
Molecular Detection and Phylogenetic Relationships of Honey Bee-Associated Viruses in Bee Products

 , ,
, ,  , , and
, , and 
Abstract
Simple Summary
Abstract
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Type | Order | Family | Virus | Acronym | Ref. Sequence Acc. No. | References |
|---|---|---|---|---|---|---|
| ssRNA(+) | Picornavirales | Dicistroviridae | Acute bee paralysis virus | ABPV | NC 002548 | Govan et al. [36] |
| KBV | NC 004807 | de Miranda et al. [37] | ||||
| IAPV | NC 009025 | Maori et al. [38] | ||||
| Black queen cell virus | BQCV | NC 003784 | Leat et al. [39] | |||
| ssRNA(+) | Picornavirales | Iflaviridae | Deformed wing virus | DWV-A | NC 004830 | Lanzi et al. [40] |
| DWV-B | NC 006494 | Ongus et al. [31] | ||||
| Egypt bee virus | DWV-C | ERS 657948 | Mordecai et al. [32] | |||
| DWV-D | MT 504363 | de Miranda et al. [41] | ||||
| Moku virus | MV | KU 645789 | Mordecai et al. [42] | |||
| Sacbrood virus | SBV | NC 002066 | Ghosh et al. [43] | |||
| Slow bee paralysis virus | SBPV | NC 014137 | de Miranda et al. [44] | |||
| ssRNA(+) | Tymovirales | Tymoviridae | Bee Macula-like virus | BeeMLV | KT 162924 | de Miranda et al. [45] |
| ? | ? | Cloudy wing virus * | CWV | n.a. | - | |
| ? | ? | Chronic bee paralysis virus | CBPV | NC 010711 | Olivier et al. [46] | |
| ssRNA(+) | Nodamuvirales | Sinhaliviridae | Lake Sinai virus | LSV-1 | HQ 871931 | Runckel et al. [47] |
| LSV-2 | HQ 888865 | |||||
| LSV-3 | MH 267700 | Thaduri et al. [48] | ||||
| LSV-4 | KM 886903 | Ravoet et al. [49] | ||||
| dsDNA | Megavirales | Baculoviridae | Apis mellifera filamentous virus a | AmFV | MH 243376 | Gauthier et al. [50] |
2. Materials and Methods
2.1. Ethical Statement
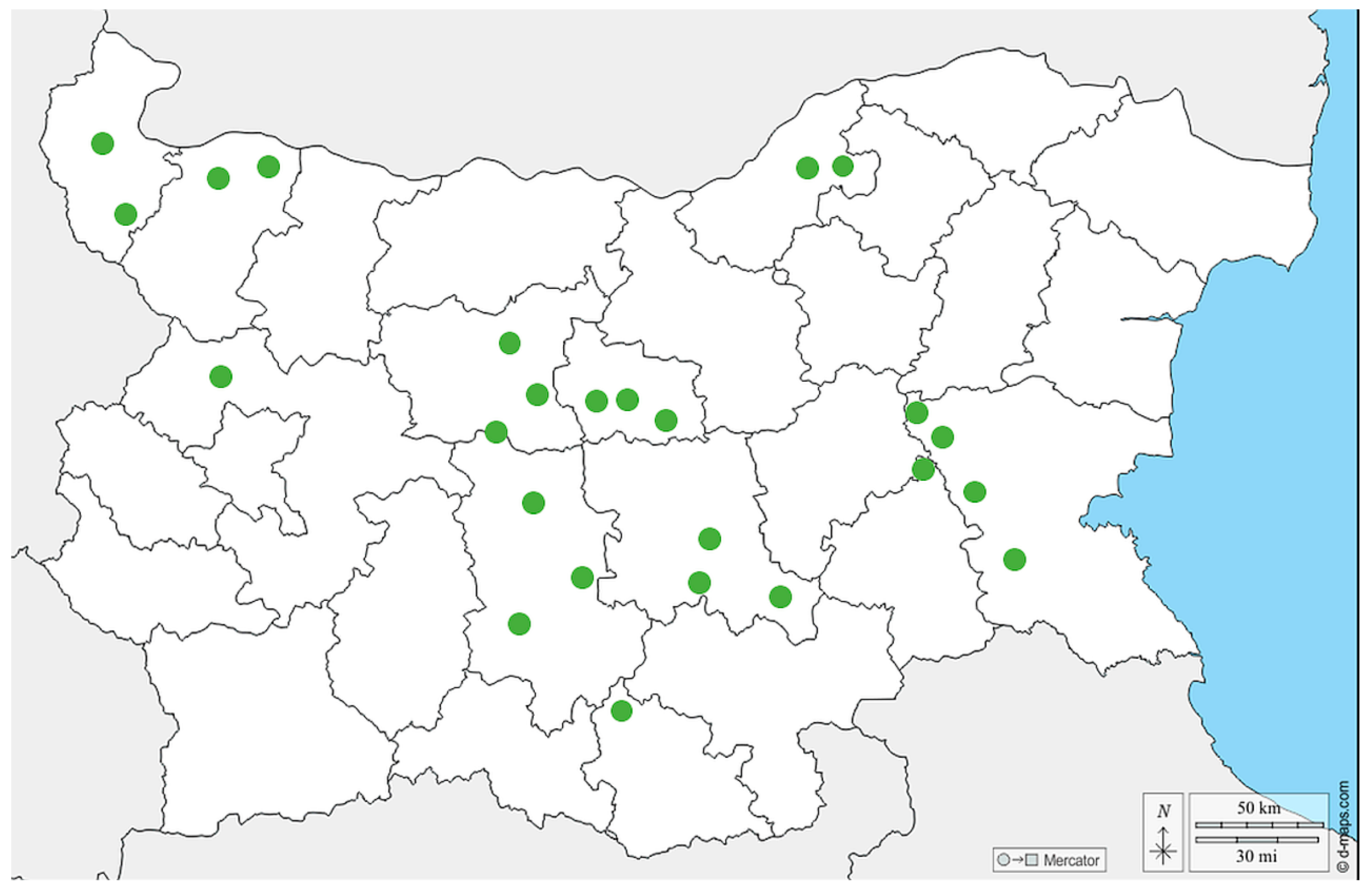
2.2. Geographic Distribution of the Collected Samples
2.3. Gene Selection
2.4. Total RNA Extraction, Copy DNA (cDNA) Synthesis, and RT-PCR Amplification
2.5. Bioinformatics and Molecular Phylogenetic Analysis of Honey Bee-Associated Viruses
3. Results
3.1. Prevalence of Detected Honey Bee-Associated Viruses in Different Regions of Bulgaria
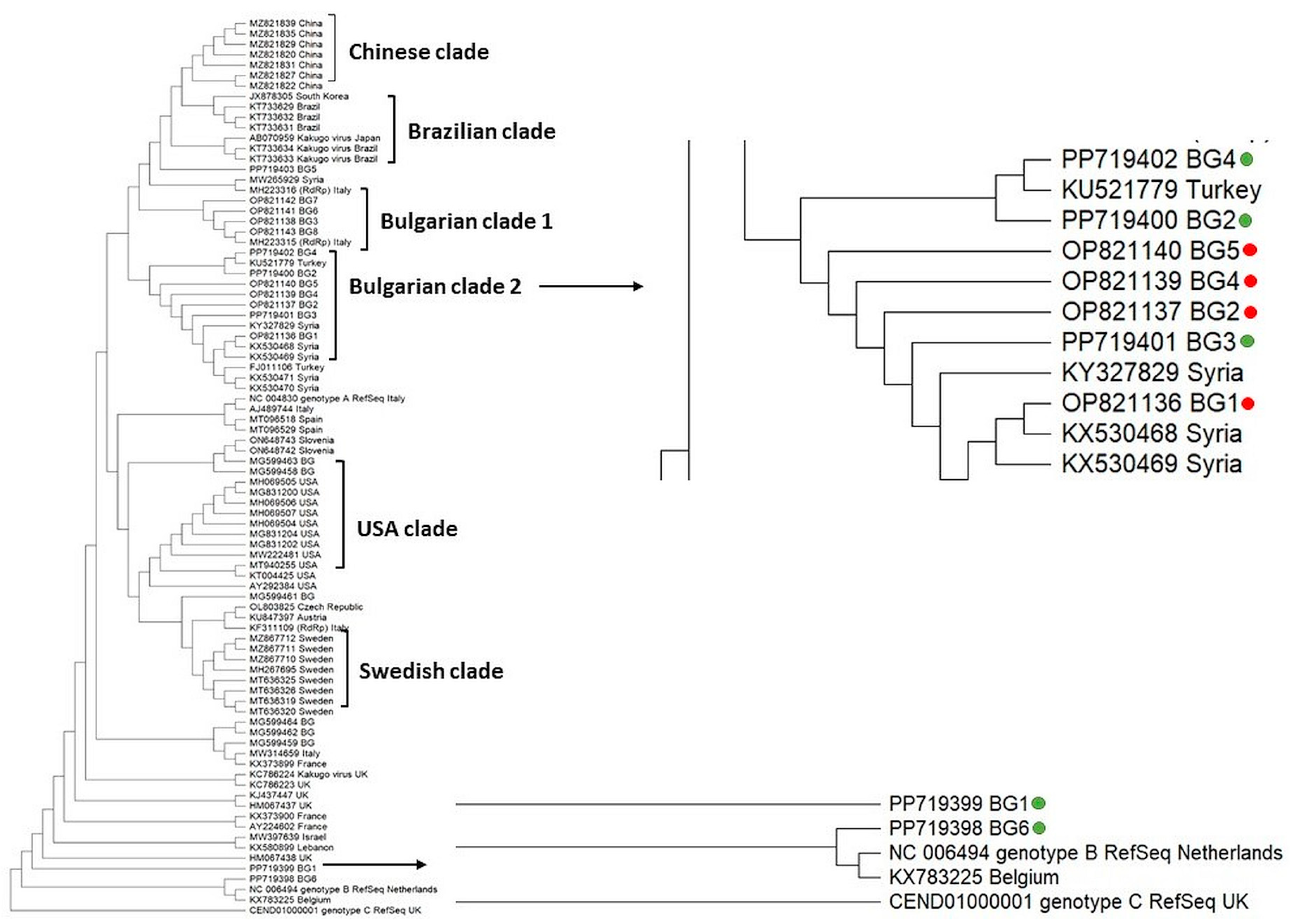
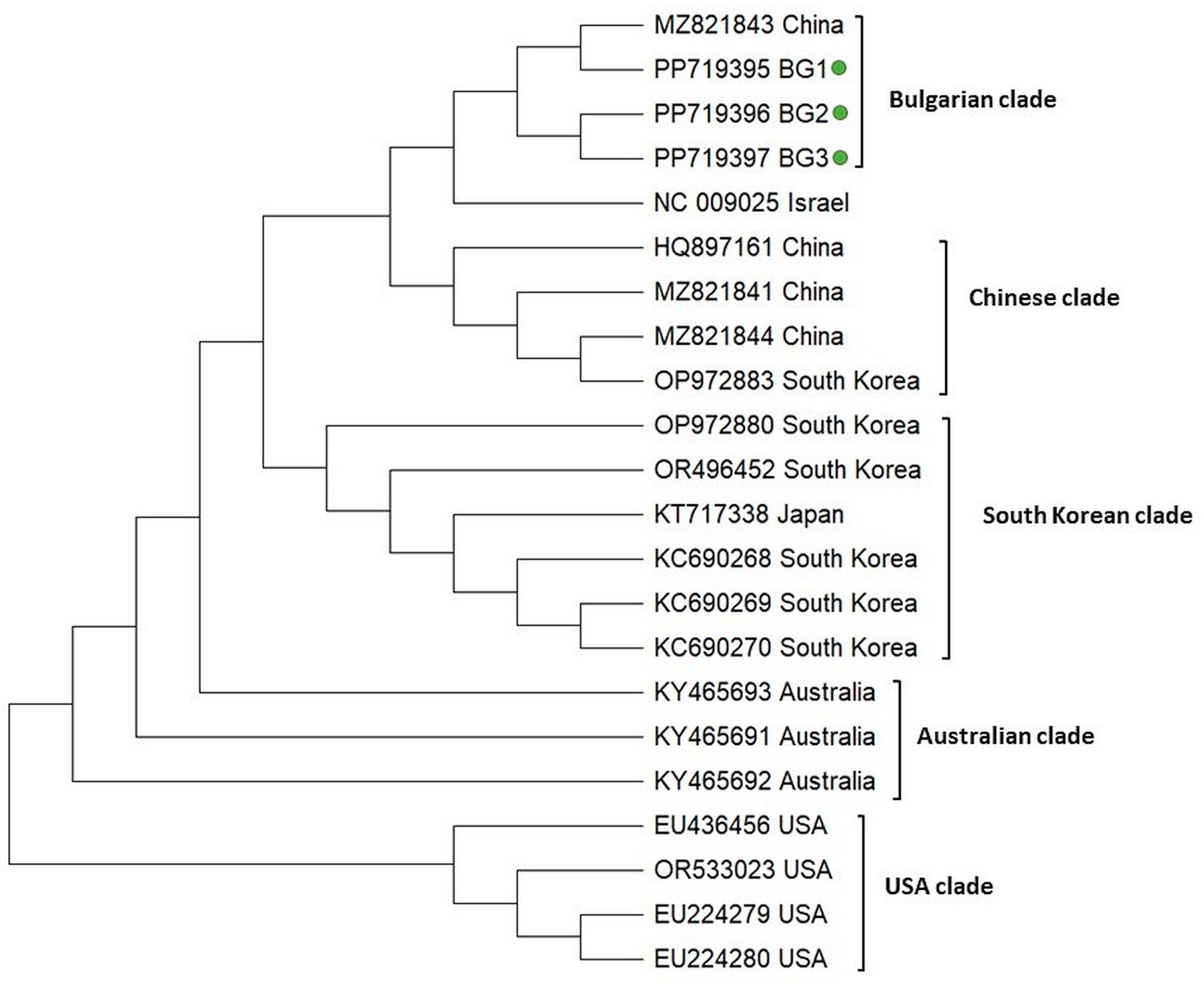
3.2. Molecular Phylogenetic Relationships of Honey Bee-Associated Viruses
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barnes, M.A.; Turner, C.R. The ecology of environmental DNA and implications for conservation genetics. Conserv. Genet. 2016, 17, 1–17. [Google Scholar] [CrossRef]
- Freeland, J.R. The importance of molecular markers and primer design when characterizing biodiversity from environmental DNA. Genome 2017, 60, 358–374. [Google Scholar] [CrossRef] [PubMed]
- Sigsgaard, E.E.; Jensen, M.R.; Winkelmann, I.E.; Møller, P.R.; Hansen, M.M.; Thomsen, P.F. Population-level inferences from environmental DNA—Current status and future perspectives. Evol. Appl. 2020, 13, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Cantera, I.; Decotte, J.B.; Dejean, T.; Murienne, J.; Vigouroux, R.; Valentini, A.; Brosse, S. Characterizing the spatial signal of environmental DNA in river systems using a community ecology approach. Mol. Ecol. Resour. 2022, 22, 1274–1283. [Google Scholar] [CrossRef]
- Bovo, S.; Ribani, A.; Utzeri, V.J.; Schiavo, G.; Bertolini, F.; Fontanesi, L. Shotgun metagenomics of honey DNA: Evaluation of a methodological approach to describe a multi-kingdom honey bee derived environmental DNA signature. PLoS ONE 2018, 13, e0205575. [Google Scholar] [CrossRef] [PubMed]
- Bovo, S.; Utzeri, V.J.; Ribani, A.; Cabbri, R.; Fontanesi, L. Shotgun sequencing of honey DNA can describe honey bee derived environmental signatures and the honey bee hologenome complexity. Sci. Rep. 2020, 10, 9279. [Google Scholar] [CrossRef] [PubMed]
- Utzeri, V.J.; Schiavo, G.; Ribani, A.; Bertolini, F.; Bovo, S.; Fontanesi, L. A next generation sequencing approach for targeted Varroa destructor (Acari: Varroidae) mitochondrial DNA analysis based on honey derived environmental DNA. J. Invertebr. Pathol. 2019, 161, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Ribani, A.; Utzeri, V.J.; Taurisano, V.; Galuppi, R.; Fontanesi, L. Analysis of honey environmental DNA indicates that the honey bee (Apis mellifera L.) trypanosome parasite Lotmaria passim is widespread in the apiaries of the North of Italy. J. Invertebr. Pathol. 2021, 184, 107628. [Google Scholar] [CrossRef]
- Schittny, D.; Yañez, O.; Neumann, P. Honey bee virus transmission via hive products. Vet. Sci. 2020, 7, 96. [Google Scholar] [CrossRef]
- Ribani, A.; Utzeri, V.J.; Taurisano, V.; Fontanesi, L. Honey as a source of environmental DNA for the detection and monitoring of honey bee pathogens and parasites. Vet. Sci. 2020, 7, 113. [Google Scholar] [CrossRef]
- Ribani, A.; Taurisano, V.; Utzeri, V.J.; Fontanesi, L. Honey environmental DNA can be used to detect and monitor honey bee pests: Development of methods useful to identify Aethina tumida and Galleria mellonella infestations. Vet. Sci. 2022, 9, 213. [Google Scholar] [CrossRef] [PubMed]
- Balkanska, R.; Shumkova, R.; Atsenova, N.; Salkova, D.; Dundarova, H.; Radoslavov, G.; Hristov, P. Molecular Detection and Phylogenetic Analysis of Deformed Wing Virus and Sacbrood Virus Isolated from Pollen. Vet. Sci. 2023, 10, 140. [Google Scholar] [CrossRef]
- Čukanová, E.; Prodělalová, J.; Palíková, M.; Kováčová, K.; Linhart, P.; Papežíková, I. Can the examination of different types of hive samples be a non-invasive method for detection and quantification of viruses in honey bee (Apis mellifera L.) colonies? J. Vet. Res. 2023, 67, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Guimarães-Cestaro, L.; Serrão, J.E.; Message, D.; Martins, M.F.; Teixeira, E.W. Simultaneous detection of Nosema spp., Ascosphaera apis and Paenibacillus larvae in honey bee products. J. Hymenopt. Res. 2016, 49, 43–50. [Google Scholar] [CrossRef]
- Boardman, L.; Marcelino, J.A.; Valentin, R.E.; Boncristiani, H.; Standley, J.M.; Ellis, J.D. Novel eDNA approaches to monitor Western honey bee (Apis mellifera L.) microbial and arthropod communities. Environ. DNA 2024, 6, e419. [Google Scholar] [CrossRef]
- Yakubu, A.; Sahabi, S.; Sani, G.D.; Faruku, S. Determination of sugar adulteration in honey using conductivity meter and pH meter. Res. J. Environ. Sci. 2021, 11, 50–57. [Google Scholar]
- Muhammad, N.I.I.; Sarbon, N.M. Physicochemical profile, antioxidant activity and mineral contents of honey from stingless bee and honey bee species. J. Apic. Res. 2023, 62, 394–401. [Google Scholar] [CrossRef]
- Kunugi, H.; Mohammed Ali, A. Royal jelly and its components promote healthy aging and longevity: From animal models to humans. Int. J. Mol. Sci. 2019, 20, 4662. [Google Scholar] [CrossRef] [PubMed]
- McMenamin, A.J.; Flenniken, M.L. Recently identified bee viruses and their impact on bee pollinators. Curr. Opin. Insect Sci. 2018, 26, 120–129. [Google Scholar] [CrossRef]
- Grozinger, C.M.; Flenniken, M.L. Bee viruses: Ecology, pathogenicity, and impacts. Annu. Rev. Entomol. 2019, 64, 205–226. [Google Scholar] [CrossRef]
- Hristov, P.; Shumkova, R.; Palova, N.; Neov, B. Factors associated with honey bee colony losses: A mini-review. Vet. Sci. 2020, 7, 166. [Google Scholar] [CrossRef] [PubMed]
- Kadlečková, D.; Tachezy, R.; Erban, T.; Deboutte, W.; Nunvář, J.; Saláková, M.; Matthijnssens, J. The virome of healthy honey bee colonies: Ubiquitous occurrence of known and new viruses in bee populations. mSystems 2022, 7, e00072-22. [Google Scholar] [CrossRef] [PubMed]
- Sarwar, M.; Gupta, P. Prevalence of multiple viral diseases associated with honey bees colony collapse and control of disorders. IJZS 2016, 1, 2455–7269. [Google Scholar]
- Stanimirović, Z.; Glavinić, U.; Ristanić, M.; Aleksić, N.; Jovanović, N.; Vejnović, B.; Stevanović, J. Looking for the causes of and solutions to the issue of honey bee colony losses. Acta Vet. 2019, 69, 1–31. [Google Scholar] [CrossRef]
- Salina, M.D.; Garcia, M.L.G.; Bais, B.; Bravi, M.E.; Brasesco, C.; Maggi, M.; Reynaldi, F.J. Viruses that affect Argentinian honey bees (Apis mellifera). Arch. Virol. 2021, 166, 1533–1545. [Google Scholar] [CrossRef] [PubMed]
- Brutscher, L.M.; McMenamin, A.J.; Flenniken, M.L. The buzz about honey bee viruses. PLoS Pathog. 2016, 12, e1005757. [Google Scholar] [CrossRef] [PubMed]
- Procházková, M.; Škubník, K.; Füzik, T.; Mukhamedova, L.; Přidal, A.; Plevka, P. Virion structures and genome delivery of honeybee viruses. Curr. Opin. Virol. 2020, 45, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, G.B. Phylogeographic relationship of honey bee dicistroviruses. Bee World 2022, 99, 99–102. [Google Scholar] [CrossRef]
- Li, J.; Wang, T.; Evans, J.D.; Rose, R.; Zhao, Y.; Li, Z.; Li, J.; Huang, S.; Heerman, M.; Rodríguez-García, C.; et al. The phylogeny and pathogenesis of sacbrood virus (SBV) infection in European honey bees, Apis mellifera. Viruses 2019, 11, 61. [Google Scholar] [CrossRef]
- McMahon, D.P.; Natsopoulou, M.E.; Doublet, V.; Fürst, M.; Weging, S.; Brown, M.J.F.; Gogol-Döring, A.; Paxton, R.J. Elevated virulence of an emerging viral genotype as a driver of honeybee loss. Proc. R. Soc. Lond. B Biol. Sci. 2016, 283, 20160811. [Google Scholar] [CrossRef]
- Ongus, J.R.; Peters, D.; Bonmatin, J.M.; Bengsch, E.; Vlak, J.M.; van Oers, M.M. Complete sequence of a picorna-like virus of the genus Iflavirus replicating in the mite Varroa destructor. J. Gen. Virol. 2004, 85, 3747–3755. [Google Scholar] [CrossRef] [PubMed]
- Mordecai, G.J.; Wilfert, L.; Martin, S.J.; Jones, I.M.; Schroeder, D.C. Diversity in a honey bee pathogen: First report of a third master variant of the Deformed Wing Virus quasispecies. ISME J. 2016, 10, 1264–1273. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, J.R.; Bailey, L.; Ball, B.V.; Blanchard, P.; Budge, G.E.; Chejanovsky, N.; Chen, Y.P.; Gauthier, L.; Genersch, E.; De Graaf, D.C.; et al. Standard methods for virus research in Apis mellifera. J. Apic. Res. 2013, 52, 1–56. [Google Scholar] [CrossRef]
- Milićević, V.; Radojičić, S.; Kureljušić, J.; Šekler, M.; Nešić, K.; Veljović, L.; Zorić, J.M.; Radosavljević, V. Molecular detection of black queen cell virus and Kashmir bee virus in honey. AMB Express 2018, 8, 128. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Levitt, A.L.; Rajotte, E.G.; Holmes, E.C.; Ostiguy, N.; Vanengelsdorp, D.; Lipkin, W.I.; Depamphilis, C.W.; Toth, A.L.; Cox-Foster, D.L. RNA viruses in hymenopteran pollinators: Evidence of inter-Taxa virus transmission via pollen and potential impact on non-Apis hymenopteran species. PLoS ONE 2010, 5, e14357. [Google Scholar] [CrossRef] [PubMed]
- Govan, V.A.; Leat, N.; Allsopp, M.; Davison, S. Analysis of the complete genome sequence of acute bee paralysis virus shows that it belongs to the novel group of insect-infecting RNA viruses. Virology 2000, 277, 457–463. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, J.R.; Drebot, M.; Tyler, S.; Shen, M.; Cameron, C.E.; Stoltz, D.B.; Camazine, S.M. Complete nucleotide sequence of Kashmir bee virus and comparison with acute bee paralysis virus. J. Gen. Virol. 2004, 85, 2263–2270. [Google Scholar] [CrossRef] [PubMed]
- Maori, E.; Lavi, S.; Mozes-Koch, R.; Gantman, Y.; Peretz, Y.; Edelbaum, O.; Sela, I. Isolation and characterization of Israeli acute paralysis virus, a dicistrovirus affecting honeybees in Israel: Evidence for diversity due to intra- and inter-species recombination. J. Gen. Virol. 2007, 88, 3428–3438. [Google Scholar] [CrossRef] [PubMed]
- Leat, N.; Ball, B.; Govan, V.; Davison, S. Analysis of the complete genome sequence of black queen-cell virus, a picorna-like virus of honey bees. J. Gen. Virol. 2000, 81, 2111–2119. [Google Scholar] [CrossRef]
- Lanzi, G.; de Miranda, J.R.; Boniotti, M.B.; Cameron, C.E.; Lavazza, A.; Capucci, L.; Camazine, S.M.; Rossi, C. Molecular and biological characterization of Deformed wing virus of honeybees (Apis mellifera L.). J. Virol. 2006, 80, 4998–5009. [Google Scholar] [CrossRef]
- de Miranda, J.R.; Brettell, L.E.; Chejanovsky, N.; Childers, A.K.; Dalmon, A.; Deboutte, W.; de Graaf, D.C.; Doublet, V.; Gebremedhn, H.; Genersch, E.; et al. Cold case: The disappearance of Egypt bee virus, a fourth distinct master strain of deformed wing virus linked to honeybee mortality in 1970’s Egypt. Virol. J. 2022, 19, 12. [Google Scholar] [CrossRef] [PubMed]
- Mordecai, G.J.; Brettell, L.E.; Pachori, P.; Villalobos, E.M.; Martin, S.J.; Jones, I.M.; Schroeder, D.C. Moku virus; a new Iflavirus found in wasps, honey bees and Varroa. Sci. Rep. 2016, 6, 34983. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.C.; Ball, B.V.; Willcocks, M.M.; Carter, M.J. The nucleotide sequence of sacbrood virus of the honey bee: An insect picorna-like virus. J. Gen. Virol. 1999, 80, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, J.R.; Dainat, B.; Locke, B.; Cordoni, G.; Berthoud, H.; Gauthier, L.; Neumann, P.; Budge, G.E.; Ball, B.V.; Stoltz, D.B. Genetic characterization of slow bee paralysis virus of the honeybee (Apis mellifera L.). J. Gen. Virol. 2010, 91, 2524–2530. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, J.; Cornman, R.; Evans, J.; Semberg, E.; Haddad, N.; Neumann, P.; Gauthier, L. Genome characterization, prevalence and distribution of a macula-like virus from Apis mellifera and Varroa destructor. Viruses 2015, 7, 3586–3602. [Google Scholar] [CrossRef] [PubMed]
- Olivier, V.; Blanchard, P.; Chaouch, S.; Lallemand, P.; Schurr, F.; Celle, O.; Dubois, E.; Tordo, N.; Thiéry, R.; Houlgatte, R.; et al. Molecular characterisation and phylogenetic analysis of Chronic bee paralysis virus, a honey bee virus. Virus Res. 2008, 132, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Runckel, C.; Flenniken, M.L.; Engel, J.C.; Ruby, J.G.; Ganem, D.; Andino, R.; DeRisi, J.L. Temporal analysis of the honey bee microbiome reveals four novel viruses and seasonal prevalence of known viruses, Nosema, and Crithidia. PLoS ONE 2011, 6, e20656. [Google Scholar] [CrossRef]
- Thaduri, S.; Locke, B.; Granberg, F.; De Miranda, J.R. Temporal changes in the viromes of Swedish Varroa-resistant and Varroa-susceptible honeybee populations. PLoS ONE 2018, 13, e0206938. [Google Scholar] [CrossRef] [PubMed]
- Ravoet, J.; De Smet, L.; Wenseleers, T.; De Graaf, D.C. Genome sequence heterogeneity of Lake Sinai Virus found in honey bees and Orf1/RdRP-based polymorphisms in a single host. Virus Res. 2015, 201, 67–72. [Google Scholar] [CrossRef]
- Gauthier, L.; Cornman, S.; Hartmann, U.; Cousserans, F.; Evans, J.; De Miranda, J.; Neumann, P. The Apis mellifera filamentous virus genome. Viruses 2015, 7, 3798–3815. [Google Scholar] [CrossRef]
- Stoltz, D.; Shen, X.-R.; Boggis, C.; Sisson, G. Molecular diagnostic of Kashmir bee virus infection. J. Apic. Res. 1995, 34, 153–165. [Google Scholar] [CrossRef]
- Tentcheva, D.; Gauthier, L.; Zappulla, N.; Dainat, B.; Cousserans, F.; Colin, M.E.; Bergoin, M. Prevalence and seasonal variations of six bee viruses in Apis mellifera L. and Varroa destructor mite populations in France. Appl. Environ. Microbiol. 2004, 70, 7185–7191. [Google Scholar] [CrossRef]
- Ribière, M.; Faucon, J.P.; Pépin, M. Detection of chronic honey bee (Apis mellifera L.) paralysis virus infection: Application to a field survey. Apidologie 2000, 31, 567–577. [Google Scholar] [CrossRef]
- Shumkova, R.; Neov, B.; Sirakova, D.; Georgieva, A.; Gadjev, D.; Teofanova, D.; Radoslavov, G.; Bouga, M.; Hristov, P. Molecular detection and phylogenetic assessment of six honeybee viruses in Apis mellifera L. colonies in Bulgaria. PeerJ 2018, 6, e5077. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Tamura, K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Mol. Biol. Evol. 1992, 9, 678–687. [Google Scholar] [CrossRef]
- Veilleux, H.D.; Misutka, M.D.; Glover, C.N. Environmental DNA and environmental RNA: Current and prospective applications for biological monitoring. Sci. Total Environ. 2021, 782, 146891. [Google Scholar] [CrossRef]
- Laroche, O.; Wood, S.A.; Tremblay, L.A.; Ellis, J.I.; Lejzerowicz, F.; Pawlowski, J.; Lear, G.; Atalah, J.; Pochon, X. First evaluation of foraminiferal metabarcoding for monitoring environmental impact from an offshore oil drilling site. Mar. Environ. Res. 2016, 120, 225–235. [Google Scholar] [CrossRef]
- Kim, H.J.; Hwang, J.; Ullah, Z.; Mustafa, B.; Kwon, H.W. Comparison of physicochemical properties of pollen substitute diet for honey bee (Apis mellifera). J. Asia-Pac. Entomol. 2022, 25, 101967. [Google Scholar] [CrossRef]
- Bleha, R.; Shevtsova, T.; Kružik, V.; Šorpilová, T.; Saloň, I.; Erban, E.; Brindza, J.; Brovarskyi, V.; Sinica, A. Bee breads from two regions of Eastern Ukraine: Composition, physical properties and biological activities. Czech J. Food Sci. 2019, 37, 9–20. [Google Scholar] [CrossRef]
- Bernhardt, H.S.; Tate, W.P. Primordial soup or vinaigrette: Did the RNA world evolve at acidic pH? Biol. Direct 2012, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Silva de Oliveira, V.H.; Nilsson, A.; Kim, H.; Hallgren, G.; Frössling, J.; Kristiansen, L.F.; Forsgren, E. Honey bee pathogens and parasites in Swedish apiaries: A baseline study. J. Apic. Res. 2021, 60, 697–706. [Google Scholar] [CrossRef]
- Ullah, A.; Gajger, I.T.; Majoros, A.; Dar, S.A.; Khan, S.; Shah, A.H.; Khabir, M.N.; Hussain, R.; Khan, H.U.; Hameed, M.; et al. Viral impacts on honey bee populations: A review. Saudi J. Biol. Sci. 2021, 28, 523–530. [Google Scholar] [CrossRef]
- Bruckner, S.; Wilson, M.; Aurell, D.; Rennich, K.; Vanengelsdorp, D.; Steinhauer, N.; Williams, G.R. A national survey of managed honey bee colony losses in the USA: Results from the Bee Informed Partnership for 2017–18, 2018–19, and 2019–20. J. Apic. Res. 2023, 62, 429–443. [Google Scholar] [CrossRef]
- Stevens, J.D.; Parsley, M.B. Environmental RNA applications and their associated gene targets for management and conservation. Environ. DNA 2023, 5, 227–239. [Google Scholar] [CrossRef]
- Chen, Y.P.; Siede, R. Honey bee viruses. Adv. Virus Res. 2007, 70, 33–80. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, J.R.; Genersch, E. Deformed wing virus. J. Invertebr. Pathol. 2010, 103, S48–S61. [Google Scholar] [CrossRef] [PubMed]
- Bakonyi, T.; Grabensteiner, E.; Kolodziejek, J.; Rusvai, M.; Topolska, G.; Ritter, W.; Nowotny, N. Phylogenetic analysis of acute bee paralysis virus strains. Appl. Environ. Microbiol. 2002, 68, 6446–6450. [Google Scholar] [CrossRef]
- Griffiths, D.A.; Bowman, C.E. World distribution of the mite Varroa jacobsoni, a parasite of honeybees. Bee World 1981, 62, 154–163. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salkova, D.; Balkanska, R.; Shumkova, R.; Lazarova, S.; Radoslavov, G.; Hristov, P. Molecular Detection and Phylogenetic Relationships of Honey Bee-Associated Viruses in Bee Products. Vet. Sci. 2024, 11, 369. https://doi.org/10.3390/vetsci11080369
Salkova D, Balkanska R, Shumkova R, Lazarova S, Radoslavov G, Hristov P. Molecular Detection and Phylogenetic Relationships of Honey Bee-Associated Viruses in Bee Products. Veterinary Sciences. 2024; 11(8):369. https://doi.org/10.3390/vetsci11080369
Chicago/Turabian StyleSalkova, Delka, Ralitsa Balkanska, Rositsa Shumkova, Stela Lazarova, Georgi Radoslavov, and Peter Hristov. 2024. "Molecular Detection and Phylogenetic Relationships of Honey Bee-Associated Viruses in Bee Products" Veterinary Sciences 11, no. 8: 369. https://doi.org/10.3390/vetsci11080369
APA StyleSalkova, D., Balkanska, R., Shumkova, R., Lazarova, S., Radoslavov, G., & Hristov, P. (2024). Molecular Detection and Phylogenetic Relationships of Honey Bee-Associated Viruses in Bee Products. Veterinary Sciences, 11(8), 369. https://doi.org/10.3390/vetsci11080369