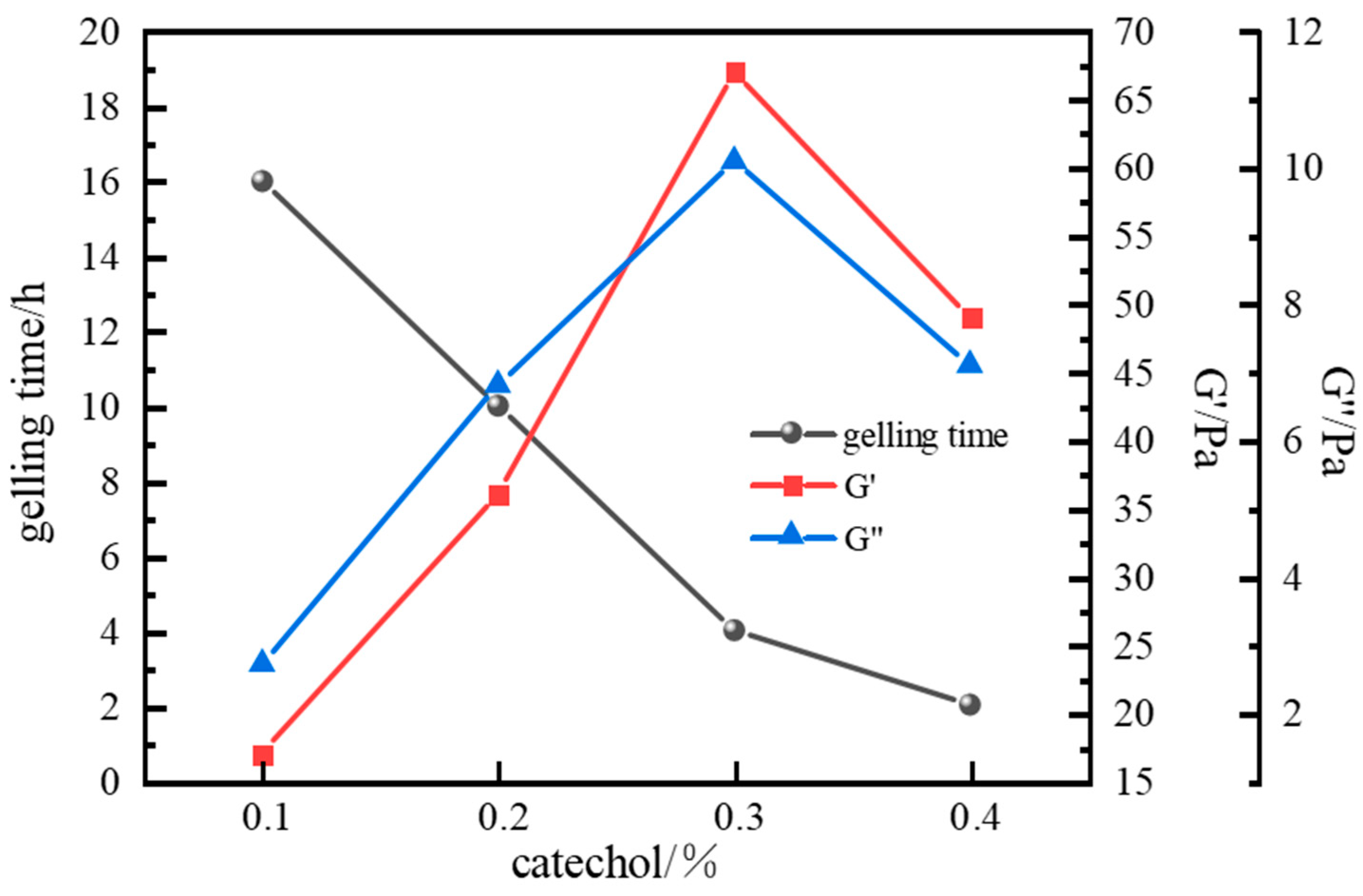
Optimization of Phenolic Crosslinker
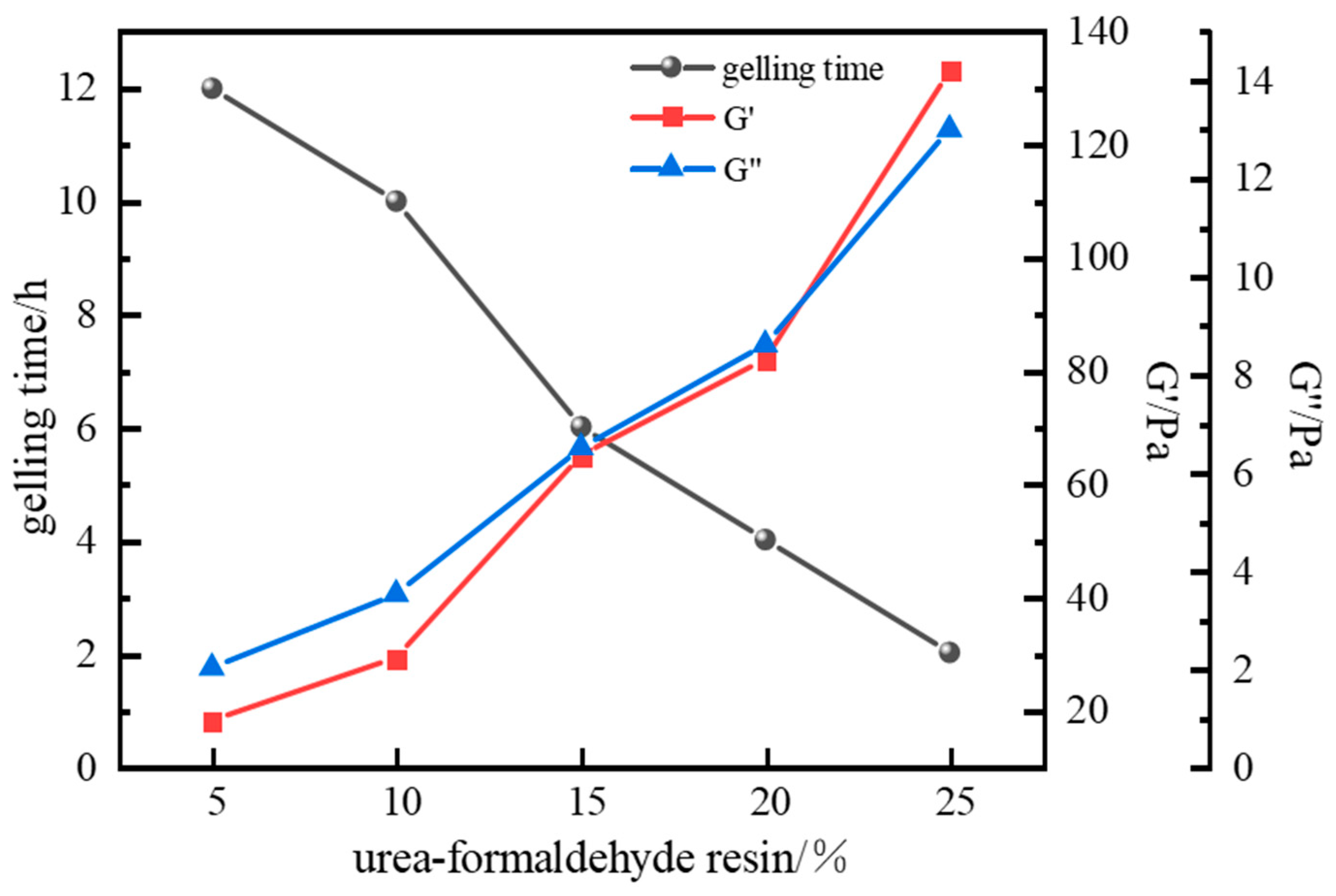
2.1.3. Optimization of Resin Hardener Type and Concentration
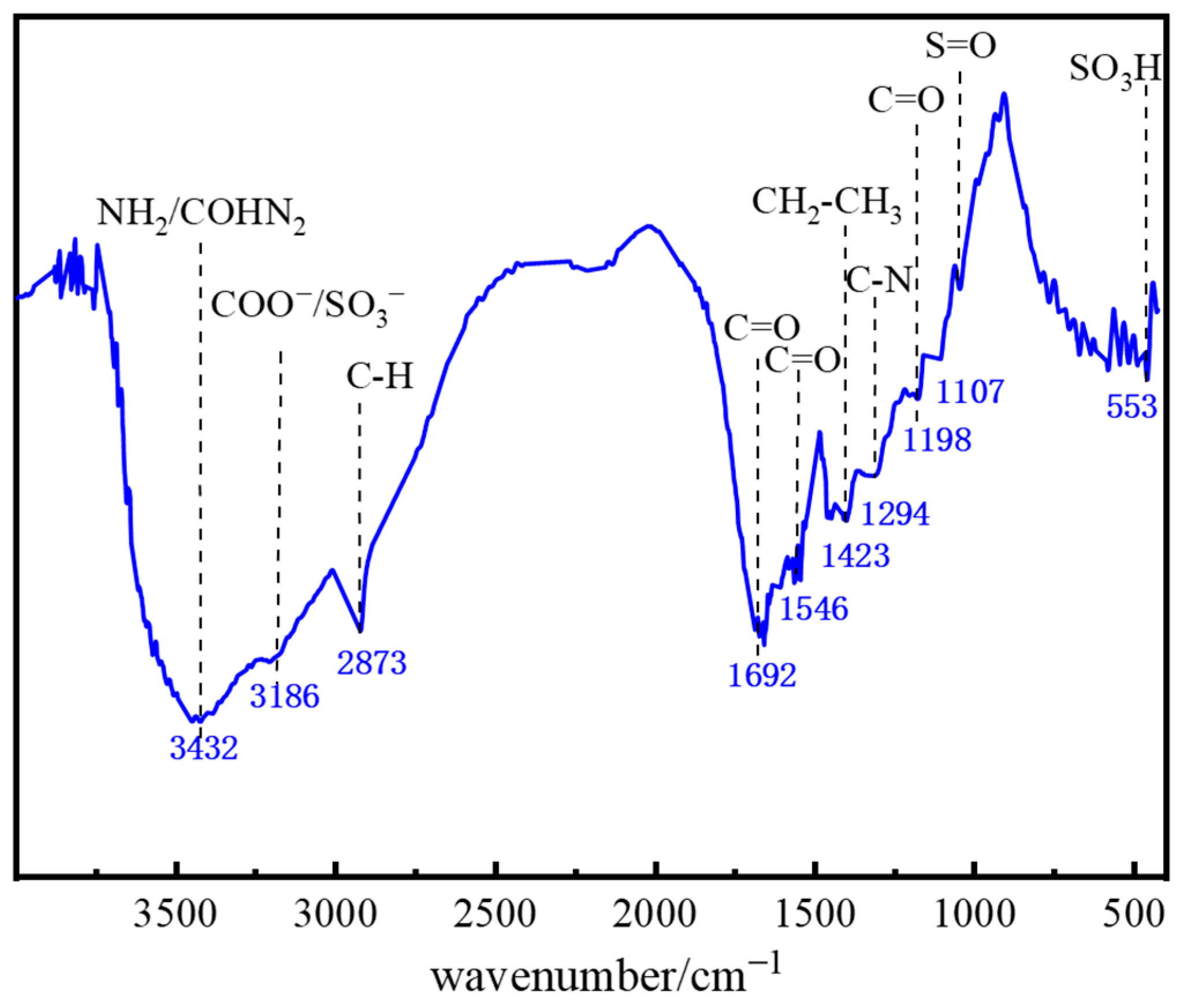
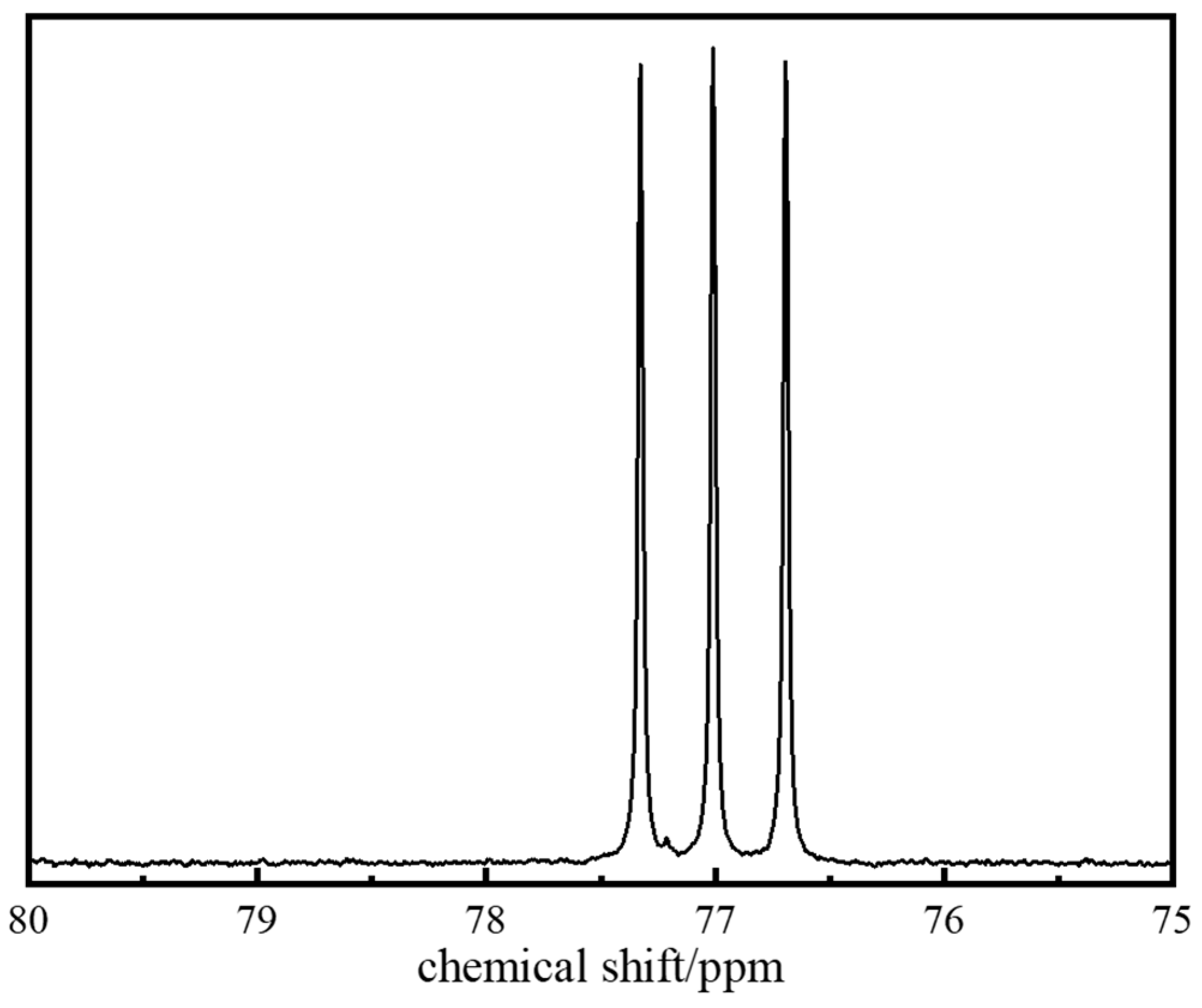
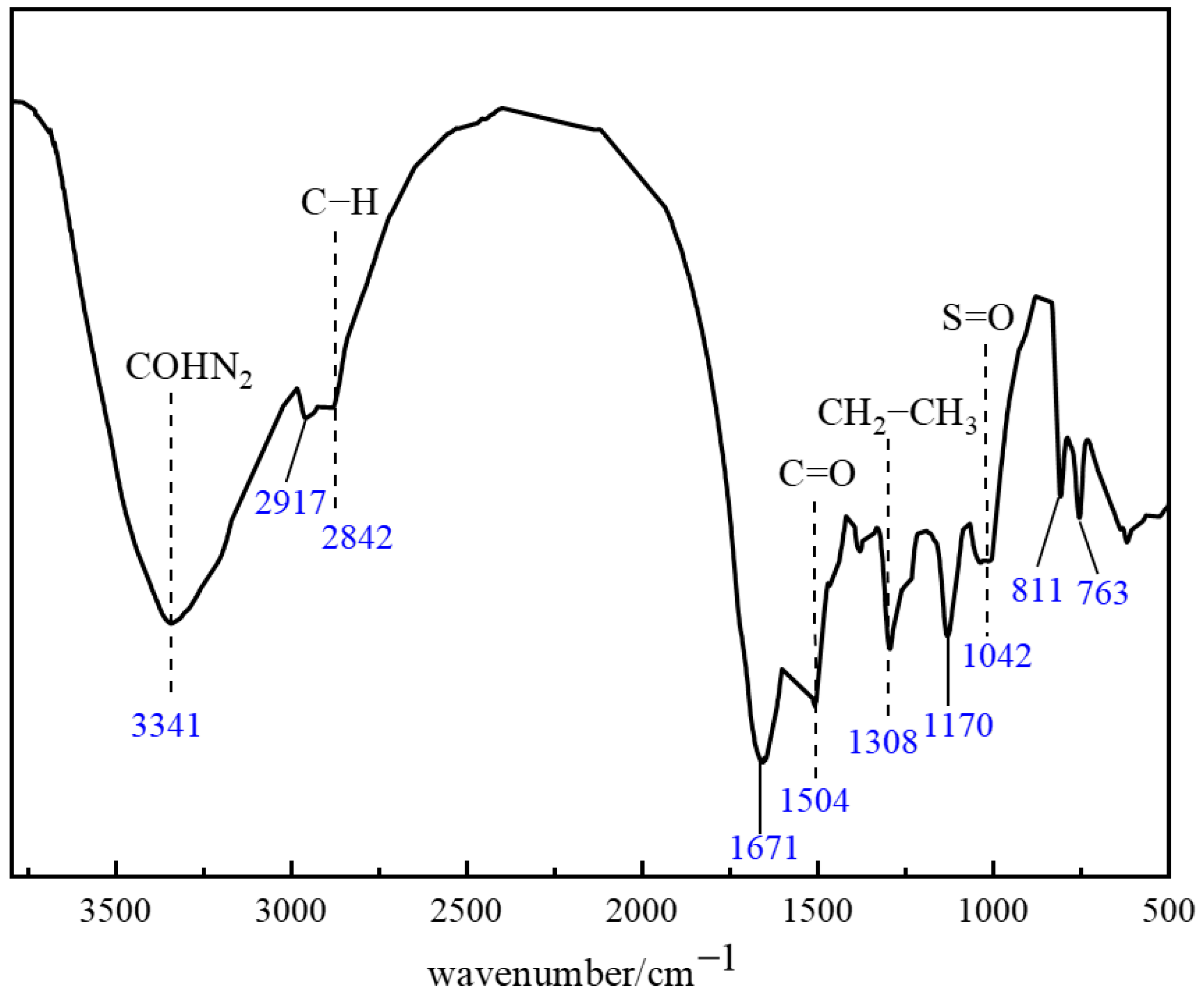
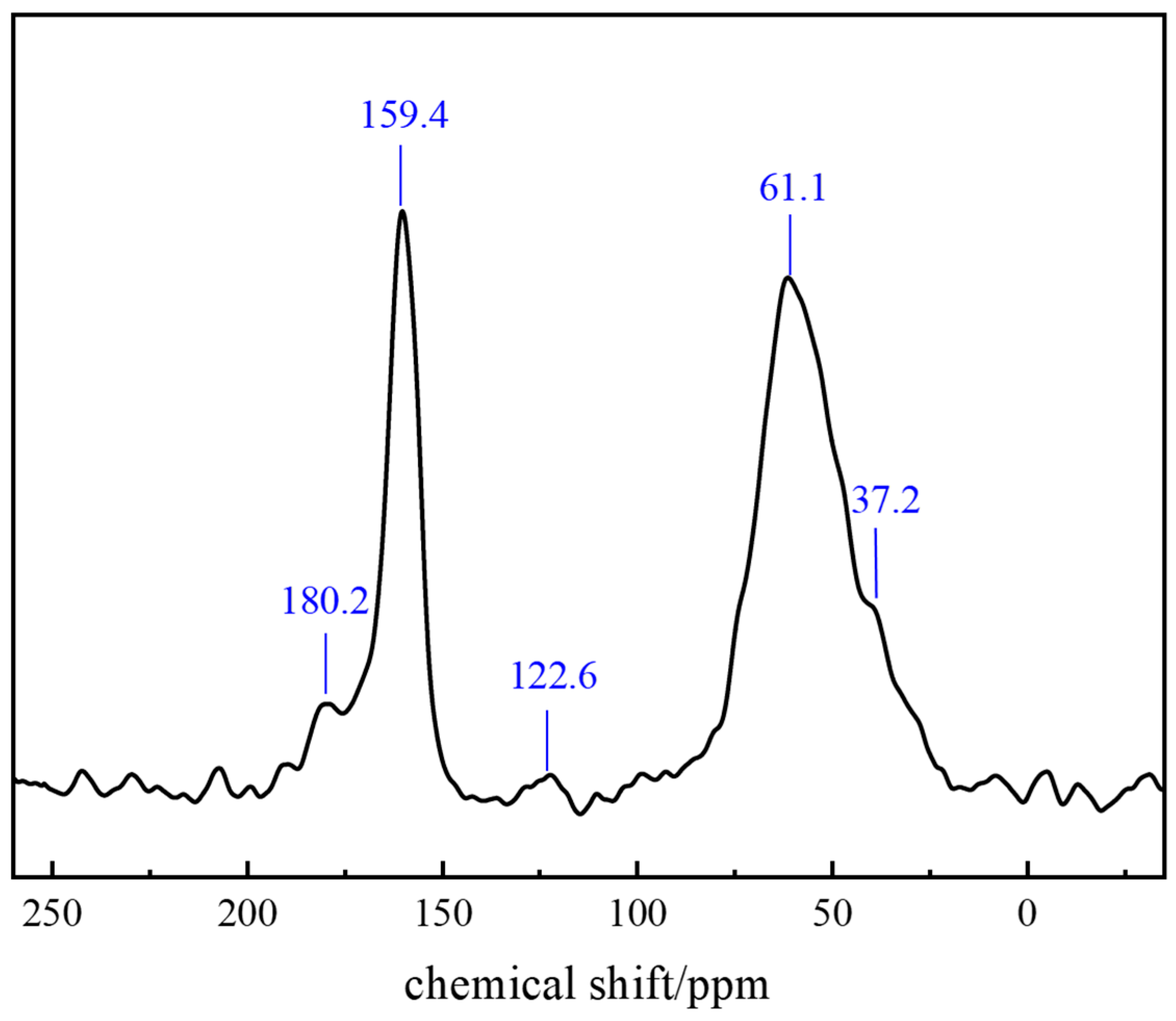
2.1.4. Molecular Structure Characterization of the Polymer Gel System
2.2. Study on Gelation Performance of Polymer Gel Systems
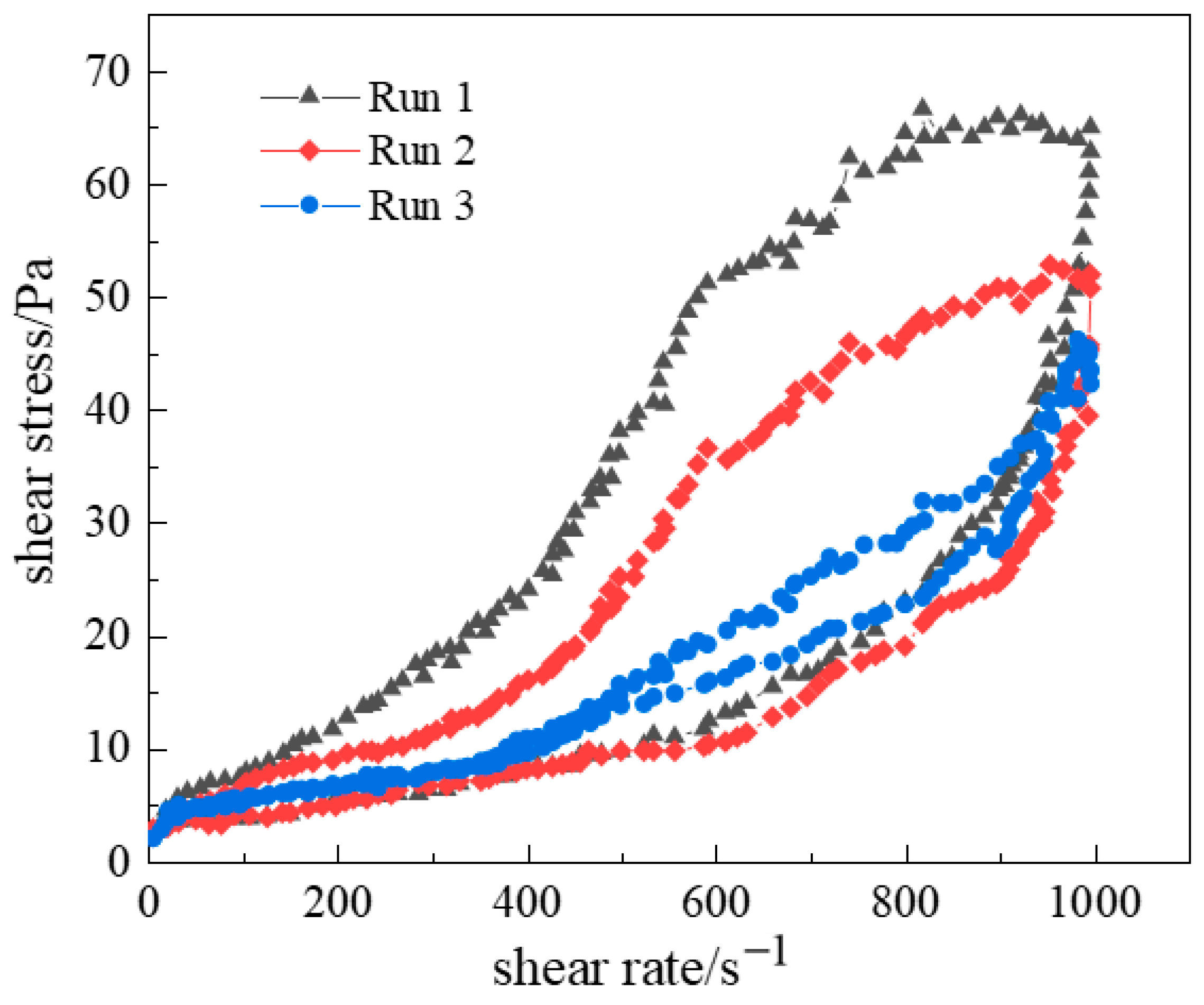
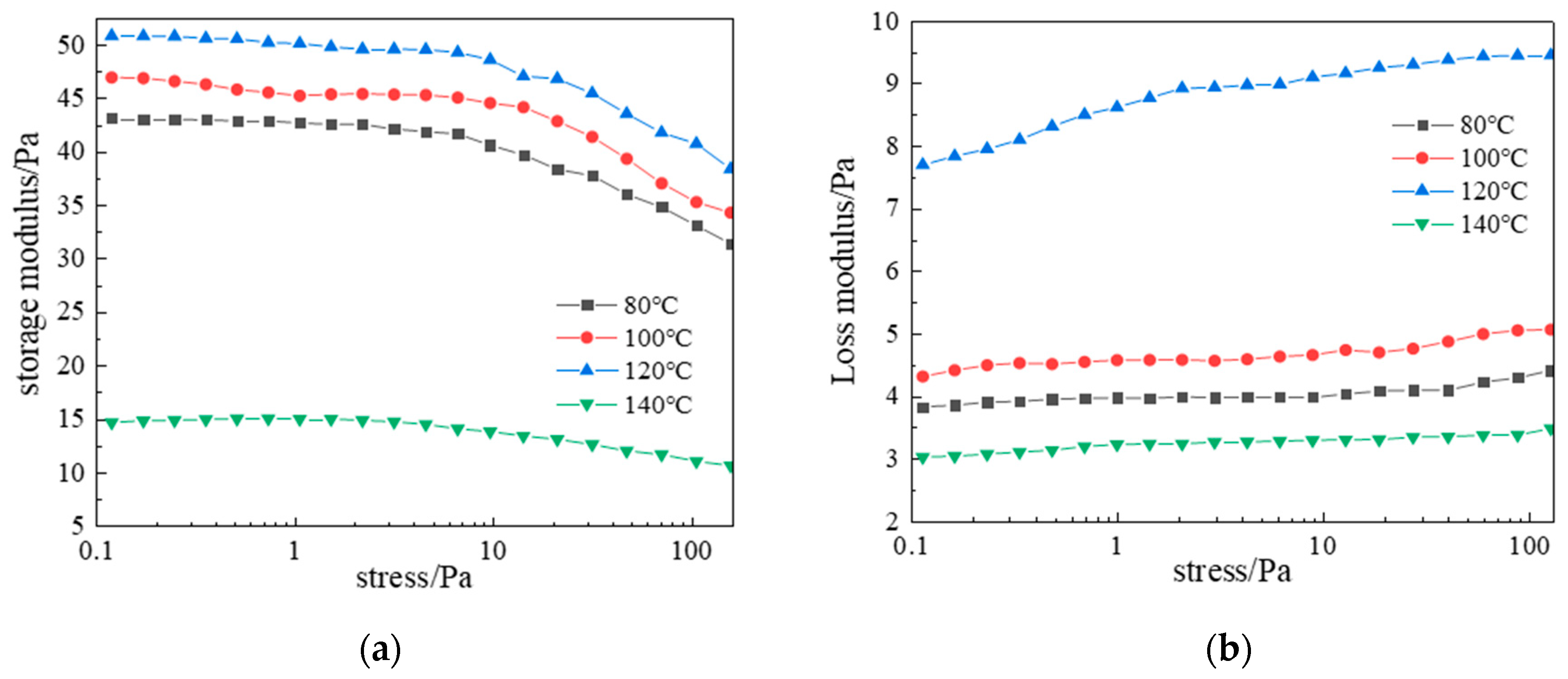
2.2.1. Shear Resistance of Polymer Gel System
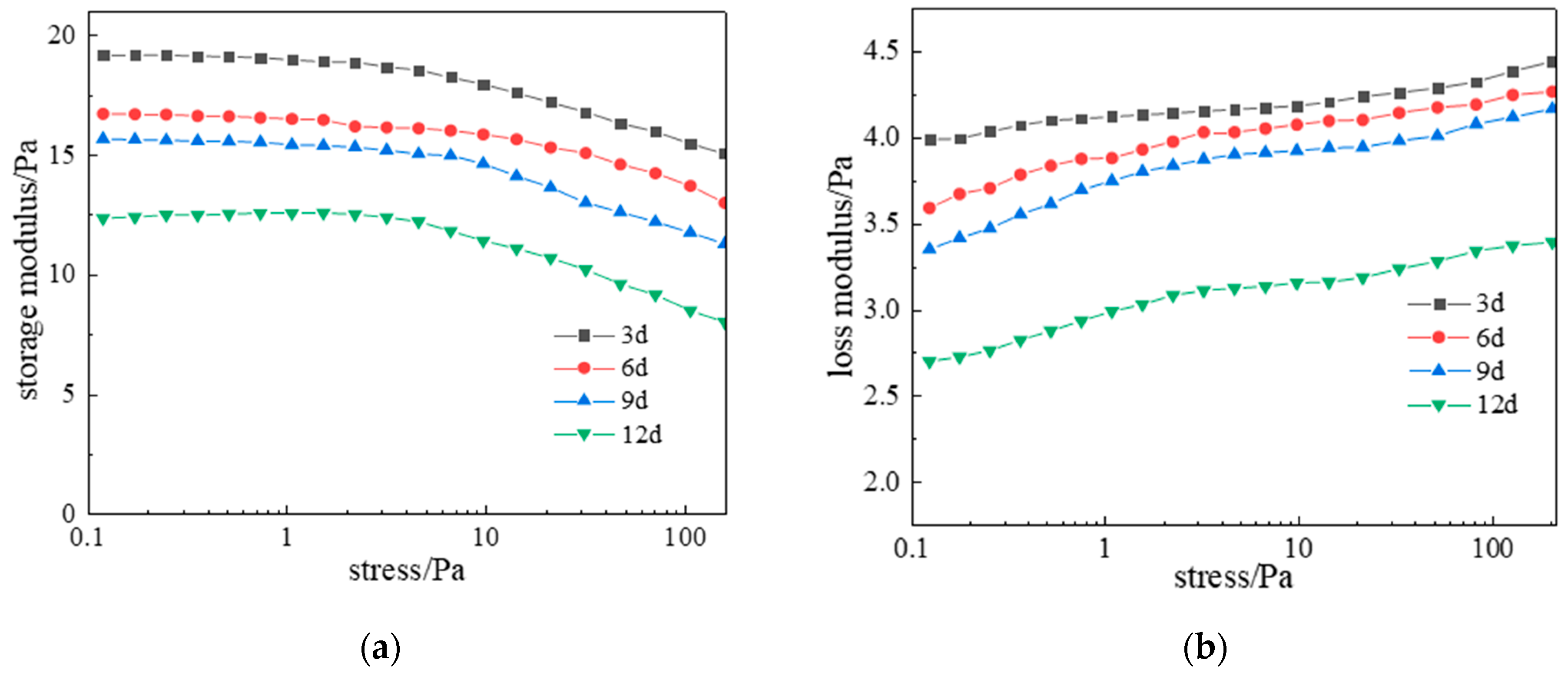
2.2.2. Thixotropic Behavior of Polymer Gel System
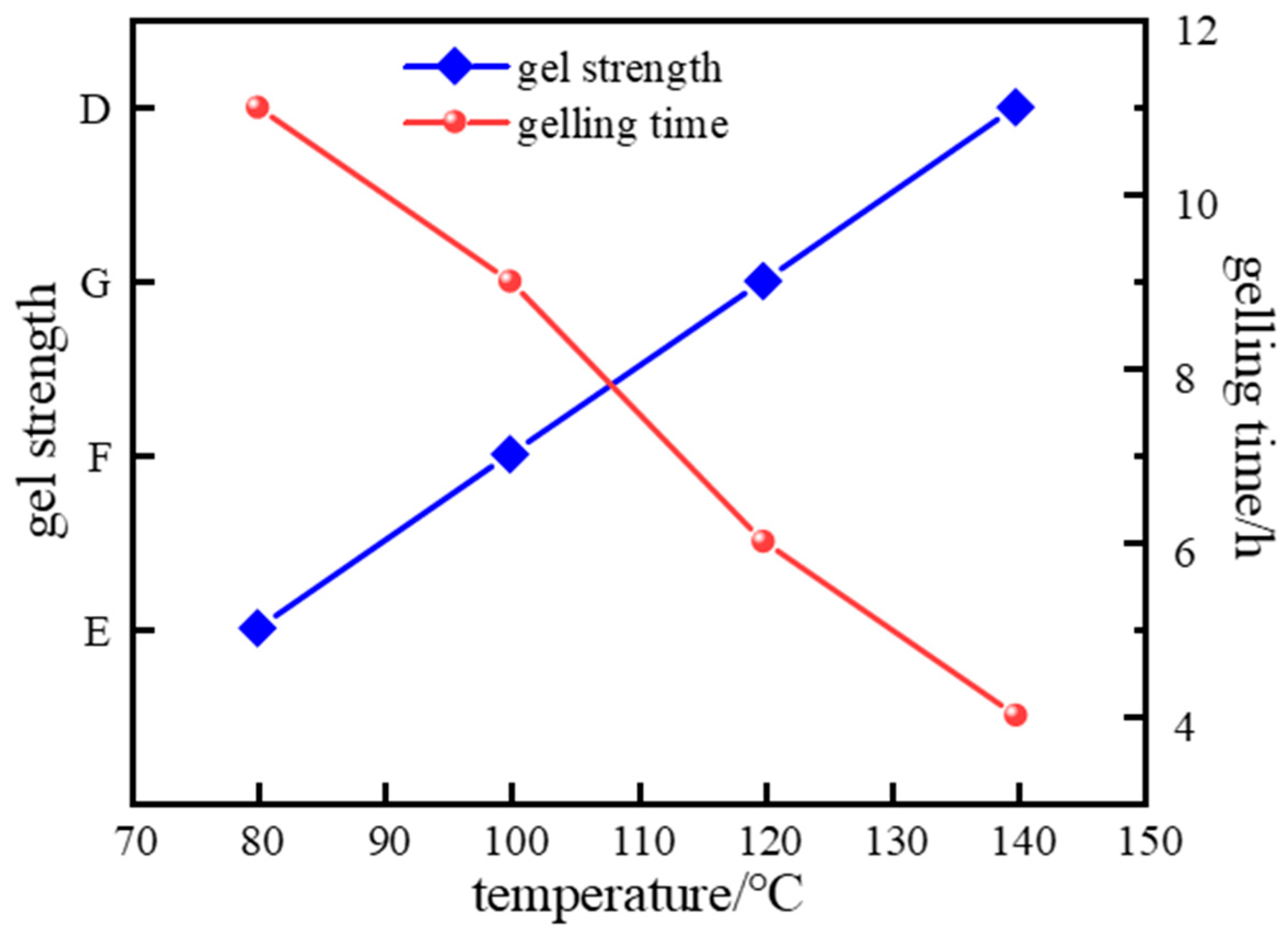
2.2.3. High-Temperature Gelation Performance
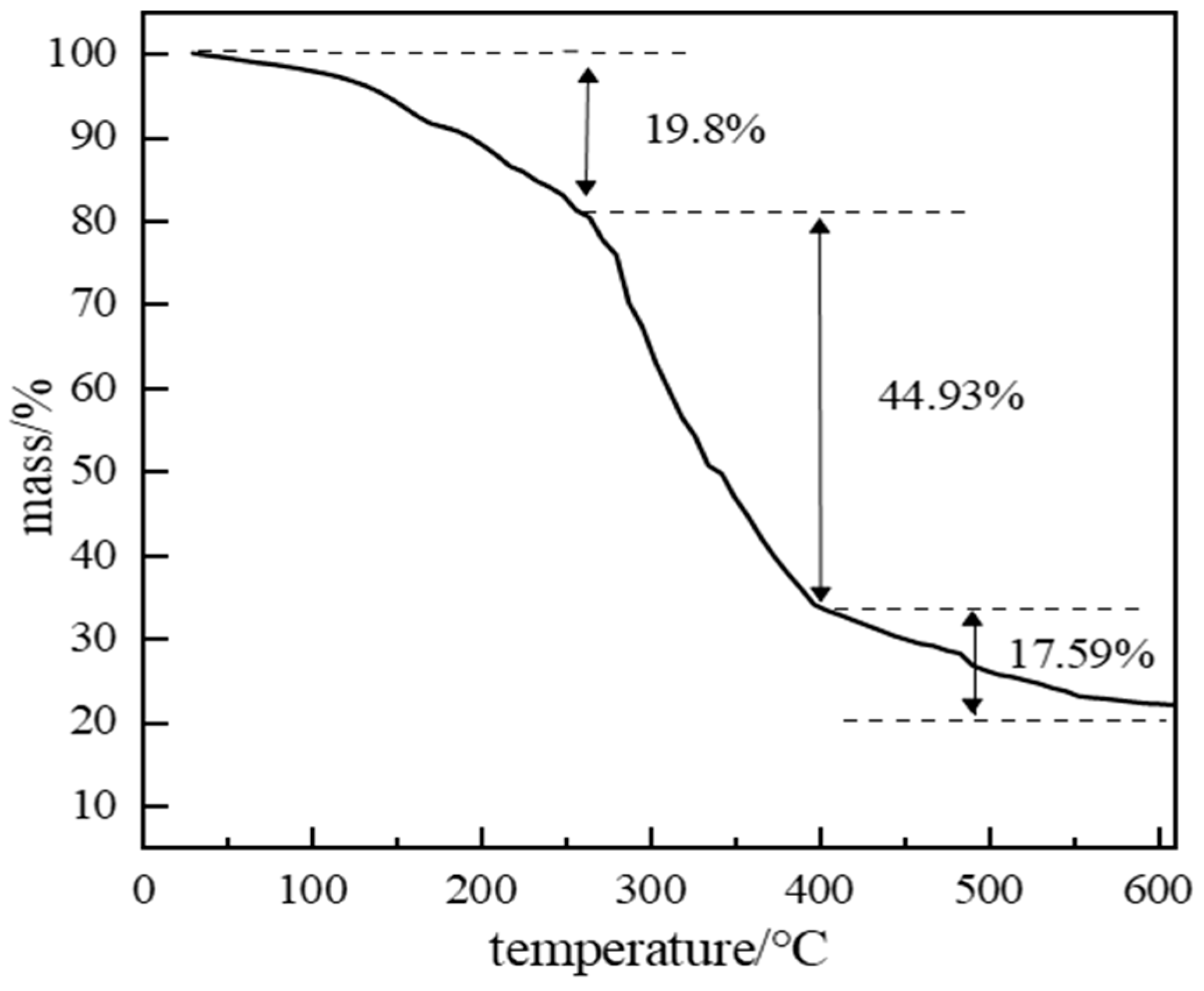
2.2.4. High-Temperature Stability of Polymer Gel System
2.2.5. Self-Filling Capacity of Polymer Gel Systems
2.2.6. Plugging Performance of Polymer Gel System
3. Conclusions
- (1)
- This study introduced the concept of thixotropy to develop a novel polymer gel plugging system. Through optimization of polymer, crosslinker, and resin hardener types/concentrations, the gel formulation was refined to the following: 1% polymer J-1 + 0.3% catechol + 0.6% hexamethylenetetramine (HMTA) + 15% resin hardener.
- (2)
- Rheological characterization demonstrated exceptional performance: At 120 °C, apparent viscosity reached 1471 mPa·s under low shear (0.1 s−1) and maintained 931 mPa·s at high shear (100 s−1), confirming superior shear resistance. Hysteresis in cyclic shear tests validated excellent thixotropy through reversible structure breakdown/rebuilding. After 9 days of aging at high temperatures, minimal strength reduction (G′ decreased from 17.5 to 16 Pa) demonstrated outstanding thermal stability.
- (3)
- The engineered system exhibited uniform migration with negligible gravitational effects, enabling complete filling of fractures across dimensions. Plugging capacities of 166 kPa/m (water flooding) and 122 kPa/m (gas flooding) after 1 day of aging at 120 °C, with significantly enhanced pressure-bearing capability when weighted. Effective plugging in vertical fractures, achieving 105.6 kPa pressure capacity in 6 mm fractures through the formation of intact gel barriers. This thixotropic polymer gel system provides a technically robust solution for controlling lost circulation in challenging high-temperature formations.
4. Experimental Materials and Methods
4.1. Experimental Materials
4.1.1. Experimental Reagents
4.1.2. Experimental Instruments
4.2. Experimental Methods
4.2.1. Preparation of Polymer Solution
4.2.2. Evaluation of Gelling Time and Strength
4.2.3. Rheological Strength Testing
4.2.4. Microstructural Analysis
4.2.5. FTIR Characterization
4.2.6. Thermogravimetric Analysis (TGA)
4.2.7. Nuclear Magnetic Resonance (NMR)
4.2.8. Plugging Performance Evaluation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yan, X.; Kang, Y.; Xu, C.; Shang, X.; You, Z.; Zhang, J. Fracture plugging zone for lost circulation control in fractured reservoirs: Multiscale structure and structure characterization methods. Powder Technol. 2020, 370, 159–175. [Google Scholar] [CrossRef]
- Fang, J.; Zhang, X.; Li, L.; Zhang, J.; Shi, X.; Hu, G. Research progress of high-temperature resistant functional gel materials and their application in oil and gas drilling. Gels 2023, 9, 34. [Google Scholar] [CrossRef]
- Guo, P.; Qiu, Z.; Zang, X.; Zhong, H.; Zhao, X.; Zhang, Y.; Mu, T. Epoxy resin microencapsulated by complex coacervation as physical-chemical synergetic lost circulation control material. Energy 2024, 293, 130630. [Google Scholar] [CrossRef]
- He, Z.; Fan, S.; Fang, J.; Yu, Y.; Zhang, J.; Li, S.; Xu, P. Research and application of fast plugging method for fault zone formation in Tarim Basin, China. Energies 2023, 16, 4330. [Google Scholar] [CrossRef]
- Bai, Y.; Liu, Y.; Sun, J.; Lv, K. Plugging mechanism of rigid and flexible composite plugging materials for millimeter-scale fractures. SPE J. 2024, 29, 1786–1801. [Google Scholar] [CrossRef]
- Wang, K.; Guo, Y.; Wen, J.; Yang, H.; Zhang, H. Magnetic smart polymer gel with directional plugging for conformance control in oil reservoirs. J. Mol. Liq. 2024, 405, 125046. [Google Scholar] [CrossRef]
- Bai, Y.; Lang, Y.; Wang, P.; Wang, R.; Bai, Y.; Sun, J. Fracture plugging performance and mechanism of adhesive material in bridging system during well drilling. Geoenergy Sci. Eng. 2025, 247, 213729. [Google Scholar] [CrossRef]
- Pu, W.; Tian, K.; Wang, H. Synthesis of a low viscosity amphoteric polyacrylamide and its application in oilfield profile control. J. Appl. Polym. Sci. 2022, 139, e52981. [Google Scholar] [CrossRef]
- Seright, R. Gel propagation through fractures. SPE Prod. Facil. 2001, 16, 225–231. [Google Scholar] [CrossRef]
- Chen, L.; Huang, F.; Li, G.; Mao, Z.; Hu, Y.; Liu, L.; Zeng, H.; Xu, S. Experimental study on fiber balls for bridging in fractured-vuggy reservoir. SPE J. 2023, 28, 1880–1894. [Google Scholar] [CrossRef]
- Xu, Z.; Zhao, M.; Sun, N.; Meng, X.; Yang, Z.; Xie, Y.; Ding, F.; Dong, Y.; Gao, M.; Wu, Y.; et al. Delayed crosslinking gel fracturing fluid with dually crosslinked polymer network for ultra-deep reservoir: Performance and delayed crosslinking mechanism. Colloids Surf. A Physicochem. Eng. Asp. 2025, 708, 135967. [Google Scholar] [CrossRef]
- Zhai, K.; Yi, H.; Liu, Y.; Geng, Y.; Fan, S.; Zhu, D. Experimental evaluation of the shielded temporary plugging system composed of calcium carbonate and acid-soluble preformed particle gels (ASPPG) for petroleum drilling. Energy Fuels 2020, 34, 14023–14033. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Y.; Huang, W.; Yang, X.; Liu, Z.; Zhang, X. Preparation and performance evaluation of a plugging agent with an interpenetrating polymer network. Gels 2023, 9, 205. [Google Scholar] [CrossRef]
- Bu, Y.; Wang, G.; Ren, M.; Wang, D.; Du, J.; Guo, S.; Liu, H. Development of a plugging material featuring external flexibility and internal rigidity. Colloids Surf. A Physicochem. Eng. Asp. 2024, 697, 134294. [Google Scholar] [CrossRef]
- Ye, S.; Yang, Y.; Wu, Z.; Yang, J.; Liang, K. Preparation and performance evaluation of self-degrading temporary plugging agent for well workover of fractured reservoir. J. Phys. Conf. Ser. 2024, 2834, 012031. [Google Scholar] [CrossRef]
- Feng, J.; Zheng, L.; Hao, Y.; Hui, B.; Yang, H.; Hao, H.; Liu, F.; Li, L.; Li, Y. Development and field application of downhole crosslinking plugging agent. Mater. Express 2021, 11, 1887–1891. [Google Scholar] [CrossRef]
- Bai, Y.; Zhang, Q.; Sun, J.; Jiang, G.; Lv, k. Double network self-healing hydrogel based on hydrophobic association and ionic bond for formation plugging. Pet. Sci. 2022, 19, 2150–2164. [Google Scholar] [CrossRef]
- Chen, J.; Qiu, H.; Djouonkep, L.D.W.; Lv, J.; Xie, B. Preparation, evaluation and field application of thermally induced crosslinked polymer gel leakage plugging agent. J. Polym. Environ. 2024, 32, 5677–5688. [Google Scholar] [CrossRef]
- Albonico, P.; Lockhart, T. Divalent ion-resistant polymer gels for high-temperature applications: Syneresis inhibiting additives. In Proceedings of the SPE International Symposium on Oilfield Chemistry, Richardson, TX, USA, 2–5 March 1993. [Google Scholar]
- Moradi-Araghi, A. A review of thermally stable gels for fluid diversion in petroleum production. J. Pet. Sci. Eng. 2000, 26, 1–10. [Google Scholar] [CrossRef]
- Sengupta, B.; Sharma, V.; Udayabhanu, G. Gelation studies of an organically cross-linked polyacrylamide water shut-off gel system at different temperatures and pH. J. Pet. Sci. Eng. 2012, 81, 145–150. [Google Scholar] [CrossRef]
- Liu, X.; Li, Q.; Li, B.; Chen, W. A high-efficiency self-healing cementitious material based on supramolecular hydrogels impregnated with phosphate and ammonium. Cem. Concr. Res. 2021, 144, 106427. [Google Scholar] [CrossRef]
- Bai, Y.; Liu, Y.; Yang, K.; Lang, Y. Application and research prospect of functional polymer gels in oil and gas drilling and development engineering. Gels 2023, 9, 413. [Google Scholar] [CrossRef]
- Feng, X.; Cao, X.; Li, L.; Li, Z.; Zhang, Q.; Sun, W.; Hou, B.; Liu, C.; Shi, Z. Study on performance and engineering application of novel expansive superfine cement slurry. Materials 2024, 17, 5597. [Google Scholar] [CrossRef]
- Du, G.; Peng, Y.; Pei, Y.; Zhao, L.; Wen, Z.; Hu, Z. Thermo-responsive temporary plugging agent based on multiple phase transition supramolecular gel. Energy Fuels 2017, 31, 9283–9289. [Google Scholar] [CrossRef]
- Bai, X.; Wang, M.; Chen, Y.; Wu, L.; Yu, J.; Luo, Y. Synthesis and properties of self-healing hydrogel plugging agent. J. Appl. Polym. Sci. 2024, 141, e55005. [Google Scholar] [CrossRef]
- Degaki, H.; Jorgensen, L.; Koga, T.; Narita, T. Molecular weight determination of Kuhn monomers from a dynamic property of polymers. Macromolecules 2025, 58, 314–320. [Google Scholar] [CrossRef]
- Shin, W.; Ko, W.; Jin, S.-H.; Earmme, T.; Hwang, Y.-J. Reproducible and rapid synthesis of a conjugated polymer by Stille polycondensation in flow: Effects of reaction parameters on molecular weight. Chem. Eng. J. 2021, 412, 128572. [Google Scholar] [CrossRef]
- Matsuzawa, T.; Tai, R.; Mano, H.; Ogura, I. Applicability of enzymatic and phenol–sulfuric acid methods for determination of cellulose nanofibers in ecotoxicity testing. J. Wood Sci. 2024, 70, 17. [Google Scholar] [CrossRef]
- Cao, W.; Xie, K.; Lu, X.; Liu, Y.; Zhang, Y. Effect of profile-control oil-displacement agent on increasing oil recovery and its mechanism. Fuel 2019, 237, 1151–1160. [Google Scholar] [CrossRef]
- Li, Z.; Ma, H.; Zheng, H.; Li, Z.; Meng, F.; Liu, R.; Oguzie, E.E.; Liu, L. Urea-formaldehyde resin covered etched basalt as durable composite coating with antibacterial activity and corrosion resistance. Corros. Sci. 2022, 209, 110760. [Google Scholar] [CrossRef]
- Sydansk, R.D.; Argabright, P.A. Conformance Improvement in a Subterranean Hydrocarbon-Bearing Formation Using a Polymer Gel. U.S. Patent US4683949A, 4 August 1987. [Google Scholar]
- Zhao, G.; Dai, C.; Chen, A.; Yan, Z.; Zhao, M. Experimental study and application of gels formed by nonionic polyacrylamide and phenolic resin for in-depth profile control. J. Pet. Sci. Eng. 2015, 135, 552–560. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aldehyde Crosslinker and Concentration (%) | Catechol Concentration (%) | Gelling Time (h) | Gelation Quality and Stability | |
|---|---|---|---|---|
| Formaldehyde | 0.1 | 0.3 | 2 | Gel strength reached Grade G; Severe dehydration and degradation after 6 h aging |
| 0.3 | 1 | Gel strength reached Grade G; Severe dehydration and degradation after 5 h aging | ||
| 0.6 | 1 | Gel strength reached Grade G; Severe dehydration and degradation after 5 h aging | ||
| Paraformaldehyde | 0.1 | 0.3 | No gelation | - |
| 0.3 | No gelation | - | ||
| 0.6 | No gelation | - | ||
| HMTA | 0.1 | 0.3 | 15 | Gel strength Grade G; <10% dehydration after 7 d |
| 0.3 | 11 | Gel strength Grade H; <10% dehydration after 7 d | ||
| 0.6 | 6 | Gel strength Grade H; <10% dehydration after 7 d | ||
| Crosslinker and Concentration (%) | Polymer Concentration (%) | Gelling Time (h) | Gelation Quality and Stability |
|---|---|---|---|
| 0.3% Phenol + 0.6% HMTA | 0.2 | - | Strength too weak |
| 0.4 | - | Strength too weak | |
| 0.6 | - | Strength too weak | |
| 0.8 | - | Strength too weak | |
| 1.0 | - | Strength too weak | |
| 0.3% Hydroquinone + 0.6% HMTA | 0.2 | 15 | Gel strength Grade D; <35% dehydration after 7 d |
| 0.4 | 14 | Gel strength Grade D; <35% dehydration after 7 d | |
| 0.6 | 12 | Gel strength Grade D; <35% dehydration after 7 d | |
| 0.8 | 10 | Gel strength Grade E; <30% dehydration after 7 d | |
| 1.0 | 8 | Gel strength Grade E; <30% dehydration after 7 d | |
| 0.3% Catechol + 0.6% HMTA | 0.2 | 16 | Gel strength Grade D; <10% dehydration after 7 d |
| 0.4 | 14 | Gel strength Grade D; <10% dehydration after 7 d | |
| 0.6 | 11 | Gel strength Grade E; <10% dehydration after 7 d | |
| 0.8 | 8 | Gel strength Grade F; <10% dehydration after 7 d | |
| 1.0 | 6 | Gel strength Grade H; <10% dehydration after 7 d |
| Fracture Width | Injection Volume | Primary Filling Zone | Filling Degree |
|---|---|---|---|
| 5 mm | 0.2 PV | Mid-fracture | 24% |
| 0.3 PV | Mid-fracture | 32% | |
| 0.4 PV | Mid-fracture | 45% | |
| 0.5 PV | Mid-fracture | 53% | |
| 0.6 PV | Bottom | 62% | |
| 0.7 PV | Bottom | 75% | |
| 0.8 PV | Bottom | 90% | |
| 1.0 PV | Mid-fracture | 100% |
| Sample | Length (cm) | Temp (°C) | Aging (d) | Pressure Gradient. (kPa/m) |
|---|---|---|---|---|
| 1 (Water flood) | 50 | 120 | 1.0 | 166 |
| 2 (Water flood) | 50 | 120 | 3.0 | 148 |
| 3 (Water flood) | 50 | 140 | 1.0 | 142 |
| 4 (Water flood) | 50 | 140 | 3.0 | 108 |
| 5 (Gas flood) | 50 | 120 | 1.0 | 122 |
| 6 (Gas flood) | 50 | 120 | 3.0 | 100 |
| 7 (Gas flood) | 50 | 140 | 1.0 | 84 |
| 8 (Gas flood) | 50 | 140 | 3.0 | 66 |
| ID | Fracture Width (mm) | Plugging Strength (kPa) |
|---|---|---|
| 1 | 6 | 105.6 |
| 2 | 8 | 88.3 |
| 3 | 10 | 69.7 |
| ID | Instrument Name | Manufacturer |
|---|---|---|
| 1 | TGA/DTA Thermogravimetric Analyzer | METTLER TOLEDO (Greifensee, Switzerland) |
| 2 | High-Temperature/High-Pressure Leak-Plugging and Displacement Apparatus | Nantong Xinhua Cheng Scientific Instrument Co., Ltd. (Nantong, China) |
| 3 | Quanta 200F Field Emission Scanning Electron Microscope (FE-SEM) | FEI (Morristown, NJ, USA) |
| 4 | HAAKE MARS 60 Rheometer | Thermo Fisher Scientific (Dreieich, Germany) |
| 5 | Fourier Transform Infrared Spectrometer (FTIR-7600) | Shanghai Precision Instrument Co., Ltd. (Shanghai, China) |
| 6 | Constant Temperature Blast Drying Oven | Beijing Heng Aode Instrument (Beijing, China) |
| 7 | Visualized Fracture Model | Nantong Xinhua Cheng Scientific Instrument Co., Ltd. (Nantong, China) |
| 8 | Magnetic Stirrer | Shanghai NIYUE Instrument Co., Ltd. (Shanghai, China) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, Y.; Tang, T.; Ou, B.; Wu, J.; Liu, Y.; Yang, J. High Strength and Strong Thixotropic Gel Suitable for Oil and Gas Drilling in Fractured Formation. Gels 2025, 11, 578. https://doi.org/10.3390/gels11080578
Yan Y, Tang T, Ou B, Wu J, Liu Y, Yang J. High Strength and Strong Thixotropic Gel Suitable for Oil and Gas Drilling in Fractured Formation. Gels. 2025; 11(8):578. https://doi.org/10.3390/gels11080578
Chicago/Turabian StyleYan, Yancheng, Tao Tang, Biao Ou, Jianzhong Wu, Yuan Liu, and Jingbin Yang. 2025. "High Strength and Strong Thixotropic Gel Suitable for Oil and Gas Drilling in Fractured Formation" Gels 11, no. 8: 578. https://doi.org/10.3390/gels11080578
APA StyleYan, Y., Tang, T., Ou, B., Wu, J., Liu, Y., & Yang, J. (2025). High Strength and Strong Thixotropic Gel Suitable for Oil and Gas Drilling in Fractured Formation. Gels, 11(8), 578. https://doi.org/10.3390/gels11080578