Nutrients and Pathways that Regulate Health Span and Life Span

 ,
,  and
and 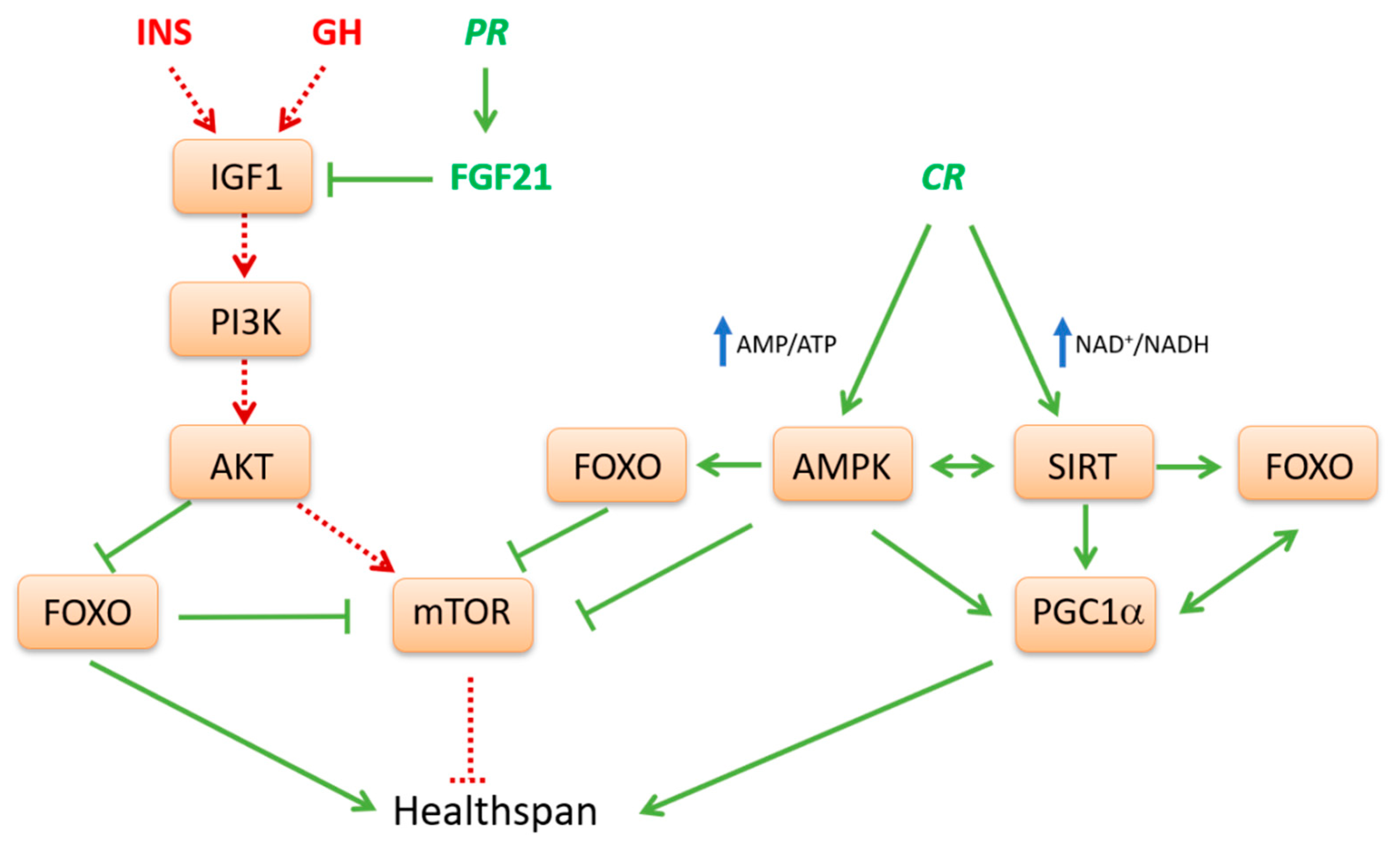{kind=link}
{kind=link}
Abstract
1. Introduction
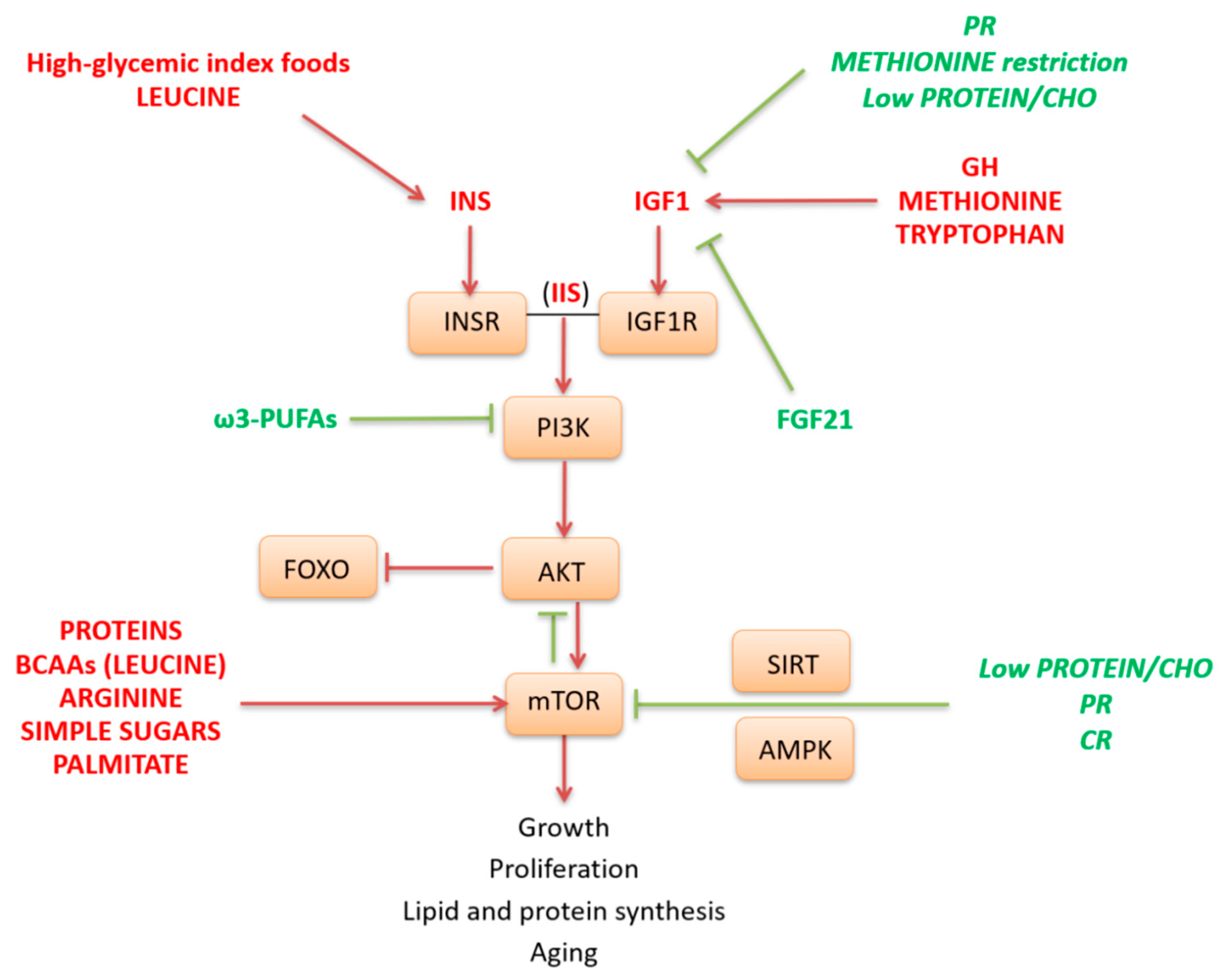
2. Nutrient Sensors that Potentially Affect Health Span
2.1. Sirtuin 1 (SIRT1)
2.2. AMP-Activated Protein Kinase (AMPK)
2.3. Forkhead Box O transcription Factors (FOXOs)
2.4. Fibroblast Growth Factor 21 (FGF21)
2.5. Insulin-Like Growth Factor 1 (IGF1) and Growth Hormone (GH)
2.6. Mammalian Target of Rapamycin (mTOR)
3. Interplay Between Nutrient-Sensing Pathways
4. Dietary Composition and Health Span
4.1. Caloric Restriction (CR)
4.2. Caloric Restriction Mimetic (CRM) Compounds
- (1)
- The classical mTOR inhibitor, rapamycin and novel drugs as rapalogs, having the same molecular scaffold as rapamycin, but with different physiochemical properties [126].
- (2)
- Drugs targeting the NAD+-dependent SIRTs, such as the polyphenol resveratrol, activators of SIRTs and AMPK; other plant-derived metabolites or synthetic drugs having better selectivity for SIRTs named SIRT-activating compounds (STACs). Natural STACs include flavones, chalcones and anthocyanidins [127].
- (3)
- Drugs targeting NAD+ biosynthesis, as nicotinamide mononucleotide (NMN) and nicotinamide riboside (NR), recently tested in humans [128].
- (4)
- Drugs targeting insulin signaling pathways, CHO and fat metabolism, as the biguanide class of antidiabetic drug metformin, known activator of AMPK. Activators of AMPK are also most of the above cited phytonutrients, including resveratrol, green tea polyphenols, quercetin, etc.
- (5)
- Drugs targeting autophagy and inducing epigenetic changes, such as spermidine. This compound is a ubiquitous natural polyamine involved in a wide range of cellular processes thanks to the endogenous synthesis which occurs in all living species. Spermidine has been shown to extend life span in several animal species [129]. In humans, spermidine supplementation resulted to be safe and to have positive effects on cognitive function of older adults [130]. Polyamines, and in particular spermidine, can be found both in animal and vegetable foods such as wheat, mushrooms, fish and cheese and such a dietary component could be particularly useful during aging, when de novo synthesis tends to decrease. Human epidemiological studies correlated dietary polyamine intake with reduced cardiovascular and cancer-related mortality [131]. Moreover, emerging evidence suggests that spermidine may exert a protective effect against an age-related pathology, such as osteoarthritis [132]. Madeo and Eisenberg described the role of this polyamine in aging and diseases [133]. More recently the health benefits of CRM compounds and nutraceuticals and their specific targets have been extensively summarized in a review [134].
4.3. Protein Requirement
5. Nutrients as Epigenetic Modulators of Health Span and Longevity
6. Specific Dietary Pattern and Health Span
Dietary Models
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ekmekcioglu, C. Nutrition and longevity—From mechanisms to uncertainties. Crit. Rev. Food Sci. Nutr. 2019, 60, 3063–3082. [Google Scholar] [CrossRef] [PubMed]
- Heiss, C.; Spyridopoulos, I.; Haendeler, J. Interventions to slow cardiovascular aging: Dietary restriction, drugs and novel molecules. Exp. Gerontol. 2018, 109, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D. Programmed longevity, youthspan, and juventology. Aging Cell 2019, 18, e12843. [Google Scholar] [CrossRef]
- Li, X.; Kazgan, N. Mammalian sirtuins and energy metabolism. Int. J. Biol. Sci. 2011, 7, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Pande, S.; Kratasyuk, V.A.; Medvedeva, N.N.; Kolenchukova, O.A.; Salmina, A.B. Nutritional biomarkers: Current view and future perspectives. Crit. Rev. Food Sci. Nutr. 2017, 58, 3055–3069. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Xu, W.; Jiang, W.; Yu, W.; Lin, Y.; Zhang, T.; Yao, J.; Zhou, L.; Zeng, Y.; Li, H.; et al. Regulation of Cellular Metabolism by Protein Lysine Acetylation. Science 2010, 327, 1000–1004. [Google Scholar] [CrossRef]
- Ponugoti, B.; Kim, D.-H.; Xiao, Z.; Smith, Z.; Miao, J.; Zang, M.; Wu, S.-Y.; Chiang, C.-M.; Veenstra, T.D.; Kemper, J.K. SIRT1 Deacetylates and Inhibits SREBP-1C Activity in Regulation of Hepatic Lipid Metabolism. J. Biol. Chem. 2010, 285, 33959–33970. [Google Scholar] [CrossRef]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; Machado de Oliveira, R.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-γ. Nature 2004, 429, 771–776. [Google Scholar] [CrossRef]
- Baur, J.A.; Ungvari, Z.; Minor, R.K.; Le Couteur, D.G.; de Cabo, R. Are sirtuins viable targets for improving healthspan and lifespan? Nat. Rev. Drug Discov. 2012, 11, 443–461. [Google Scholar] [CrossRef]
- Guarente, L. Calorie restriction and sirtuins revisited. Genes Dev. 2013, 27, 2072–2085. [Google Scholar] [CrossRef]
- Bai, X.; Yao, L.; Ma, X.; Xu, X. Small Molecules as SIRT Modulators. Mini-Rev. Med. Chem. 2018, 18, 1151–1157. [Google Scholar] [CrossRef]
- Escobar, K.A.; Cole, N.H.; Mermier, C.M.; VanDusseldorp, T.A. Autophagy and aging: Maintaining the proteome through exercise and caloric restriction. Aging Cell 2019, 18, e12876. [Google Scholar] [CrossRef]
- Ng, F.; Tang, B.L. Sirtuins’ modulation of autophagy. J. Cell. Physiol. 2013, 228, 2262–2270. [Google Scholar] [CrossRef]
- Ruetenik, A.; Barrientos, A. Dietary restriction, mitochondrial function and aging: From yeast to humans. Biochim. Biophys. Acta 2015, 1847, 1434–1447. [Google Scholar] [CrossRef]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie Restriction Reduces Oxidative Stress by SIRT3-Mediated SOD2 Activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef]
- Donniacuo, M.; Urbanek, K.; Nebbioso, A.; Sodano, L.; Gallo, L.; Altucci, L.; Rinaldi, B. Cardioprotective effect of a moderate and prolonged exercise training involves sirtuin pathway. Life Sci. 2019, 222, 140–147. [Google Scholar] [CrossRef]
- Barja, G. Towards a unified mechanistic theory of aging. Exp. Gerontol. 2019, 124. [Google Scholar] [CrossRef]
- Bai, P.; Canto, C.; Oudart, H.; Brunyanszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef]
- Xu, C.; Wang, L.; Fozouni, P.; Evjen, G.; Chandra, V.; Jiang, J.; Lu, C.; Nicastri, M.; Bretz, C.; Winkler, J.D.; et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nat. Cell Biol. 2020, 22, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Guigas, B.; Leclerc, J.; Hébrard, S.; Lantier, L.; Mounier, R.; Andreelli, F.; Foretz, M. AMP-activated protein kinase in the regulation of hepatic energy metabolism: From physiology to therapeutic perspectives. Acta Physiol. 2009, 196, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Oskoui, P.R.; Banko, M.R.; Maniar, J.M.; Gygi, M.P.; Gygi, S.P.; Brunet, A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem. 2007, 282, 30107–30119. [Google Scholar] [CrossRef] [PubMed]
- Burkewitz, K.; Zhang, Y.; Mair, W.B. AMPK at the Nexus of Energetics and Aging. Cell Metab. 2014, 20, 10–25. [Google Scholar] [CrossRef]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef]
- Piskovatska, V.; Stefanyshyn, N.; Storey, K.B.; Vaiserman, A.M.; Lushchak, O. Metformin as a geroprotector: Experimental and clinical evidence. Biogerontology 2018, 20, 33–48. [Google Scholar] [CrossRef]
- Brown, A.K.; Webb, A.E. Regulation of FOXO Factors in Mammalian Cells. Curr. Top. Dev. Biol. 2018, 127, 165–192. [Google Scholar] [CrossRef]
- Murtaza, G.; Khan, A.K.; Rashid, R.; Muneer, S.; Hasan, S.M.F.; Chen, J. FOXO Transcriptional Factors and Long-Term Living. Oxid. Med. Cell Longev. 2017, 2017, 3494289. [Google Scholar] [CrossRef] [PubMed]
- Link, W. Introduction to FOXO Biology. Methods Mol. Biol. 2019, 1890, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Partridge, L. Promoting health and longevity through diet: From model organisms to humans. Cell 2015, 161, 106–118. [Google Scholar] [CrossRef]
- Mercken, E.M.; Crosby, S.D.; Lamming, D.W.; JeBailey, L.; Krzysik-Walker, S.; Villareal, D.T.; Capri, M.; Franceschi, C.; Zhang, Y.; Becker, K.; et al. Calorie restriction in humans inhibits the PI3K/AKT pathway and induces a younger transcription profile. Aging Cell 2013, 12, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Urbánek, P.; Klotz, L.O. Posttranscriptional regulation ofFOXOexpression: MicroRNAs and beyond. Br. J. Pharmacol. 2017, 174, 1514–1532. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Regulation of longevity by FGF21: Interaction between energy metabolism and stress responses. Ageing Res. Rev. 2017, 37, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xie, Y.; Berglund, E.D.; Coate, K.C.; He, T.T.; Katafuchi, T.; Xiao, G.; Potthoff, M.J.; Wei, W.; Wan, Y.; et al. The starvation hormone, fibroblast growth factor-21, extends lifespan in mice. Elife 2012, 1, e00065. [Google Scholar] [CrossRef]
- Lees, E.K.; Król, E.; Grant, L.; Shearer, K.; Wyse, C.; Moncur, E.; Bykowska, A.S.; Mody, N.; Gettys, T.W.; Delibegovic, M. Methionine restriction restores a younger metabolic phenotype in adult mice with alterations in fibroblast growth factor 21. Aging Cell 2014, 13, 817–827. [Google Scholar] [CrossRef]
- Hill, C.M.; Berthoud, H.-R.; Münzberg, H.; Morrison, C.D. Homeostatic sensing of dietary protein restriction: A case for FGF21. Front. Neuroendocrinol. 2018, 51, 125–131. [Google Scholar] [CrossRef]
- Solon-Biet, S.M.; Cogger, V.C.; Pulpitel, T.; Heblinski, M.; Wahl, D.; McMahon, A.C.; Warren, A.; Durrant-Whyte, J.; Walters, K.A.; Krycer, J.R.; et al. Defining the Nutritional and Metabolic Context of FGF21 Using the Geometric Framework. Cell Metab. 2016, 24, 555–565. [Google Scholar] [CrossRef]
- BonDurant, L.D.; Potthoff, M.J. Fibroblast Growth Factor 21: A Versatile Regulator of Metabolic Homeostasis. Annu. Rev. Nutr. 2018, 38, 173–196. [Google Scholar] [CrossRef]
- Sonoda, J.; Chen, M.Z.; Baruch, A. FGF21-receptor agonists: An emerging therapeutic class for obesity-related diseases. Horm. Mol. Biol. Clin. Investig. 2017, 30. [Google Scholar] [CrossRef] [PubMed]
- Perez-Marti, A.; Garcia-Guasch, M.; Tresserra-Rimbau, A.; Carrilho-Do-Rosario, A.; Estruch, R.; Salas-Salvado, J.; Martinez-Gonzalez, M.A.; Lamuela-Raventos, R.; Marrero, P.F.; Haro, D.; et al. A low-protein diet induces body weight loss and browning of subcutaneous white adipose tissue through enhanced expression of hepatic fibroblast growth factor 21 (FGF21). Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Vainshtein, A.; Sandri, M. Signaling Pathways That Control Muscle Mass. Int. J. Mol. Sci. 2020, 21, 4759. [Google Scholar] [CrossRef]
- Tezze, C.; Romanello, V.; Sandri, M. FGF21 as Modulator of Metabolism in Health and Disease. Front. Physiol. 2019, 10. [Google Scholar] [CrossRef]
- Oost, L.J.; Kustermann, M.; Armani, A.; Blaauw, B.; Romanello, V. Fibroblast growth factor 21 controls mitophagy and muscle mass. J. Cachexia Sarcopenia Muscle 2019, 10, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Hanks, L.J.; Gutiérrez, O.M.; Bamman, M.M.; Ashraf, A.; McCormick, K.L.; Casazza, K. Circulating levels of fibroblast growth factor-21 increase with age independently of body composition indices among healthy individuals. J. Clin. Transl. Endocrinol. 2015, 2, 77–82. [Google Scholar] [CrossRef]
- Tezze, C.; Romanello, V.; Desbats, M.A.; Fadini, G.P.; Albiero, M.; Favaro, G.; Ciciliot, S.; Soriano, M.E.; Morbidoni, V.; Cerqua, C.; et al. Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence. Cell Metab. 2017, 25, 1374–1389. [Google Scholar] [CrossRef]
- Ji, K.; Zheng, J.; Lv, J.; Xu, J.; Ji, X.; Luo, Y.-B.; Li, W.; Zhao, Y.; Yan, C. Skeletal muscle increases FGF21 expression in mitochondrial disorders to compensate for energy metabolic insufficiency by activating the mTOR–YY1–PGC1α pathway. Free Radic. Biol. Med. 2015, 84, 161–170. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Warfel, J.D.; Wicks, S.E.; Ghosh, S.; Salbaum, J.M.; Burk, D.; Dubuisson, O.S.; Mendoza, T.M.; Zhang, J.; Noland, R.C.; et al. Impaired Mitochondrial Fat Oxidation Induces FGF21 in Muscle. Cell Rep. 2016, 15, 1686–1699. [Google Scholar] [CrossRef]
- Haywood, N.J.; Slater, T.A.; Matthews, C.J.; Wheatcroft, S.B. The insulin like growth factor and binding protein family: Novel therapeutic targets in obesity & diabetes. Mol. Metab. 2019, 19, 86–96. [Google Scholar] [CrossRef]
- Junnila, R.K.; List, E.O.; Berryman, D.E.; Murrey, J.W.; Kopchick, J.J. The GH/IGF-1 axis in ageing and longevity. Nat. Rev. Endocrinol. 2013, 9, 366–376. [Google Scholar] [CrossRef]
- Lapierre, L.R.; Hansen, M. Lessons from C. elegans: Signaling pathways for longevity. Trends Endocrinol. Metab. 2012, 23, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Vitale, G.; Pellegrino, G.; Vollery, M.; Hofland, L.J. ROLE of IGF-1 System in the Modulation of Longevity: Controversies and New Insights From a Centenarians’ Perspective. Front. Endocrinol. 2019, 10, 27. [Google Scholar] [CrossRef]
- Yuan, R.; Tsaih, S.-W.; Petkova, S.B.; De Evsikova, C.M.; Xing, S.; Marion, M.A.; Bogue, M.A.; Mills, K.D.; Peters, L.L.; Bult, C.J.; et al. Aging in inbred strains of mice: Study design and interim report on median lifespans and circulating IGF1 levels. Aging Cell 2009, 8, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Milman, S.; Atzmon, G.; Huffman, D.M.; Wan, J.; Crandall, J.P.; Cohen, P.; Barzilai, N. Low insulin-like growth factor-1 level predicts survival in humans with exceptional longevity. Aging Cell 2014, 13, 769–771. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef]
- Villela, T.R.; Freire, B.L.; Braga, N.T.P.; Arantes, R.R.; Funari, M.F.A.; Alexander, J.A.L.; Silva, I.N. Growth Hormone insensitivity (Laron syndrome): Report of a new family and review of Brazilian patients. Genet. Mol. Biol. 2020, 42, e20180197. [Google Scholar] [CrossRef]
- Laron, Z.; Kauli, R. Fifty seven years of follow-up of the Israeli cohort of Laron Syndrome patients-From discovery to treatment. Growth Horm. IGF Res. 2016, 28, 53–56. [Google Scholar] [CrossRef]
- Laron, Z. Epilogue: The future of Laron syndrome—The need for changes. Growth Horm. IGF Res. 2016, 28, 79–80. [Google Scholar] [CrossRef] [PubMed]
- Janecka, A.; Kolodziej-Rzepa, M.; Biesaga, B. Clinical and Molecular Features of Laron Syndrome, A Genetic Disorder Protecting from Cancer. In Vivo 2016, 30, 375–381. [Google Scholar]
- Guevara-Aguirre, J.; Balasubramanian, P.; Guevara-Aguirre, M.; Wei, M.; Madia, F.; Cheng, C.W.; Hwang, D.; Martin-Montalvo, A.; Saavedra, J.; Ingles, S.; et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci. Transl. Med. 2011, 3, 70ra13. [Google Scholar] [CrossRef] [PubMed]
- Vitale, G.; Barbieri, M.; Kamenetskaya, M.; Paolisso, G. GH/IGF-I/insulin system in centenarians. Mech. Ageing Dev. 2017, 165, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Takayama, M.; Gondo, Y.; Inagaki, H.; Yamamura, K.; Nakazawa, S.; Kojima, T.; Ebihara, Y.; Shimizu, K.; Masui, Y.; et al. Adipose Endocrine Function, Insulin-Like Growth Factor-1 Axis, and Exceptional Survival Beyond 100 Years of Age. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2008, 63, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Paolisso, G.; Ammendola, S.; Del Buono, A.; Gambardella, A.; Riondino, M.; Tagliamonte, M.R.; Rizzo, M.R.; Carella, C.; Varricchio, M. Serum Levels of Insulin-Like Growth Factor-I (IGF-I) and IGF-Binding Protein-3 in Healthy Centenarians: Relationship with Plasma Leptin and Lipid Concentrations, Insulin Action, and Cognitive Function. J. Clin. Endocrinol. Metab. 1997, 82, 2204–2209. [Google Scholar] [CrossRef]
- Lytras, A.; Tolis, G. Assessment of endocrine and nutritional status in age-related catabolic states of muscle and bone. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 604–610. [Google Scholar] [CrossRef]
- Kamei, Y.; Hatazawa, Y.; Uchitomi, R.; Yoshimura, R.; Miura, S. Regulation of Skeletal Muscle Function by Amino Acids. Nutrients 2020, 12, 261. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Calorie restriction: Decelerating mTOR-driven aging from cells to organisms (including humans). Cell Cycle 2014, 9, 683–688. [Google Scholar] [CrossRef]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef]
- Weichhart, T. mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology 2018, 64, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Wipperman, M.F.; Montrose, D.C.; Gotto, A.M., Jr.; Hajjar, D.P. Mammalian Target of Rapamycin: A Metabolic Rheostat for Regulating Adipose Tissue Function and Cardiovascular Health. Am. J. Pathol. 2019, 189, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in Mice of the mTORC Components raptor, rictor, or mLST8 Reveals that mTORC2 Is Required for Signaling to Akt-FOXO and PKCα, but Not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Papadopoli, D.; Boulay, K.; Kazak, L.; Pollak, M.; Mallette, F.; Topisirovic, I.; Hulea, L. mTOR as a central regulator of lifespan and aging. F1000Research 2019, 8, 998. [Google Scholar] [CrossRef]
- Templeman, N.M.; Murphy, C.T. Regulation of reproduction and longevity by nutrient-sensing pathways. J. Cell Biol. 2018, 217, 93–106. [Google Scholar] [CrossRef]
- Simpson, S.J.; Raubenheimer, D. Macronutrient balance and lifespan. Aging 2009, 1, 875–880. [Google Scholar] [CrossRef]
- Lushchak, O.; Strilbytska, O.; Piskovatska, V.; Storey, K.B.; Koliada, A.; Vaiserman, A. The role of the TOR pathway in mediating the link between nutrition and longevity. Mech. Ageing Dev. 2017, 164, 127–138. [Google Scholar] [CrossRef]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span—From yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef]
- Albert, V.; Hall, M.N. mTOR signaling in cellular and organismal energetics. Curr. Opin. Cell Biol. 2015, 33, 55–66. [Google Scholar] [CrossRef]
- Kitada, M.; Ogura, Y.; Monno, I.; Koya, D. The impact of dietary protein intake on longevity and metabolic health. EBioMedicine 2019, 43, 632–640. [Google Scholar] [CrossRef]
- Chen, C.C.; Jeon, S.M.; Bhaskar, P.T.; Nogueira, V.; Sundararajan, D.; Tonic, I.; Park, Y.; Hay, N. FoxOs inhibit mTORC1 and activate Akt by inducing the expression of Sestrin3 and Rictor. Dev. Cell 2010, 18, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yan, F.; Feng, Z.; Lazarovici, P.; Zheng, W. Signaling Network of Forkhead Family of Transcription Factors (FOXO) in Dietary Restriction. Cells 2019, 9, 100. [Google Scholar] [CrossRef] [PubMed]
- Morris, B.J.; Willcox, D.C.; Donlon, T.A.; Willcox, B.J. FOXO3: A Major Gene for Human Longevity—A Mini-Review. Gerontology 2015, 61, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Chin, R.M.; Fu, X.; Pai, M.Y.; Vergnes, L.; Hwang, H.; Deng, G.; Diep, S.; Lomenick, B.; Meli, V.S.; Monsalve, G.C.; et al. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 2014, 510, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.L.; Soeters, M.R.; Wust, R.C.I.; Houtkooper, R.H. Metabolic Flexibility as an Adaptation to Energy Resources and Requirements in Health and Disease. Endocr. Rev. 2018, 39, 489–517. [Google Scholar] [CrossRef]
- Palacios, O.M.; Carmona, J.J.; Michan, S.; Chen, K.Y.; Manabe, Y.; Ward, J.L., 3rd; Goodyear, L.J.; Tong, Q. Diet and exercise signals regulate SIRT3 and activate AMPK and PGC-1alpha in skeletal muscle. Aging 2009, 1, 771–783. [Google Scholar] [CrossRef]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Passariello, C.L.; Zini, M.; Nassi, P.A.; Pignatti, C.; Stefanelli, C. Upregulation of SIRT1 deacetylase in phenylephrine-treated cardiomyoblasts. Biochem. Biophys. Res. Commun. 2011, 407, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lluch, G.; Navas, P. Calorie restriction as an intervention in ageing. J. Physiol. 2016, 594, 2043–2060. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [PubMed]
- Amat, R.; Planavila, A.; Chen, S.L.; Iglesias, R.; Giralt, M.; Villarroya, F. SIRT1 controls the transcription of the peroxisome proliferator-activated receptor-gamma Co-activator-1alpha (PGC-1alpha) gene in skeletal muscle through the PGC-1alpha autoregulatory loop and interaction with MyoD. J. Biol. Chem. 2009, 284, 21872–21880. [Google Scholar] [CrossRef] [PubMed]
- Terada, S.; Goto, M.; Kato, M.; Kawanaka, K.; Shimokawa, T.; Tabata, I. Effects of low-intensity prolonged exercise on PGC-1 mRNA expression in rat epitrochlearis muscle. Biochem. Biophys. Res. Commun. 2002, 296, 350–354. [Google Scholar] [CrossRef]
- Suwa, M.; Nakano, H.; Kumagai, S. Effects of chronic AICAR treatment on fiber composition, enzyme activity, UCP3, and PGC-1 in rat muscles. J. Appl. Physiol. 2003, 95, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Boutant, M.; Cantó, C. SIRT1 metabolic actions: Integrating recent advances from mouse models. Mol. Metab. 2014, 3, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.J.; Han, H.S.; Kim, M.J.; Koo, S.H. CREB and FoxO1: Two transcription factors for the regulation of hepatic gluconeogenesis. BMB Rep. 2013, 46, 567–574. [Google Scholar] [CrossRef]
- Dillon, L.M.; Rebelo, A.P.; Moraes, C.T. The role of PGC-1 coactivators in aging skeletal muscle and heart. IUBMB Life 2012, 64, 231–241. [Google Scholar] [CrossRef]
- Huh, J.Y. The role of exercise-induced myokines in regulating metabolism. Arch. Pharm. Res. 2018, 41, 14–29. [Google Scholar] [CrossRef]
- Cheng, C.F.; Ku, H.C.; Lin, H. PGC-1alpha as a Pivotal Factor in Lipid and Metabolic Regulation. Int. J. Mol. Sci. 2018, 19, 3447. [Google Scholar] [CrossRef]
- Green, C.L.; Lamming, D.W. Regulation of metabolic health by essential dietary amino acids. Mech. Ageing Dev. 2019, 177, 186–200. [Google Scholar] [CrossRef]
- Brandhorst, S.; Longo, V.D. Protein Quantity and Source, Fasting-Mimicking Diets, and Longevity. Adv. Nutr. 2019, 10, S340–S350. [Google Scholar] [CrossRef]
- Jin, J.; Zhang, T. Effects of Glucose Restriction on Replicative Senescence of Human Diploid Fibroblasts IMR-90. Cell. Physiol. Biochem. 2013, 31, 718–727. [Google Scholar] [CrossRef] [PubMed]
- Dutcher, S.K.; Roux, A.E.; Leroux, A.; Alaamery, M.A.; Hoffman, C.S.; Chartrand, P.; Ferbeyre, G.; Rokeach, L.A. Pro-Aging Effects of Glucose Signaling through a G Protein-Coupled Glucose Receptor in Fission Yeast. PLoS Genet. 2009, 5, e1000408. [Google Scholar] [CrossRef]
- Weinberger, M.; Mesquita, A.; Carroll, T.; Marks, L.; Yang, H.; Zhang, Z.; Ludovico, P.; Burhans, W.C. Growth signaling promotes chronological aging in budding yeast by inducing superoxide anions that inhibit quiescence. Aging 2010, 2, 709–726. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Murphy, C.T.; Kenyon, C. Glucose Shortens the Life Span of C. elegans by Downregulating DAF-16/FOXO Activity and Aquaporin Gene Expression. Cell Metab. 2009, 10, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose Restriction Extends Caenorhabditis elegans Life Span by Inducing Mitochondrial Respiration and Increasing Oxidative Stress. Cell Metab. 2007, 6, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.G.; Buel, G.R.; Blenis, J. Nutrient regulation of the mTOR Complex 1 signaling pathway. Mol. Cells 2013, 35, 463–473. [Google Scholar] [CrossRef]
- Bröer, S.; Bröer, A. Amino acid homeostasis and signalling in mammalian cells and organisms. Biochem. J. 2017, 474, 1935–1963. [Google Scholar] [CrossRef]
- Gu, X.; Orozco, J.M.; Saxton, R.A.; Condon, K.J.; Liu, G.Y.; Krawczyk, P.A.; Scaria, S.M.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 2017, 358, 813–818. [Google Scholar] [CrossRef]
- Sangüesa, G.; Roglans, N.; Baena, M.; Velázquez, A.; Laguna, J.; Alegret, M. mTOR is a Key Protein Involved in the Metabolic Effects of Simple Sugars. Int. J. Mol. Sci. 2019, 20, 1117. [Google Scholar] [CrossRef]
- Yasuda, M.; Tanaka, Y.; Kume, S.; Morita, Y.; Chin-Kanasaki, M.; Araki, H.; Isshiki, K.; Araki, S.-i.; Koya, D.; Haneda, M.; et al. Fatty acids are novel nutrient factors to regulate mTORC1 lysosomal localization and apoptosis in podocytes. Biochim. Biophys. Acta 2014, 1842, 1097–1108. [Google Scholar] [CrossRef]
- Chaumontet, C.; Azzout-Marniche, D.; Blais, A.; Piedcoq, J.; Tomé, D.; Gaudichon, C.; Even, P.C. Low-protein and methionine, high-starch diets increase energy intake and expenditure, increase FGF21, decrease IGF-1, and have little effect on adiposity in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 316, R486–R501. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Lin, V.Y.; Goetz, R.; Mohammadi, M.; Mangelsdorf, D.J.; Kliewer, S.A. Inhibition of Growth Hormone Signaling by the Fasting-Induced Hormone FGF21. Cell Metab. 2008, 8, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Mair, W.; Dillin, A. Aging and survival: The genetics of life span extension by dietary restriction. Annu. Rev. Biochem. 2008, 77, 727–754. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Longo, V. Dietary restriction with and without caloric restriction for healthy aging. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Weiss, E.P.; Villareal, D.T.; Klein, S.; Holloszy, J.O. Long-term effects of calorie or protein restriction on serum IGF-1 and IGFBP-3 concentration in humans. Aging Cell 2008, 7, 681–687. [Google Scholar] [CrossRef]
- Stekovic, S.; Hofer, S.J.; Tripolt, N.; Aon, M.A.; Royer, P.; Pein, L.; Stadler, J.T.; Pendl, T.; Prietl, B.; Url, J.; et al. Alternate Day Fasting Improves Physiological and Molecular Markers of Aging in Healthy, Non-obese Humans. Cell Metab. 2019, 30, 462–476.e466. [Google Scholar] [CrossRef]
- Most, J.; Tosti, V.; Redman, L.M.; Fontana, L. Calorie restriction in humans: An update. Ageing Res. Rev. 2017, 39, 36–45. [Google Scholar] [CrossRef]
- Hansen, M.; Chandra, A.; Mitic, L.L.; Onken, B.; Driscoll, M.; Kenyon, C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 2008, 4, e24. [Google Scholar] [CrossRef]
- Santos, J.; Leitao-Correia, F.; Sousa, M.J.; Leao, C. Dietary Restriction and Nutrient Balance in Aging. Oxid. Med. Cell Longev. 2016, 2016, 4010357. [Google Scholar] [CrossRef]
- Gonzalez-Freire, M.; Diaz-Ruiz, A.; Hauser, D.; Martinez-Romero, J.; Ferrucci, L.; Bernier, M.; de Cabo, R. The road ahead for health and lifespan interventions. Ageing Res. Rev. 2020, 59, 101037. [Google Scholar] [CrossRef]
- Riera, C.E.; Merkwirth, C.; De Magalhaes Filho, C.D.; Dillin, A. Signaling Networks Determining Life Span. Annu. Rev. Biochem. 2016, 85, 35–64. [Google Scholar] [CrossRef] [PubMed]
- Mattison, J.A.; Colman, R.J.; Beasley, T.M.; Allison, D.B.; Kemnitz, J.W.; Roth, G.S.; Ingram, D.K.; Weindruch, R.; de Cabo, R.; Anderson, R.M. Caloric restriction improves health and survival of rhesus monkeys. Nat. Commun. 2017, 8, 14063. [Google Scholar] [CrossRef] [PubMed]
- Shintani, H.; Shintani, T.; Ashida, H.; Sato, M. Calorie Restriction Mimetics: Upstream-Type Compounds for Modulating Glucose Metabolism. Nutrients 2018, 10, 1821. [Google Scholar] [CrossRef] [PubMed]
- Weimer, S.; Priebs, J.; Kuhlow, D.; Groth, M.; Priebe, S.; Mansfeld, J.; Merry, T.L.; Dubuis, S.; Laube, B.; Pfeiffer, A.F.; et al. D-Glucosamine supplementation extends life span of nematodes and of ageing mice. Nat. Commun. 2014, 5, 3563. [Google Scholar] [CrossRef]
- Ingram, D.K.; Roth, G.S. Calorie restriction mimetics: Can you have your cake and eat it, too? Ageing Res. Rev. 2015, 20, 46–62. [Google Scholar] [CrossRef]
- Lamming, D.W.; Ye, L.; Sabatini, D.M.; Baur, J.A. Rapalogs and mTOR inhibitors as anti-aging therapeutics. J. Clin. Investig. 2013, 123, 980–989. [Google Scholar] [CrossRef]
- Sinclair, D.A.; Guarente, L. Small-Molecule Allosteric Activators of Sirtuins. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 363–380. [Google Scholar] [CrossRef]
- Martens, C.R.; Denman, B.A.; Mazzo, M.R.; Armstrong, M.L.; Reisdorph, N.; McQueen, M.B.; Chonchol, M.; Seals, D.R. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD+ in healthy middle-aged and older adults. Nat. Commun. 2018, 9, 1286. [Google Scholar] [CrossRef]
- Minois, N. Molecular Basis of the ‘Anti-Aging’ Effect of Spermidine and Other Natural Polyamines—A Mini-Review. Gerontology 2014, 60, 319–326. [Google Scholar] [CrossRef]
- Schwarz, C.; Stekovic, S.; Wirth, M.; Benson, G.; Royer, P.; Sigrist, S.J.; Pieber, T.; Dammbrueck, C.; Magnes, C.; Eisenberg, T.; et al. Safety and tolerability of spermidine supplementation in mice and older adults with subjective cognitive decline. Aging 2018, 10, 19–33. [Google Scholar] [CrossRef]
- Eisenberg, T.; Abdellatif, M.; Schroeder, S.; Primessnig, U.; Stekovic, S.; Pendl, T.; Harger, A.; Schipke, J.; Zimmermann, A.; Schmidt, A.; et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 2016, 22, 1428–1438. [Google Scholar] [CrossRef]
- D’Adamo, S.; Cetrullo, S.; Guidotti, S.; Silvestri, Y.; Minguzzi, M.; Santi, S.; Cattini, L.; Filardo, G.; Flamigni, F.; Borzì, R.M. Spermidine rescues the deregulated autophagic response to oxidative stress of osteoarthritic chondrocytes. Free Radic. Biol. Med. 2020, 153, 159–172. [Google Scholar] [CrossRef]
- Madeo, F.; Eisenberg, T.; Pietrocola, F.; Kroemer, G. Spermidine in health and disease. Science 2018, 359. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Carmona-Gutierrez, D.; Hofer, S.J.; Kroemer, G. Caloric Restriction Mimetics against Age-Associated Disease: Targets, Mechanisms, and Therapeutic Potential. Cell Metab. 2019, 29, 592–610. [Google Scholar] [CrossRef] [PubMed]
- Solon-Biet, S.M.; Mitchell, S.J.; de Cabo, R.; Raubenheimer, D.; Le Couteur, D.G.; Simpson, S.J. Macronutrients and caloric intake in health and longevity. J. Endocrinol. 2015, 226, R17–R28. [Google Scholar] [CrossRef] [PubMed]
- Westerterp-Plantenga, M.S.; Nieuwenhuizen, A.; Tome, D.; Soenen, S.; Westerterp, K.R. Dietary protein, weight loss, and weight maintenance. Annu. Rev. Nutr. 2009, 29, 21–41. [Google Scholar] [CrossRef]
- van den Brandt, P.A. Red meat, processed meat, and other dietary protein sources and risk of overall and cause-specific mortality in The Netherlands Cohort Study. Eur. J. Epidemiol. 2019, 34, 351–369. [Google Scholar] [CrossRef]
- Etemadi, A.; Sinha, R.; Ward, M.H.; Graubard, B.I.; Inoue-Choi, M.; Dawsey, S.M.; Abnet, C.C. Mortality from different causes associated with meat, heme iron, nitrates, and nitrites in the NIH-AARP Diet and Health Study: Population based cohort study. Br. Med. J. 2017, 357, j1957. [Google Scholar] [CrossRef]
- Song, M.; Fung, T.T.; Hu, F.B.; Willett, W.C.; Longo, V.D.; Chan, A.T.; Giovannucci, E.L. Association of Animal and Plant Protein Intake With All-Cause and Cause-Specific Mortality. JAMA Intern. Med. 2016, 176, 1453. [Google Scholar] [CrossRef]
- Huang, J.; Liao, L.M.; Weinstein, S.J.; Sinha, R.; Graubard, B.I.; Albanes, D. Association Between Plant and Animal Protein Intake and Overall and Cause-Specific Mortality. JAMA Intern. Med. 2020, 180, 1173–1184. [Google Scholar] [CrossRef]
- Fontana, L.; Cummings, N.E.; Arriola Apelo, S.I.; Neuman, J.C.; Kasza, I.; Schmidt, B.A.; Cava, E.; Spelta, F.; Tosti, V.; Syed, F.A.; et al. Decreased Consumption of Branched-Chain Amino Acids Improves Metabolic Health. Cell Rep. 2016, 16, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Brown-Borg, H.M.; Buffenstein, R. Cutting back on the essentials: Can manipulating intake of specific amino acids modulate health and lifespan? Ageing Res. Rev. 2017, 39, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Kaya, A.; Gladyshev, V.N. Methionine restriction and life-span control. Ann. N. Y. Acad. Sci. 2016, 1363, 116–124. [Google Scholar] [CrossRef]
- Malloy, V.L.; Krajcik, R.A.; Bailey, S.J.; Hristopoulos, G.; Plummer, J.D.; Orentreich, N. Methionine restriction decreases visceral fat mass and preserves insulin action in aging male Fischer 344 rats independent of energy restriction. Aging Cell 2006, 5, 305–314. [Google Scholar] [CrossRef]
- Elshorbagy, A.K.; Valdivia-Garcia, M.; Mattocks, D.A.; Plummer, J.D.; Smith, A.D.; Drevon, C.A.; Refsum, H.; Perrone, C.E. Cysteine supplementation reverses methionine restriction effects on rat adiposity: Significance of stearoyl-coenzyme A desaturase. J. Lipid Res. 2011, 52, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Perrone, C.E.; Mattocks, D.A.; Hristopoulos, G.; Plummer, J.D.; Krajcik, R.A.; Orentreich, N. Methionine restriction effects on 11 -HSD1 activity and lipogenic/lipolytic balance in F344 rat adipose tissue. J. Lipid Res. 2008, 49, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Ables, G.P.; Perrone, C.E.; Orentreich, D.; Orentreich, N. Methionine-restricted C57BL/6J mice are resistant to diet-induced obesity and insulin resistance but have low bone density. PLoS ONE 2012, 7, e51357. [Google Scholar] [CrossRef] [PubMed]
- Wanders, D.; Forney, L.A.; Stone, K.P.; Burk, D.H.; Pierse, A.; Gettys, T.W. FGF21 Mediates the Thermogenic and Insulin-Sensitizing Effects of Dietary Methionine Restriction but Not Its Effects on Hepatic Lipid Metabolism. Diabetes 2017, 66, 858–867. [Google Scholar] [CrossRef]
- Mirzaei, H.; Suarez, J.A.; Longo, V.D. Protein and amino acid restriction, aging and disease: From yeast to humans. Trends Endocrinol. Metab. 2014, 25, 558–566. [Google Scholar] [CrossRef]
- Zapata, R.C.; Singh, A.; Ajdari, N.M.; Chelikani, P.K. Dietary Tryptophan Restriction Dose-Dependently Modulates Energy Balance, Gut Hormones, and Microbiota in Obesity-Prone Rats. Obesity 2018, 26, 730–739. [Google Scholar] [CrossRef]
- Xiao, F.; Huang, Z.; Li, H.; Yu, J.; Wang, C.; Chen, S.; Meng, Q.; Cheng, Y.; Gao, X.; Li, J.; et al. Leucine deprivation increases hepatic insulin sensitivity via GCN2/mTOR/S6K1 and AMPK pathways. Diabetes 2011, 60, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, A.; Oki, N.; Takahashi, S.I.; Noguchi, T. Dietary restriction of single essential amino acids reduces plasma insulin-like growth factor-I (IGF-I) but does not affect plasma IGF-binding protein-1 in rats. J. Nutr. 2000, 130, 2910–2914. [Google Scholar] [CrossRef] [PubMed]
- Strasser, B.; Volaklis, K.; Fuchs, D.; Burtscher, M. Role of Dietary Protein and Muscular Fitness on Longevity and Aging. Aging Dis. 2018, 9, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, P.; Longo, V.D. Growth factors, aging and age-related diseases. Growth Horm. IGF Res. 2016, 28, 66–68. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.E.; Suarez, J.A.; Brandhorst, S.; Balasubramanian, P.; Cheng, C.W.; Madia, F.; Fontana, L.; Mirisola, M.G.; Guevara-Aguirre, J.; Wan, J.; et al. Low protein intake is associated with a major reduction in IGF-1, cancer, and overall mortality in the 65 and younger but not older population. Cell Metab. 2014, 19, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Naghshi, S.; Sadeghi, O.; Willett, W.C.; Esmaillzadeh, A. Dietary intake of total, animal, and plant proteins and risk of all cause, cardiovascular, and cancer mortality: Systematic review and dose-response meta-analysis of prospective cohort studies. Br. Med. J. 2020, 370, m2412. [Google Scholar] [CrossRef]
- Bell, J.T.; Spector, T.D. A twin approach to unraveling epigenetics. Trends Genet. 2011, 27, 116–125. [Google Scholar] [CrossRef]
- Gadecka, A.; Bielak-Zmijewska, A. Slowing Down Ageing: The Role of Nutrients and Microbiota in Modulation of the Epigenome. Nutrients 2019, 11, 1251. [Google Scholar] [CrossRef]
- Rinninella, E.; Cintoni, M.; Raoul, P.; Lopetuso, L.R.; Scaldaferri, F.; Pulcini, G.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. Food Components and Dietary Habits: Keys for a Healthy Gut Microbiota Composition. Nutrients 2019, 11, 2393. [Google Scholar] [CrossRef]
- Lees-Murdock, D.J.; Walsh, C.P.; Dollin, C.; Hughes, C.F.; McNulty, H.; Strain, J.J.; Ward, M.; Amenyah, S.D. Nutritional Epigenomics and Age-Related Disease. Curr. Dev. Nutr. 2020, 4. [Google Scholar] [CrossRef]
- McKay, J.A.; Mathers, J.C. Diet induced epigenetic changes and their implications for health. Acta Physiol. 2011, 202, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Kok, D.E.G.; Dhonukshe-Rutten, R.A.M.; Lute, C.; Heil, S.G.; Uitterlinden, A.G.; van der Velde, N.; van Meurs, J.B.J.; van Schoor, N.M.; Hooiveld, G.J.E.J.; de Groot, L.C.P.G.M.; et al. The effects of long-term daily folic acid and vitamin B12 supplementation on genome-wide DNA methylation in elderly subjects. Clin. Epigenetics 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, J.A.; Dugué, P.-A.; Bassett, J.K.; Hodge, A.M.; Brinkman, M.T.; Joo, J.E.; Jung, C.-H.; Makalic, E.; Schmidt, D.F.; Hopper, J.L.; et al. Dietary intake of one-carbon metabolism nutrients and DNA methylation in peripheral blood. Am. J. Clin. Nutr. 2018, 108, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Metes-Kosik, N.; Luptak, I.; DiBello, P.M.; Handy, D.E.; Tang, S.-S.; Zhi, H.; Qin, F.; Jacobsen, D.W.; Loscalzo, J.; Joseph, J. Both selenium deficiency and modest selenium supplementation lead to myocardial fibrosis in mice via effects on redox-methylation balance. Mol. Nutr. Food Res. 2012, 56, 1812–1824. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, V.; Vaid, M.; Katiyar, S.K. (-)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p16INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis 2011, 32, 537–544. [Google Scholar] [CrossRef]
- Zhao, Z.; Shilatifard, A. Epigenetic modifications of histones in cancer. Genome Biol. 2019, 20. [Google Scholar] [CrossRef]
- Bouras, T.; Fu, M.; Sauve, A.A.; Wang, F.; Quong, A.A.; Perkins, N.D.; Hay, R.T.; Gu, W.; Pestell, R.G. SIRT1 Deacetylation and Repression of p300 Involves Lysine Residues 1020/1024 within the Cell Cycle Regulatory Domain 1. J. Biol. Chem. 2005, 280, 10264–10276. [Google Scholar] [CrossRef]
- Cetrullo, S.; D’Adamo, S.; Tantini, B.; Borzi, R.M.; Flamigni, F. mTOR, AMPK, and Sirt1: Key Players in Metabolic Stress Management. Crit. Rev. Eukaryot. Gene Expr. 2015, 25, 59–75. [Google Scholar] [CrossRef]
- Boccardi, V.; Paolisso, G.; Mecocci, P. Nutrition and lifestyle in healthy aging: The telomerase challenge. Aging 2016, 8, 12–15. [Google Scholar] [CrossRef]
- Davinelli, S.; Trichopoulou, A.; Corbi, G.; De Vivo, I.; Scapagnini, G. The potential nutrigeroprotective role of Mediterranean diet and its functional components on telomere length dynamics. Ageing Res. Rev. 2019, 49, 1–10. [Google Scholar] [CrossRef]
- García-Calzón, S.; Moleres, A.; Martínez-González, M.A.; Martínez, J.A.; Zalba, G.; Marti, A. Dietary total antioxidant capacity is associated with leukocyte telomere length in a children and adolescent population. Clin. Nutr. 2015, 34, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sinnott-Armstrong, N.; Wagschal, A.; Wark, A.R.; Camporez, J.-P.; Perry, R.J.; Ji, F.; Sohn, Y.; Oh, J.; Wu, S.; et al. A MicroRNA Linking Human Positive Selection and Metabolic Disorders. Cell 2020, 183, 684–701.e14. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.; Herrick, J.; Mullany, L.; Stevens, J.; Wolff, R. Diet and lifestyle factors associated with miRNA expression in colorectal tissue. Pharm. Pers. Med. 2016, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.D.; Ross, S.A. Evidence for dietary regulation of microRNA expression in cancer cells. Nutr. Rev. 2008, 66, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Lançon, A.; Michaille, J.-J.; Latruffe, N. Effects of dietary phytophenols on the expression of microRNAs involved in mammalian cell homeostasis. J. Sci. Food Agric. 2013, 93, 3155–3164. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K. Do the microRNAs we eat affect gene expression? Nature 2020, 582, S10–S11. [Google Scholar] [CrossRef]
- Medina-Remón, A.; Kirwan, R.; Lamuela-Raventós, R.M.; Estruch, R. Dietary patterns and the risk of obesity, type 2 diabetes mellitus, cardiovascular diseases, asthma, and neurodegenerative diseases. Crit. Rev. Food Sci. Nutr. 2017, 58, 262–296. [Google Scholar] [CrossRef]
- Buettner, D.; Skemp, S. Blue Zones. Am. J. Lifestyle Med. 2016, 10, 318–321. [Google Scholar] [CrossRef]
- Rizzuto, D.; Fratiglioni, L. Lifestyle Factors Related to Mortality and Survival: A Mini-Review. Gerontology 2014, 60, 327–335. [Google Scholar] [CrossRef]
- Pignolo, R.J. Exceptional Human Longevity. Mayo Clin. Proc. 2019, 94, 110–124. [Google Scholar] [CrossRef]
- Willcox, B.J.; Willcox, D.C. Caloric restriction, caloric restriction mimetics, and healthy aging in Okinawa. Curr. Opin. Clin. Nutr. Metab. Care 2013, 17, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Vasto, S.; Buscemi, S.; Barera, A.; Di Carlo, M.; Accardi, G.; Caruso, C. Mediterranean Diet and Healthy Ageing: A Sicilian Perspective. Gerontology 2014, 60, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Del Bo, C.; Marino, M.; Martini, D.; Tucci, M.; Ciappellano, S.; Riso, P.; Porrini, M. Overview of Human Intervention Studies Evaluating the Impact of the Mediterranean Diet on Markers of DNA Damage. Nutrients 2019, 11, 391. [Google Scholar] [CrossRef] [PubMed]
- Ros, M.; Carrascosa, J.M. Current nutritional and pharmacological anti-aging interventions. Biochim. Biophys. Acta 2020, 1866. [Google Scholar] [CrossRef] [PubMed]
- Peyrol, J.; Riva, C.; Amiot, M. Hydroxytyrosol in the Prevention of the Metabolic Syndrome and Related Disorders. Nutrients 2017, 9, 306. [Google Scholar] [CrossRef] [PubMed]
- Angeloni, C.; Malaguti, M.; Barbalace, M.; Hrelia, S. Bioactivity of Olive Oil Phenols in Neuroprotection. Int. J. Mol. Sci. 2017, 18, 2230. [Google Scholar] [CrossRef] [PubMed]
- Facchini, A.; Cetrullo, S.; D’Adamo, S.; Guidotti, S.; Minguzzi, M.; Facchini, A.; Borzi, R.M.; Flamigni, F. Hydroxytyrosol prevents increase of osteoarthritis markers in human chondrocytes treated with hydrogen peroxide or growth-related oncogene alpha. PLoS ONE 2014, 9, e109724. [Google Scholar] [CrossRef]
- Cetrullo, S.; D’Adamo, S.; Guidotti, S.; Borzi, R.M.; Flamigni, F. Hydroxytyrosol prevents chondrocyte death under oxidative stress by inducing autophagy through sirtuin 1-dependent and -independent mechanisms. Biochim. Biophys. Acta 2016, 1860, 1181–1191. [Google Scholar] [CrossRef]
- Sofi, F.; Macchi, C.; Abbate, R.; Gensini, G.F.; Casini, A. Mediterranean diet and health. BioFactors 2013, 39, 335–342. [Google Scholar] [CrossRef]
- Estruch, R. Anti-inflammatory effects of the Mediterranean diet: The experience of the PREDIMED study. Proc. Nutr. Soc. 2010, 69, 333–340. [Google Scholar] [CrossRef]
- Schwingshackl, L.; Morze, J.; Hoffmann, G. Mediterranean diet and health status: Active ingredients and pharmacological mechanisms. Br. J. Pharmacol. 2019, 177, 1241–1257. [Google Scholar] [CrossRef] [PubMed]
- Martel, J.; Ojcius, D.M.; Ko, Y.-F.; Ke, P.-Y.; Wu, C.-Y.; Peng, H.-H.; Young, J.D. Hormetic Effects of Phytochemicals on Health and Longevity. Trends Endocrinol. Metab. 2019, 30, 335–346. [Google Scholar] [CrossRef]
- Soultoukis, G.A.; Partridge, L. Dietary Protein, Metabolism, and Aging. Annu. Rev. Biochem. 2016, 85, 5–34. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.S.; Longo, V.D. A protein restriction-dependent sulfur code for longevity. Cell 2015, 160, 15–17. [Google Scholar] [CrossRef]
- Lynch, C.J.; Adams, S.H. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat. Rev. Endocrinol. 2014, 10, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Waern, R.V.; Cumming, R.G.; Blyth, F.; Naganathan, V.; Allman-Farinelli, M.; Le Couteur, D.; Simpson, S.J.; Kendig, H.; Hirani, V. Adequacy of nutritional intake among older men living in Sydney, Australia: Findings from the Concord Health and Ageing in Men Project (CHAMP). Br. J. Nutr. 2015, 114, 812–821. [Google Scholar] [CrossRef]
- Le Couteur, D.G.; Solon-Biet, S.; Cogger, V.C.; Mitchell, S.J.; Senior, A.; de Cabo, R.; Raubenheimer, D.; Simpson, S.J. The impact of low-protein high-carbohydrate diets on aging and lifespan. Cell Mol. Life Sci. 2016, 73, 1237–1252. [Google Scholar] [CrossRef]
- Liao, C.-Y.; Rikke, B.A.; Johnson, T.E.; Diaz, V.; Nelson, J.F. Genetic variation in the murine lifespan response to dietary restriction: From life extension to life shortening. Aging Cell 2010, 9, 92–95. [Google Scholar] [CrossRef]
- Murphy, C.T.; Perez-Matos, M.C.; Mair, W.B. Predicting longevity responses to dietary restriction: A stepping stone toward precision geroscience. PLoS Genet. 2020, 16. [Google Scholar] [CrossRef]
- Murphy, C.T.; Jin, K.; Wilson, K.A.; Beck, J.N.; Nelson, C.S.; Brownridge, G.W.; Harrison, B.R.; Djukovic, D.; Raftery, D.; Brem, R.B.; et al. Genetic and metabolomic architecture of variation in diet restriction-mediated lifespan extension in Drosophila. PLoS Genet. 2020, 16. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pignatti, C.; D’Adamo, S.; Stefanelli, C.; Flamigni, F.; Cetrullo, S. Nutrients and Pathways that Regulate Health Span and Life Span. Geriatrics 2020, 5, 95. https://doi.org/10.3390/geriatrics5040095
Pignatti C, D’Adamo S, Stefanelli C, Flamigni F, Cetrullo S. Nutrients and Pathways that Regulate Health Span and Life Span. Geriatrics. 2020; 5(4):95. https://doi.org/10.3390/geriatrics5040095
Chicago/Turabian StylePignatti, Carla, Stefania D’Adamo, Claudio Stefanelli, Flavio Flamigni, and Silvia Cetrullo. 2020. "Nutrients and Pathways that Regulate Health Span and Life Span" Geriatrics 5, no. 4: 95. https://doi.org/10.3390/geriatrics5040095
APA StylePignatti, C., D’Adamo, S., Stefanelli, C., Flamigni, F., & Cetrullo, S. (2020). Nutrients and Pathways that Regulate Health Span and Life Span. Geriatrics, 5(4), 95. https://doi.org/10.3390/geriatrics5040095