
Bisphenol A Coupled with a High-Fat Diet Promotes Hepatosteatosis through Reactive-Oxygen-Species-Induced CD36 Overexpression


 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical Reagents and Cell Line
2.2. Cell Viability Assay
2.3. Intracellular TG Levels and Lipid Staining
2.4. Determination of Intracellular Fatty Acid Uptake
2.5. Measurement of Intracellular ROS
2.6. Western Blotting and Immunofluorescence
2.7. RNA Extraction and Reverse Transcription–Quantitative Polymerase Chain Reaction (RT-qPCR)
2.8. Animals and Experimental Design
2.9. Staining for Fibrosis in Hepatic Tissue
2.10. Biochemical Assays
2.11. Statistical Analysis
3. Results
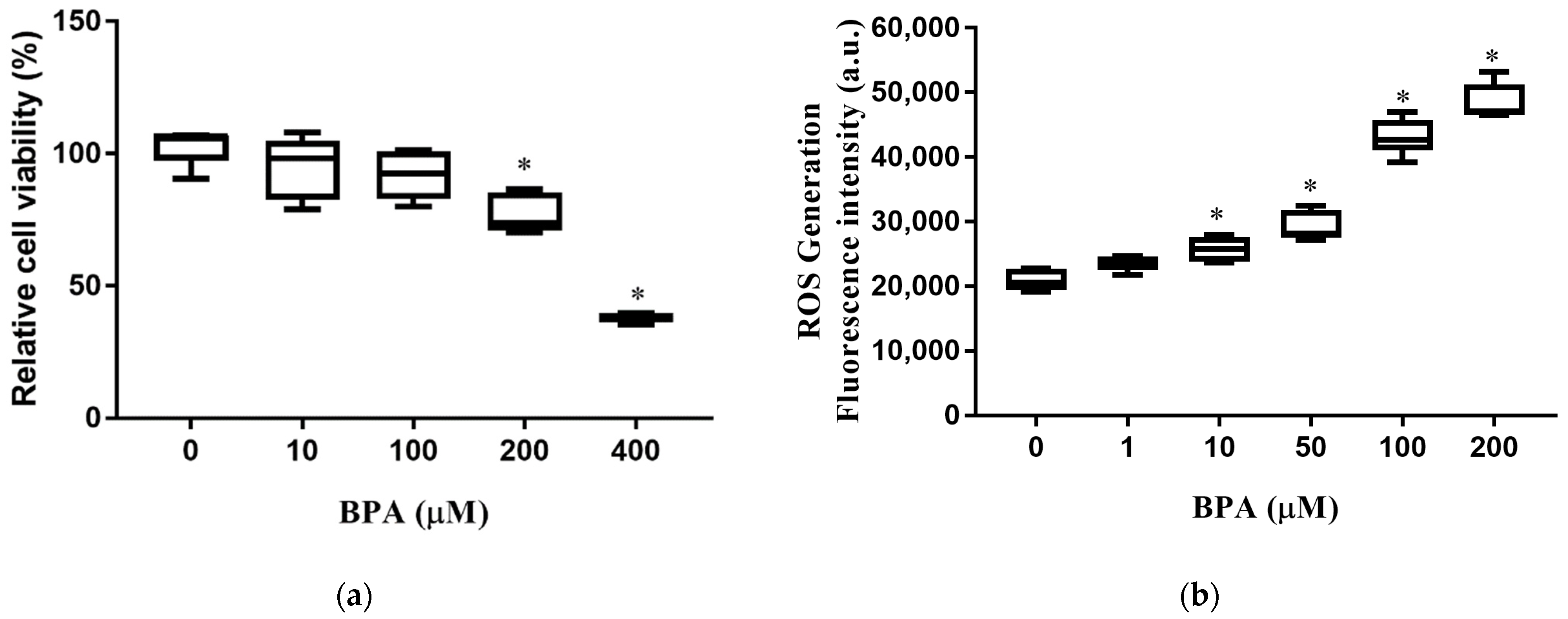
3.1. BPA Treatment Induced Cell Death and Increased Intracellular ROS Production
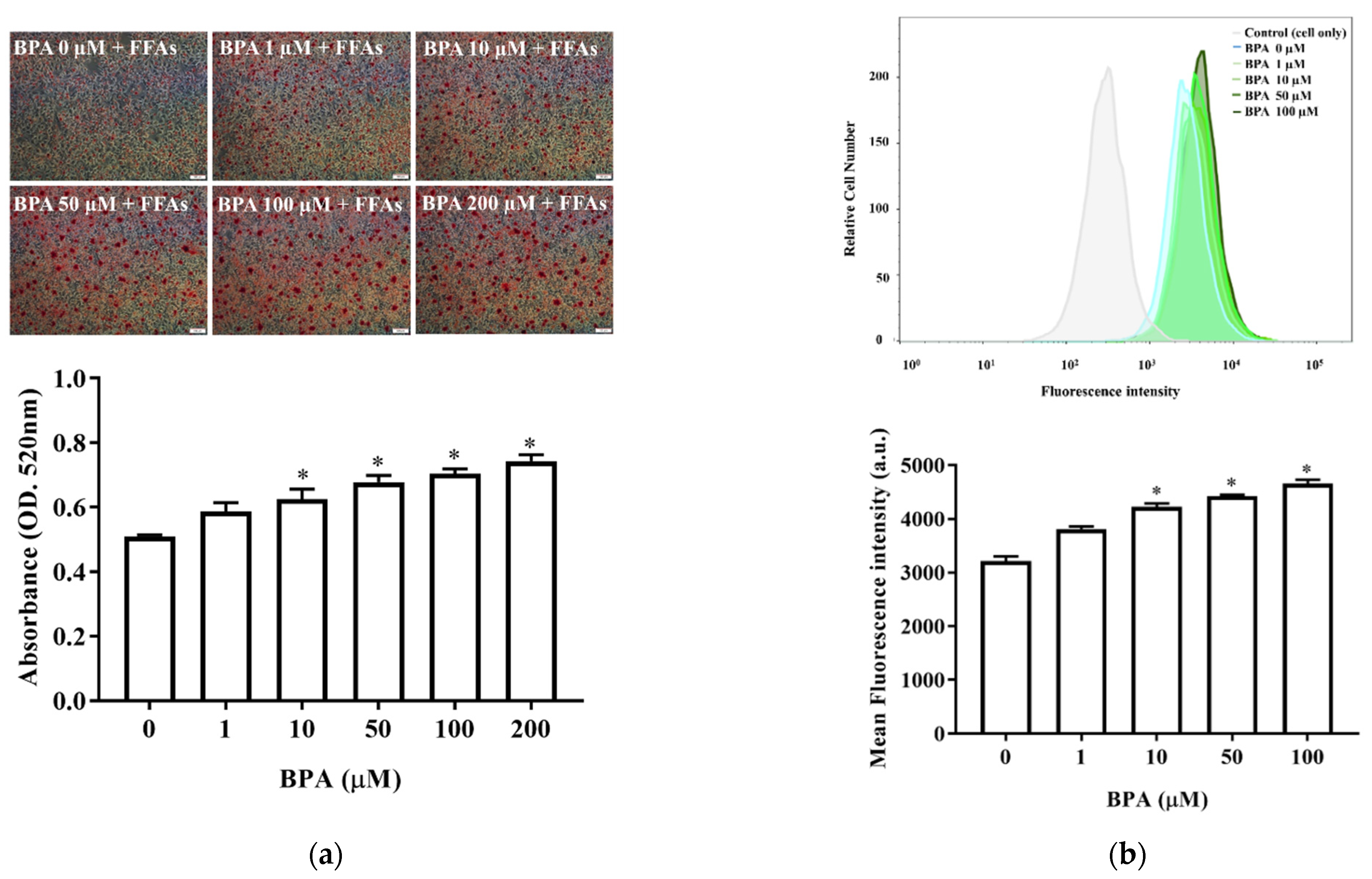
3.2. BPA Treatment Enhanced Accumulation and Uptake of Lipid Droplets
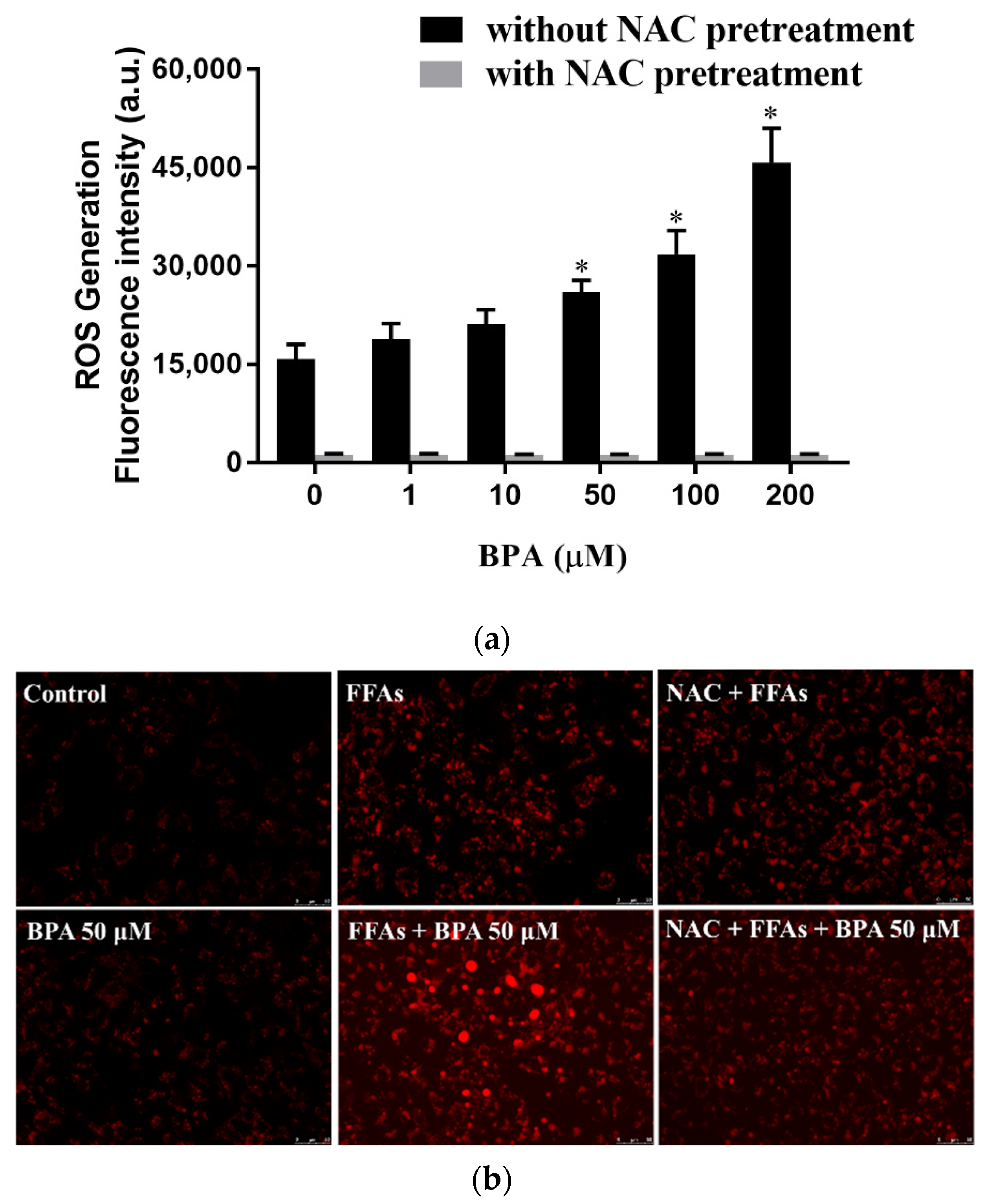
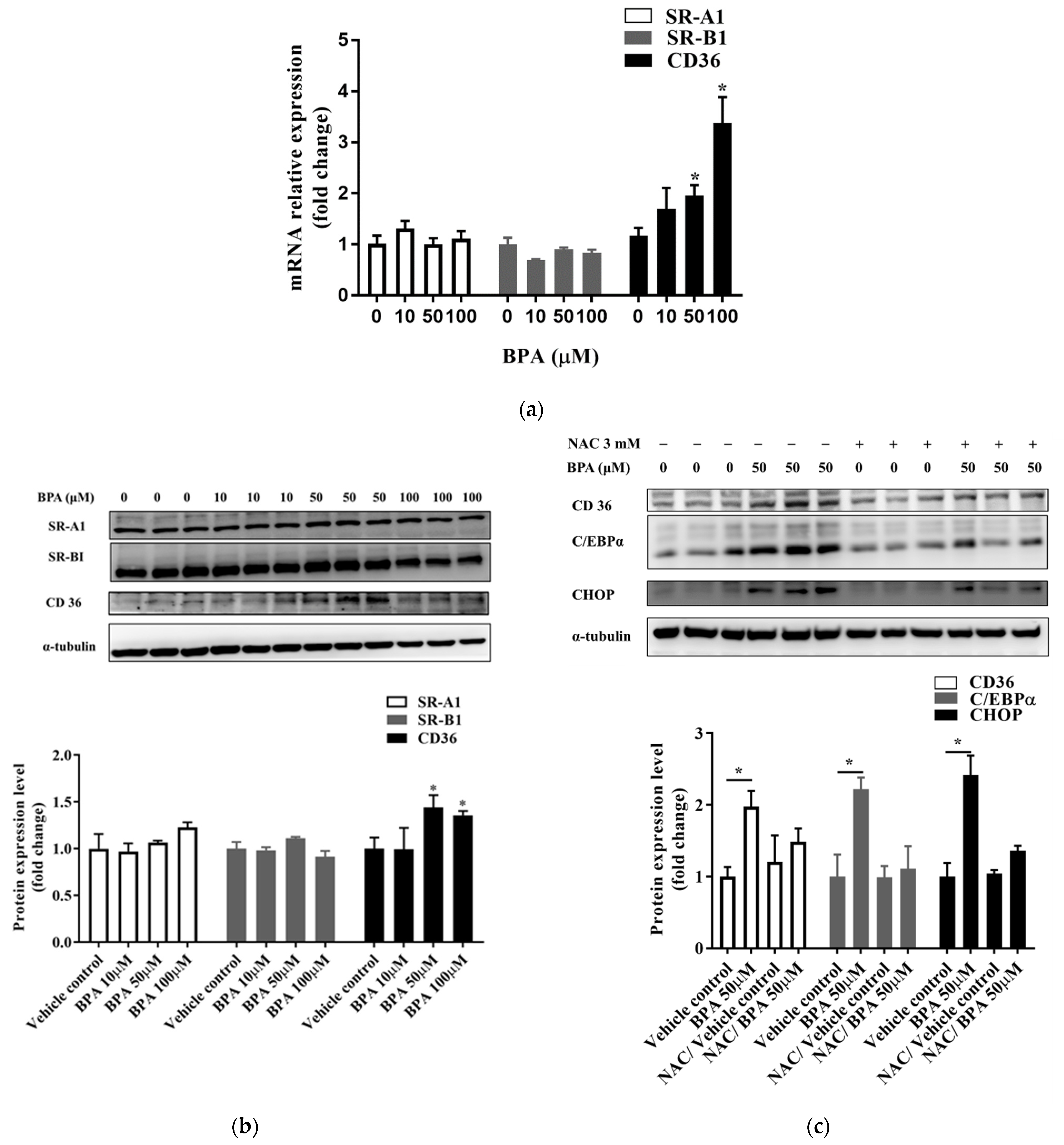
3.3. N-Acetylcysteine (NAC) Suppresses BPA-Induced Fatty Acid Uptake and Lipid Accumulation
3.4. BPA Induced Fatty Acid Uptake by Modulating CD36 Expression
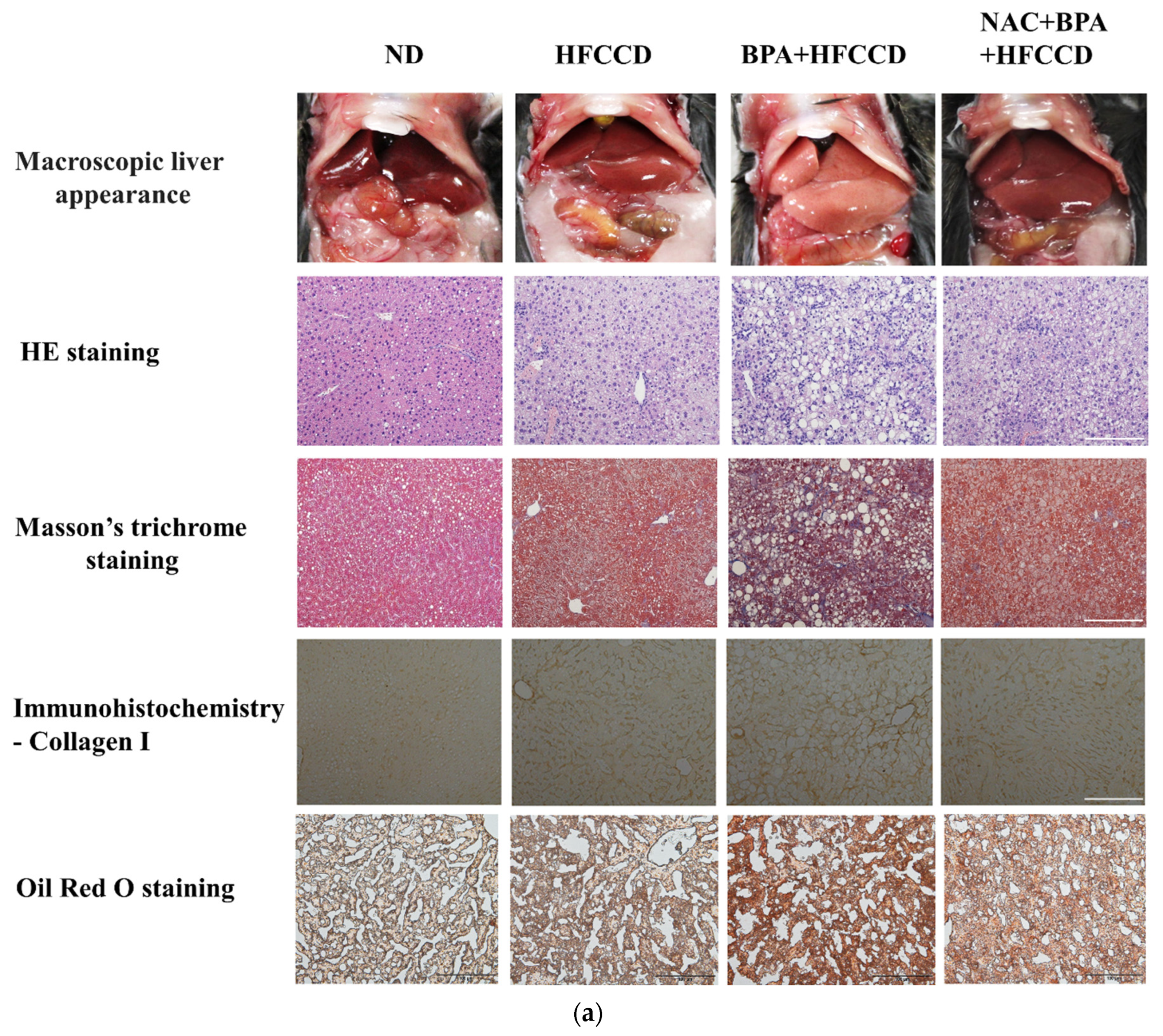
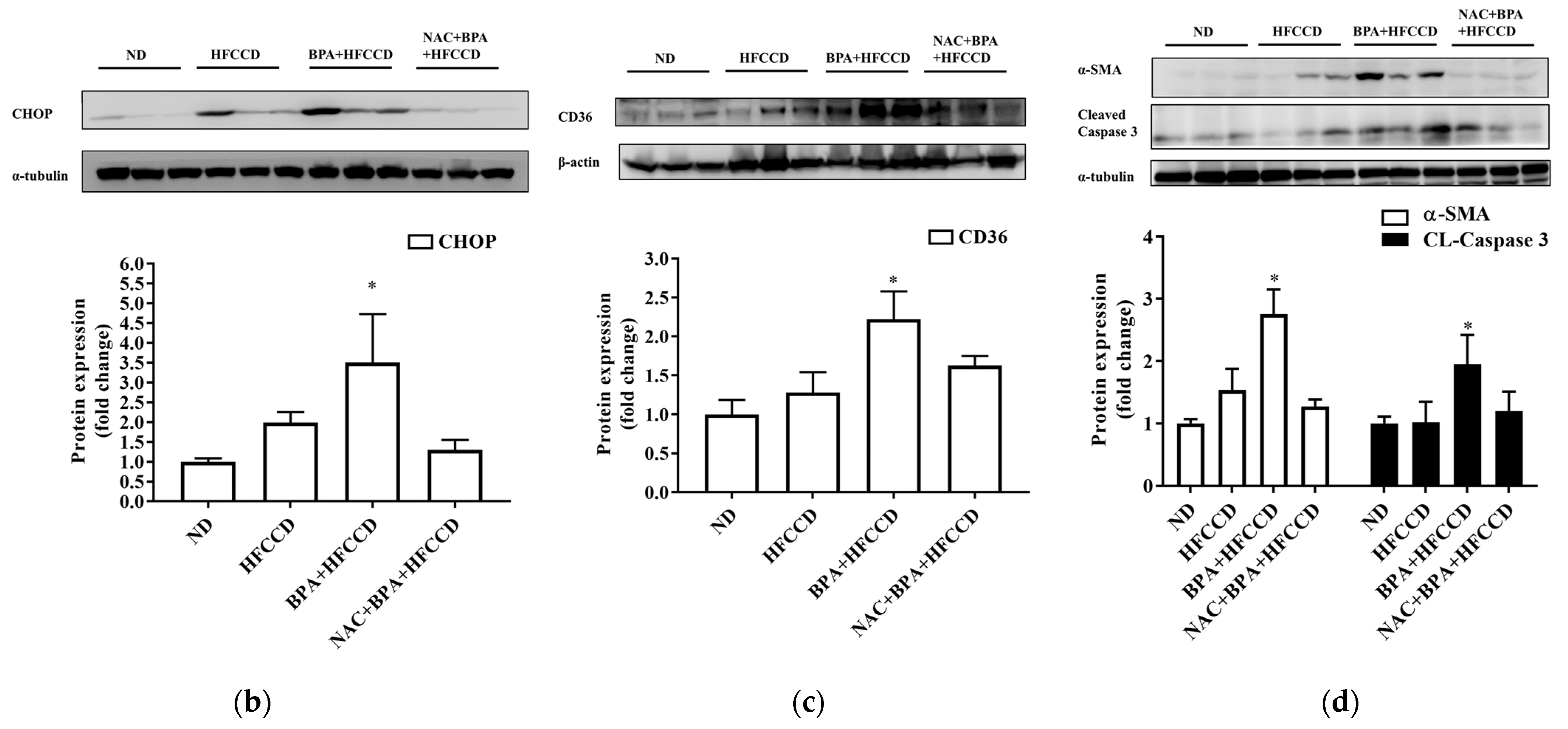
3.5. BPA Enhanced Hepatic Pathological Progression in Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Sherif, Z.A.; Saeed, A.; Ghavimi, S.; Nouraie, S.M.; Laiyemo, A.O.; Brim, H.; Ashktorab, H. Global Epidemiology of Nonalcoholic Fatty Liver Disease and Perspectives on US Minority Populations. Dig. Dis. Sci. 2016, 61, 1214–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, T.; Oakley, F.; Anstee, Q.M.; Day, C.P. Nonalcoholic Fatty Liver Disease: Pathogenesis and Disease Spectrum. Annu. Rev. Pathol. 2016, 11, 451–496. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; Broderick, L.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 627–636. [Google Scholar] [CrossRef]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef] [Green Version]
- Perla, F.M.; Prelati, M.; Lavorato, M.; Visicchio, D.; Anania, C. The Role of Lipid and Lipoprotein Metabolism in Non-Alcoholic Fatty Liver Disease. Children 2017, 4, 46. [Google Scholar] [CrossRef] [Green Version]
- De Taeye, B.M.; Novitskaya, T.; McGuinness, O.P.; Gleaves, L.; Medda, M.; Covington, J.W.; Vaughan, D.E. Macrophage TNF-alpha contributes to insulin resistance and hepatic steatosis in diet-induced obesity. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E713–E725. [Google Scholar] [CrossRef]
- Nagao, T.; Saito, Y.; Usumi, K.; Kuwagata, M.; Imai, K. Reproductive function in rats exposed neonatally to bisphenol A and estradiol benzoate. Reprod. Toxicol. 1999, 13, 303–311. [Google Scholar] [CrossRef]
- Dallio, M.; Masarone, M.; Errico, S.; Gravina, A.G.; Nicolucci, C.; Di Sarno, R.; Gionti, L.; Tuccillo, C.; Persico, M.; Stiuso, P.; et al. Role of bisphenol A as environmental factor in the promotion of non-alcoholic fatty liver disease: In vitro and clinical study. Aliment. Pharmacol. Ther. 2018, 47, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Federico, A.; Dallio, M.; Gravina, A.G.; Diano, N.; Errico, S.; Masarone, M.; Romeo, M.; Tuccillo, C.; Stiuso, P.; Morisco, F.; et al. The Bisphenol A Induced Oxidative Stress in Non-Alcoholic Fatty Liver Disease Male Patients: A Clinical Strategy to Antagonize the Progression of the Disease. Int. J. Environ. Res. Public Health 2020, 17, 3369. [Google Scholar] [CrossRef]
- Huc, L.; Lemarie, A.; Gueraud, F.; Helies-Toussaint, C. Low concentrations of bisphenol A induce lipid accumulation mediated by the production of reactive oxygen species in the mitochondria of HepG2 cells. Toxicol. In Vitro 2012, 26, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, E.; Cortese, K.; Voci, A.; Vergani, L.; Fabbri, R.; Barmo, C.; Gallo, G.; Canesi, L. Direct effects of Bisphenol A on lipid homeostasis in rat hepatoma cells. Chemosphere 2013, 91, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Gao, R.; Peng, C.; Yi, J.; Liu, L.; Yang, S.; Li, D.; Hu, J.; Luo, T.; Mei, M.; et al. Bisphenol A promotes hepatic lipid deposition involving Kupffer cells M1 polarization in male mice. J. Endocrinol. 2017, 234, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Shimpi, P.C.; More, V.R.; Paranjpe, M.; Donepudi, A.C.; Goodrich, J.M.; Dolinoy, D.C.; Rubin, B.; Slitt, A.L. Hepatic Lipid Accumulation and Nrf2 Expression following Perinatal and Peripubertal Exposure to Bisphenol A in a Mouse Model of Nonalcoholic Liver Disease. Environ. Health Perspect. 2017, 125, 087005. [Google Scholar] [CrossRef]
- Lama, S.; Vanacore, D.; Diano, N.; Nicolucci, C.; Errico, S.; Dallio, M.; Federico, A.; Loguercio, C.; Stiuso, P. Ameliorative effect of Silybin on bisphenol A induced oxidative stress, cell proliferation and steroid hormones oxidation in HepG2 cell cultures. Sci. Rep. 2019, 9, 3228. [Google Scholar] [CrossRef]
- Figueiredo, L.S.; Oliveira, K.M.; Freitas, I.N.; Silva, J.A., Jr.; Silva, J.N.; Favero-Santos, B.C.; Bonfleur, M.L.; Carneiro, E.M.; Ribeiro, R.A. Bisphenol-A exposure worsens hepatic steatosis in ovariectomized mice fed on a high-fat diet: Role of endoplasmic reticulum stress and fibrogenic pathways. Life Sci. 2020, 256, 118012. [Google Scholar] [CrossRef]
- Long, Z.; Fan, J.; Wu, G.; Liu, X.; Wu, H.; Liu, J.; Chen, Y.; Su, S.; Cheng, X.; Xu, Z.; et al. Gestational bisphenol A exposure induces fatty liver development in male offspring mice through the inhibition of HNF1b and upregulation of PPARgamma. Cell Biol. Toxicol. 2021, 37, 65–84. [Google Scholar] [CrossRef]
- Pei, K.; Gui, T.; Kan, D.; Feng, H.; Jin, Y.; Yang, Y.; Zhang, Q.; Du, Z.; Gai, Z.; Wu, J.; et al. An Overview of Lipid Metabolism and Nonalcoholic Fatty Liver Disease. Biomed. Res. Int. 2020, 2020, 4020249. [Google Scholar] [CrossRef]
- Zani, I.A.; Stephen, S.L.; Mughal, N.A.; Russell, D.; Homer-Vanniasinkam, S.; Wheatcroft, S.B.; Ponnambalam, S. Scavenger receptor structure and function in health and disease. Cells 2015, 4, 178–201. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.J.; Asthana, S.; Kraemer, F.B.; Azhar, S. Scavenger receptor B type 1: Expression, molecular regulation, and cholesterol transport function. J. Lipid Res. 2018, 59, 1114–1131. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.M. CD36, a scavenger receptor implicated in atherosclerosis. Exp. Mol. Med. 2014, 46, e99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Febbraio, M.; Reddy, S.P.; Yu, D.Y.; Yamamoto, M.; Silverstein, R.L. CD36 participates in a signaling pathway that regulates ROS formation in murine VSMCs. J. Clin. Investig. 2010, 120, 3996–4006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lett, T.A.; Wallace, T.J.; Chowdhury, N.I.; Tiwari, A.K.; Kennedy, J.L.; Muller, D.J. Pharmacogenetics of antipsychotic-induced weight gain: Review and clinical implications. Mol. Psychiatry 2012, 17, 242–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, F.; Jiang, L.; Liu, X.; Geng, C.; Wang, W.; Zhong, L.; Yang, G.; Chen, M. Bisphenol A induces oxidative stress-associated DNA damage in INS-1 cells. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2014, 769, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Dai, Y.; Cui, Z.; Jiang, X.; Liu, W.; Han, F.; Lin, A.; Cao, J.; Liu, J. The regulation of cellular apoptosis by the ROS-triggered PERK/EIF2alpha/chop pathway plays a vital role in bisphenol A-induced male reproductive toxicity. Toxicol. Appl. Pharmacol. 2017, 314, 98–108. [Google Scholar] [CrossRef]
- Seo, E.; Kang, H.; Choi, H.; Choi, W.; Jun, H.S. Reactive oxygen species-induced changes in glucose and lipid metabolism contribute to the accumulation of cholesterol in the liver during aging. Aging Cell 2019, 18, e12895. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Lechon, M.J.; Donato, M.T.; Martinez-Romero, A.; Jimenez, N.; Castell, J.V.; O’Connor, J.E. A human hepatocellular in vitro model to investigate steatosis. Chem. Biol. Interact. 2007, 165, 106–116. [Google Scholar] [CrossRef]
- Chen, H.W.; Yen, C.C.; Kuo, L.L.; Lo, C.W.; Huang, C.S.; Chen, C.C.; Lii, C.K. Benzyl isothiocyanate ameliorates high-fat/cholesterol/cholic acid diet-induced nonalcoholic steatohepatitis through inhibiting cholesterol crystal-activated NLRP3 inflammasome in Kupffer cells. Toxicol. Appl. Pharmacol. 2020, 393, 114941. [Google Scholar] [CrossRef]
- Martella, A.; Silvestri, C.; Maradonna, F.; Gioacchini, G.; Allara, M.; Radaelli, G.; Overby, D.R.; Di Marzo, V.; Carnevali, O. Bisphenol A Induces Fatty Liver by an Endocannabinoid-Mediated Positive Feedback Loop. Endocrinology 2016, 157, 1751–1763. [Google Scholar] [CrossRef] [Green Version]
- Verstraete, S.G.; Wojcicki, J.M.; Perito, E.R.; Rosenthal, P. Bisphenol a increases risk for presumed non-alcoholic fatty liver disease in Hispanic adolescents in NHANES 2003–2010. Environ. Health 2018, 17, 12. [Google Scholar] [CrossRef] [Green Version]
- Chiu, W.C.; Yang, H.H.; Chiang, S.C.; Chou, Y.X.; Yang, H.T. Auricularia polytricha aqueous extract supplementation decreases hepatic lipid accumulation and improves antioxidative status in animal model of nonalcoholic fatty liver. Biomedicine 2014, 4, 12. [Google Scholar] [CrossRef]
- Simoes, I.C.M.; Amorim, R.; Teixeira, J.; Karkucinska-Wieckowska, A.; Carvalho, A.; Pereira, S.P.; Simoes, R.F.; Szymanska, S.; Dabrowski, M.; Janikiewicz, J.; et al. The Alterations of Mitochondrial Function during NAFLD Progression-An Independent Effect of Mitochondrial ROS Production. Int. J. Mol. Sci. 2021, 22, 6848. [Google Scholar] [CrossRef]
- Gassman, N.R.; Coskun, E.; Stefanick, D.F.; Horton, J.K.; Jaruga, P.; Dizdaroglu, M.; Wilson, S.H. Bisphenol a promotes cell survival following oxidative DNA damage in mouse fibroblasts. PLoS ONE 2015, 10, e0118819. [Google Scholar] [CrossRef] [Green Version]
- Provvisiero, D.P.; Pivonello, C.; Muscogiuri, G.; Negri, M.; de Angelis, C.; Simeoli, C.; Pivonello, R.; Colao, A. Influence of Bisphenol A on Type 2 Diabetes Mellitus. Int. J. Environ. Res. Public Health 2016, 13, 989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stojanoska, M.M.; Milosevic, N.; Milic, N.; Abenavoli, L. The influence of phthalates and bisphenol A on the obesity development and glucose metabolism disorders. Endocrine 2017, 55, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Babu, S.; Uppu, S.; Claville, M.O.; Uppu, R.M. Prooxidant actions of bisphenol A (BPA) phenoxyl radicals: Implications to BPA-related oxidative stress and toxicity. Toxicol. Mech. Methods 2013, 23, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Carchia, E.; Porreca, I.; Almeida, P.J.; D’Angelo, F.; Cuomo, D.; Ceccarelli, M.; De Felice, M.; Mallardo, M.; Ambrosino, C. Evaluation of low doses BPA-induced perturbation of glycemia by toxicogenomics points to a primary role of pancreatic islets and to the mechanism of toxicity. Cell Death Dis. 2015, 6, e1959. [Google Scholar] [CrossRef] [Green Version]
- Le Magueresse-Battistoni, B. Endocrine disrupting chemicals and metabolic disorders in the liver: What if we also looked at the female side? Chemosphere 2021, 268, 129212. [Google Scholar] [CrossRef]
- Gassman, N.R. Induction of oxidative stress by bisphenol A and its pleiotropic effects. Environ. Mol. Mutagen. 2017, 58, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Sun, X.; Chen, Y.; Li, Y.; Song, L.; Zhou, Z.; Xu, B.; Lin, Y.; Xu, S. Perinatal exposure to bisphenol A exacerbates nonalcoholic steatohepatitis-like phenotype in male rat offspring fed on a high-fat diet. J. Endocrinol. 2014, 222, 313–325. [Google Scholar] [CrossRef] [Green Version]
- Moller, P.; Lohr, M.; Folkmann, J.K.; Mikkelsen, L.; Loft, S. Aging and oxidatively damaged nuclear DNA in animal organs. Free Radic. Biol. Med. 2010, 48, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- Karihtala, P.; Soini, Y.; Vaskivuo, L.; Bloigu, R.; Puistola, U. DNA adduct 8-hydroxydeoxyguanosine, a novel putative marker of prognostic significance in ovarian carcinoma. Int. J. Gynecol. Cancer 2009, 19, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Schwenk, R.W.; Holloway, G.P.; Luiken, J.J.; Bonen, A.; Glatz, J.F. Fatty acid transport across the cell membrane: Regulation by fatty acid transporters. Prostaglandins Leukot. Essent. Fat. Acids 2010, 82, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Miao, C.; Tian, H.; Sang, H.; Yang, N.; Jiao, P.; Han, J.; Zong, C.; Qin, S. Endoplasmic reticulum stress promotes macrophage-derived foam cell formation by up-regulating cluster of differentiation 36 (CD36) expression. J. Biol. Chem. 2014, 289, 4032–4042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dludla, P.V.; Nkambule, B.B.; Mazibuko-Mbeje, S.E.; Nyambuya, T.M.; Marcheggiani, F.; Cirilli, I.; Ziqubu, K.; Shabalala, S.C.; Johnson, R.; Louw, J.; et al. N-Acetyl Cysteine Targets Hepatic Lipid Accumulation to Curb Oxidative Stress and Inflammation in NAFLD: A Comprehensive Analysis of the Literature. Antioxidants 2020, 9, 1283. [Google Scholar] [CrossRef]
- Daronch, O.T.; Lima, M.M.; Gil, I.; Bittencourt, S.S.; Ingenchki, V.; Maluf, E. Large-scale contamination by Bisphenol A: Are we aware of the risk and forms of exposure? Cienc. Saude Colet. 2020, 25, 4339–4345. [Google Scholar] [CrossRef]
- Cao, X.L.; Corriveau, J.; Popovic, S. Migration of bisphenol A from can coatings to liquid infant formula during storage at room temperature. J. Food Prot. 2009, 72, 2571–2574. [Google Scholar] [CrossRef]
- Hammarling, L.; Gustavsson, H.; Svensson, K.; Oskarsson, A. Migration of bisphenol-A diglycidyl ether (BADGE) and its reaction products in canned foods. Food Addit. Contam. 2000, 17, 937–943. [Google Scholar] [CrossRef]
- Melnick, R.; Lucier, G.; Wolfe, M.; Hall, R.; Stancel, G.; Prins, G.; Gallo, M.; Reuhl, K.; Ho, S.M.; Brown, T.; et al. Summary of the National Toxicology Program’s report of the endocrine disruptors low-dose peer review. Environ. Health Perspect. 2002, 110, 427–431. [Google Scholar] [CrossRef]
- Marmugi, A.; Ducheix, S.; Lasserre, F.; Polizzi, A.; Paris, A.; Priymenko, N.; Bertrand-Michel, J.; Pineau, T.; Guillou, H.; Martin, P.G.; et al. Low doses of bisphenol A induce gene expression related to lipid synthesis and trigger triglyceride accumulation in adult mouse liver. Hepatology 2012, 55, 395–407. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, H.; Zou, J.; Mai, H.; Su, D.; Feng, X.; Feng, D. Bisphenol A exposure induces cholesterol synthesis and hepatic steatosis in C57BL/6 mice by down-regulating the DNA methylation levels of SREBP-2. Food Chem. Toxicol. 2019, 133, 110786. [Google Scholar] [CrossRef] [PubMed]
- Francque, S.M.; Marchesini, G.; Kautz, A.; Walmsley, M.; Dorner, R.; Lazarus, J.V.; Zelber-Sagi, S.; Hallsworth, K.; Busetto, L.; Fruhbeck, G.; et al. Non-alcoholic fatty liver disease: A patient guideline. JHEP Rep. 2021, 3, 100322. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Jiang, F.; Cheng, R.; Luo, Y.; Wang, J.; Luo, Z.; Li, M.; Shen, X.; He, F. A high-fat diet and high-fat and high-cholesterol diet may affect glucose and lipid metabolism differentially through gut microbiota in mice. Exp. Anim. 2021, 70, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Elias, M.S.; Smolak, R.R.; Fu, T.; Borensztajn, J.; Green, R.M. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J. Lipid Res. 2008, 49, 1068–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ND | HFCCD | BPA + HFCCD | NAC + BPA + HFCCD | |
|---|---|---|---|---|
| Initial body wt (g) | 24.0 ± 1.3 | 23.6 ± 0.6 | 24.0 ± 1.7 | 24.0 ± 0.5 |
| Body wt after 4 weeks of the HFCCD diet (g) | 27.2 ± 1.2 b | 27.2 ± 0.9 b | 29.5 ± 1.8 a | 26.2 ± 0.5 b |
| Final body wt (g) | 28.7 ± 2 b | 28.8 ± 1.3 b | 25.9 ± 0.4 a | 28.1 ± 1.6 b |
| Liver wt (g) | 1.046 ± 0.027 c | 1.301 ± 0.073 b | 1.549 ± 0.135 a | 1.403 ± 0.091 b |
| Liver wt/ body wt (%) | 3.665 ± 0.239 c | 4.539 ± 0.416 b | 6.004 ± 0.434 a | 4.997 ± 0.224 b |
| Spleen wt (g) | 0.077 ± 0.006 c | 0.099 ± 0.005 b | 0.117 ± 0.002 a | 0.098 ± 0.010 b |
| Spleen wt/ body wt (%) | 0.268 ± 0.023 c | 0.346 ± 0.028 b | 0.452 ± 0.016 a | 0.350 ± 0.052 b |
| Epididymal fat wt (g) | 0.751 ± 0.089 | 0.795 ± 0.075 | 0.864 ± 0.271 | 0.662 ± 0.091 |
| Epididymal fat wt/ body wt (%) | 2.646 ± 0.464 | 2.761 ± 0.178 | 3.353 ± 1.062 | 2.351 ± 0.239 |
| Brown fat wt (g) | 0.119 ± 0.019 a | 0.112 ± 0.009 a | 0.078 ± 0.016 b | 0.108 ± 0.009 a |
| Brown fat wt/ body wt (%) | 0.415 ± 0.065 a | 0.390 ± 0.028 a | 0.302 ± 0.028 b | 0.381 ± 0.072 a |
| ND | HFCCD | BPA + HFCCD | NAC + BPA + HFCCD | |
|---|---|---|---|---|
| Serum | ||||
| ALT (U/L) | 26.20 ± 4.21 b | 63.40 ± 15.82 a | 93.80 ± 34.58 a | 52.60 ± 11.90 a |
| Triglycerides (mg/dL) | 69.4 ± 7.83 a | 19.20 ± 4.32 b | 42.00 ± 12.14 a | 73.40 ± 32.21 a |
| Total cholesterol (mg/dL) | 134.40 ± 13.83 b | 164.40 ± 27.93 b | 280.20 ± 35.72 a | 131.20 ± 16.96 b |
| Blood glucose (mg/dL) | 152.67 ± 33.63 | 148.00 ± 5.93 | 155.00 ± 18.81 | 143.17 ± 7.60 |
| Insulin (ng/mL) | 0.177 ± 0.081 c | 0.418 ± 0.13 b | 0.651 ± 0.205 a | 0.374 ± 0.091 b |
| HOMA | 1.421 ± 0.453 c | 3.466 ± 1.023 b | 5.717 ± 2.169 a | 3.027 ± 0.774 b |
| Liver | ||||
| Triglycerides (mg/g of tissue) | 43.17 ± 9.66 | 57.36 ± 12.52 | 55.44 ± 10.40 | 47.01 ± 8.06 |
| Total cholesterol (mg/g of tissue) | 6.12 ± 0.74 c | 51.74 ± 10.18 b | 70.50 ± 5.44 a | 50.8 ± 10.81 b |
| 8-OHdG (ng/g of tissue) | 0.577 ± 0.135 c | 1.158 ± 0.148 b | 2.207±0.219 a | 1.224 ± 0.281 b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-L.; Wang, Y.-C.; Hsu, Y.-A.; Chen, C.-S.; Weng, R.-C.; Lu, Y.-P.; Chuang, C.-Y.; Wan, L. Bisphenol A Coupled with a High-Fat Diet Promotes Hepatosteatosis through Reactive-Oxygen-Species-Induced CD36 Overexpression. Toxics 2022, 10, 208. https://doi.org/10.3390/toxics10050208
Lee J-L, Wang Y-C, Hsu Y-A, Chen C-S, Weng R-C, Lu Y-P, Chuang C-Y, Wan L. Bisphenol A Coupled with a High-Fat Diet Promotes Hepatosteatosis through Reactive-Oxygen-Species-Induced CD36 Overexpression. Toxics. 2022; 10(5):208. https://doi.org/10.3390/toxics10050208
Chicago/Turabian StyleLee, Jyun-Lin, Yao-Chien Wang, Yu-An Hsu, Chih-Sheng Chen, Rui-Cian Weng, Yen-Pei Lu, Chun-Yu Chuang, and Lei Wan. 2022. "Bisphenol A Coupled with a High-Fat Diet Promotes Hepatosteatosis through Reactive-Oxygen-Species-Induced CD36 Overexpression" Toxics 10, no. 5: 208. https://doi.org/10.3390/toxics10050208
APA StyleLee, J.-L., Wang, Y.-C., Hsu, Y.-A., Chen, C.-S., Weng, R.-C., Lu, Y.-P., Chuang, C.-Y., & Wan, L. (2022). Bisphenol A Coupled with a High-Fat Diet Promotes Hepatosteatosis through Reactive-Oxygen-Species-Induced CD36 Overexpression. Toxics, 10(5), 208. https://doi.org/10.3390/toxics10050208