Abstract
Secondary interactions stabilize coordinatively demanding complexes of s-block metals. The structures of potassium fluoroalkoxides that, in addition to intra- and intermolecular K+···F contacts, also exhibit K+···Cπ interactions with tethered π ligands, are reported. A potassium–arene, a rare potassium–alkyne, and a potassium–olefin complex have been prepared by deprotonation of functionalized α,α-bis(trifluoromethyl)alcohols with KN(SiMe2R)2. They all feature a cuboid K4O4 core with μ3-bridging O atoms, and multiple stabilizing K+···F contacts in the range 2.71–3.33 Å. The potassium–arene complex shows η2, η3, and η6 K+···Cπ(arene) interactions in the range 3.35–3.47 Å. The potassium–alkyne and potassium–olefin compounds are stabilized by η2 interactions with the unsaturated carbon–carbon bond, in the range 3.17–3.49 Å and 3.15–3.19 Å, respectively. Comparison with the parent complex devoid of a flanking π ligand illustrates the role of K+···Cπ interactions.
1. Introduction
Complexes of the electropositive alkali and alkalino-earth metals are characterized by ionic bonding between the cation and the ligands and co-ligands. In addition to regular (weakly) bonding interactions in, for instance, amido or alkoxo salts of these elements, there has been a growing awareness in the past 10–15 years that non-covalent interactions can help towards the stabilization of these species, especially as the size and coordinative demand of the metal increase upon descending groups 1 or 2. The concept of so-called secondary interactions, which are essentially weak donor–acceptor interactions between the cation and charge-neutral Cπ, halide, H, or other neutral atom or group of atoms, was highlighted in a landmark article by Ruhlandt-Senge and coworkers in 2010 [1]. This and other groups have in particular prepared many a compound of alkali metals (M) featuring one or more M+···F [2,3,4,5,6] and, perhaps more prominently, M+···Cπ(arene) [4,7] intramolecular interactions. Of note, the importance of M+···Cπ(arene) in biological structures, supramolecular assemblies, and catalytic and ion transportation processes has long been established [8,9,10,11,12,13,14]. Many such complexes have been structurally characterized; there are nearly 300 referenced X-ray structures to date in the Cambridge Structural Database (CSD) for η6-coordinated K+···Cπ(arene) compounds alone.
As part of our program aimed at implementing the large alkaline earths (Ae = Ca, Sr, Ba) in molecular catalysis, we prepared some time ago several heteroleptic amido–Ae aryloxides and fluoroalkoxides stabilized by secondary interactions, particularly intramolecular Ae2+···F contacts and β-Si–H···Ae2+ agostic distortions when using the N(SiMe2H)− amido co-ligand [15,16,17,18]. More recently, we have prepared Ae–olefin and Ae–alkyne fluoroalkoxo complexes that both exhibit strong intramolecular Ae2+···Cπ in the solid state and in solution [19,20]. We have also shown that multiple Ae2+···F, β-Si–H···Ae2+, and Ae2+···Cπ secondary interactions could be combined within the same molecular structure to yield electron-deficient, yet stable, Ae complexes.
In the course of this work, we have prepared and structurally characterized several unusual homometallic potassium fluoroalkoxides that display intramolecular K+···Cπ(arene), K+···Cπ(olefin), and K+···Cπ(alkyne) interactions with tethered π ligands. K+–arene complexes are indeed well known. However, structurally authenticated K+–(η2-alkyne) complexes (13 structures in the CSD at the time of writing) are mostly limited to heterobimetallic acetylides such as [(C5HMe4)2Ti(η1-C≡C–SiMe3)2]−[K]+ [21] or [{(Me3-tacn)Cr(C≡CH)3}2K]+[CF3SO3]− bearing a N-methyl-substituted triazacyclonane ligand (Me3Tacn) [22]. The sole example of homometallic complex is the polymeric [{(C5Me4)2SiMe2C≡CPh}K·THF]∞ [23]. K+–(η2-olefin) complexes are more common (46 examples in the CSD), with representative examples including [Sn{(Me3Si)CHCH=CH(SiMe3)}3]−[K·THF]+ [24], [Zn{(Me3Si)CHCH=CH(SiMe3)}3]−[K]+ [25], [Zn(CH2SiMe3)(TMP)(CH=CH2)]−[K·PTMEDA]+ [26], or the rare homometallic [KC60(THF)5]·2 THF fulleride [27].
In this context, the structural motifs of several polymetallic potassium fluoroalkoxides displaying strong intramolecular interactions with pendant olefin, alkyne, or arene are discussed in the following. The structure of the parent complex where the ligand is devoid of dangling π groups is also presented for comparison.
2. Results
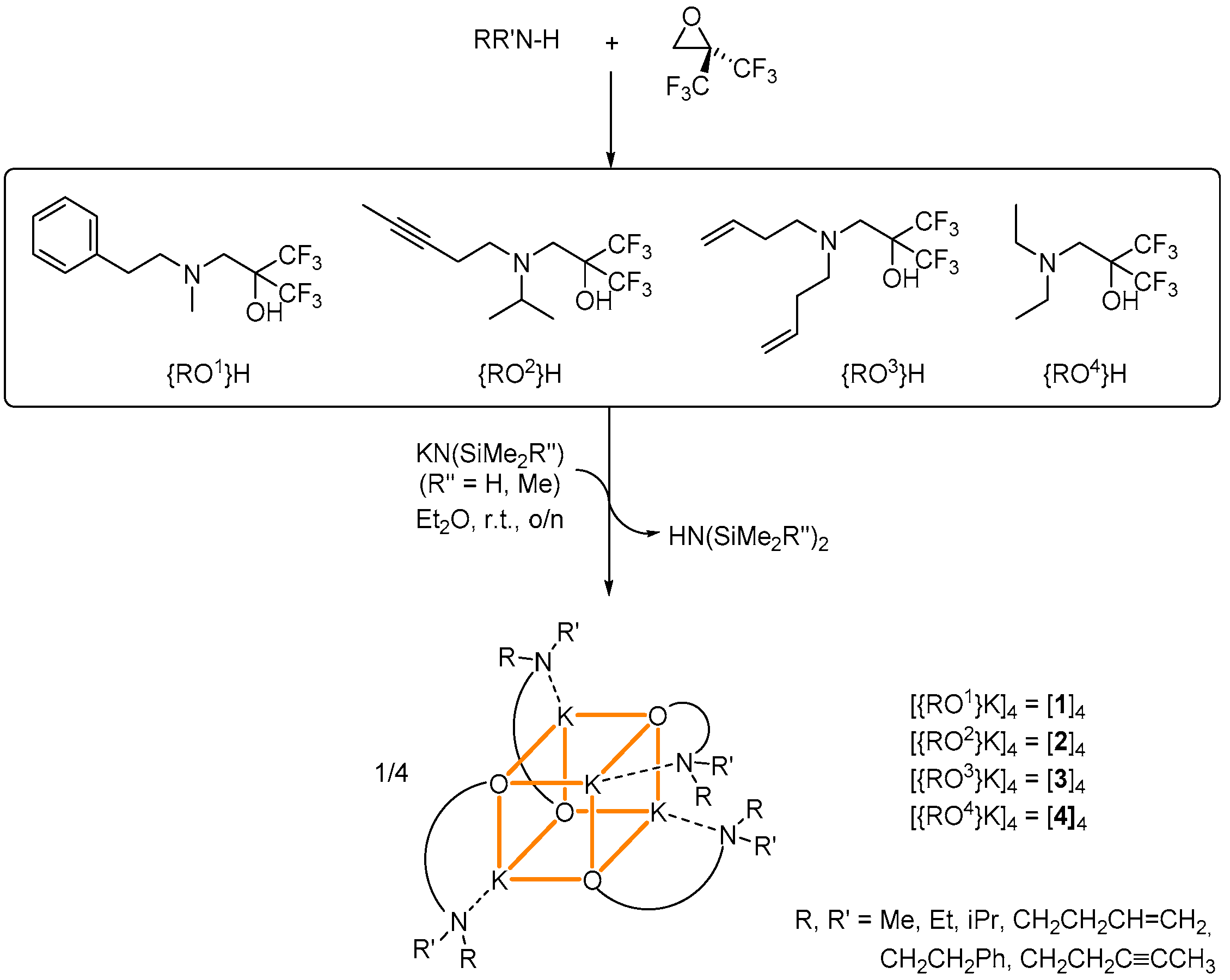
The fluoroalcohols {RO1}H–{RO4}H bearing two strongly electron-withdrawing CF3 groups in α position to the hydroxyl can be prepared in high yield by treatment of 2,2-bis(trifluoromethyl)oxirane with the appropriate amine in Et2O [16,20]. They were reacted with an equimolar amount of the potassium precursors [KN(SiMe3)2] or [KN(SiMe2H)2] to afford the corresponding potassium fluoroalkoxides in 33%–85% isolated (non-optimized) yields (Scheme 1). The resulting compounds [{ROx}K]4 were obtained as colorless, analytically pure solids (x = 1, [1]4; x = 2, [2]4; x = 3, [3]4; x = 4, [4]4). They all crystallized as tetranuclear complexes in a K4O4 cubane arrangement (vide infra). Their composition was established by X-ray crystallography and was corroborated by NMR spectroscopy. Their purity was confirmed by combustion analyses. All complexes are soluble in common organic solvents, including aliphatic hydrocarbons.
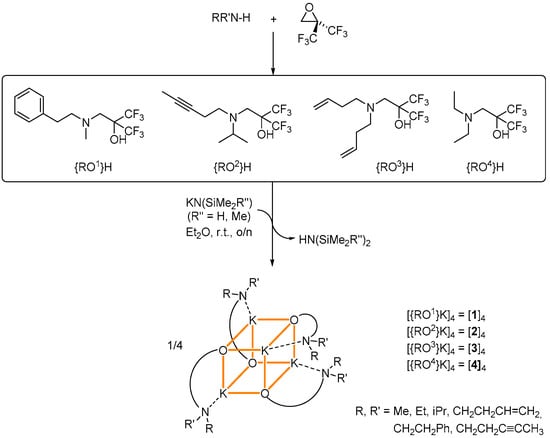
Scheme 1.
Fluoroalcohols used in this study, with a synthetic scheme for the preparation of the tetranuclear potassium fluoroalkoxides [{ROx}K] [1]4–[4]4. A representation of the cuboid structures of these complexes is given; K+···F and K+···Cπ secondary interactions not displayed.
2.1. Potassium–Arene Complex [{RO1}K]4 ([1]4)
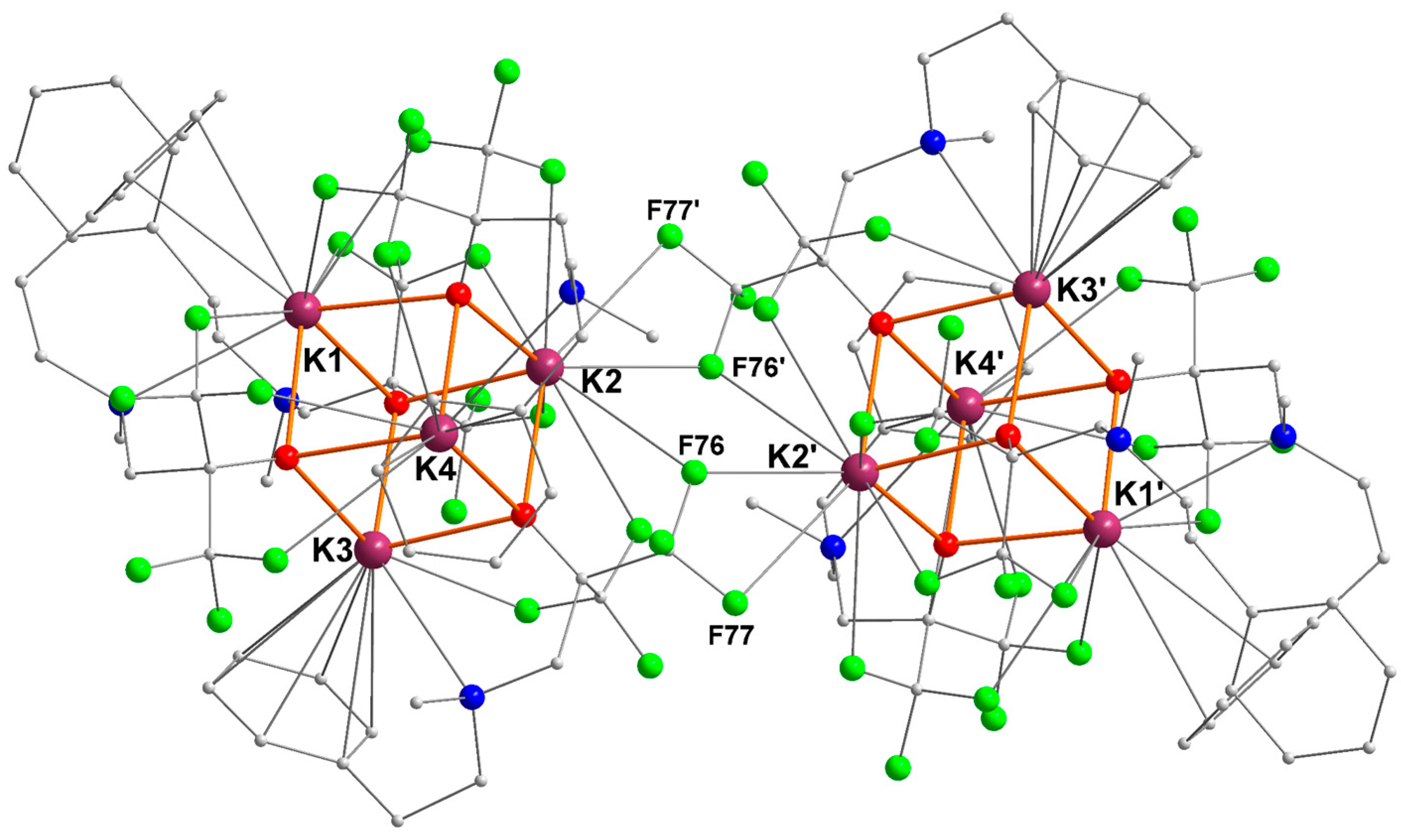
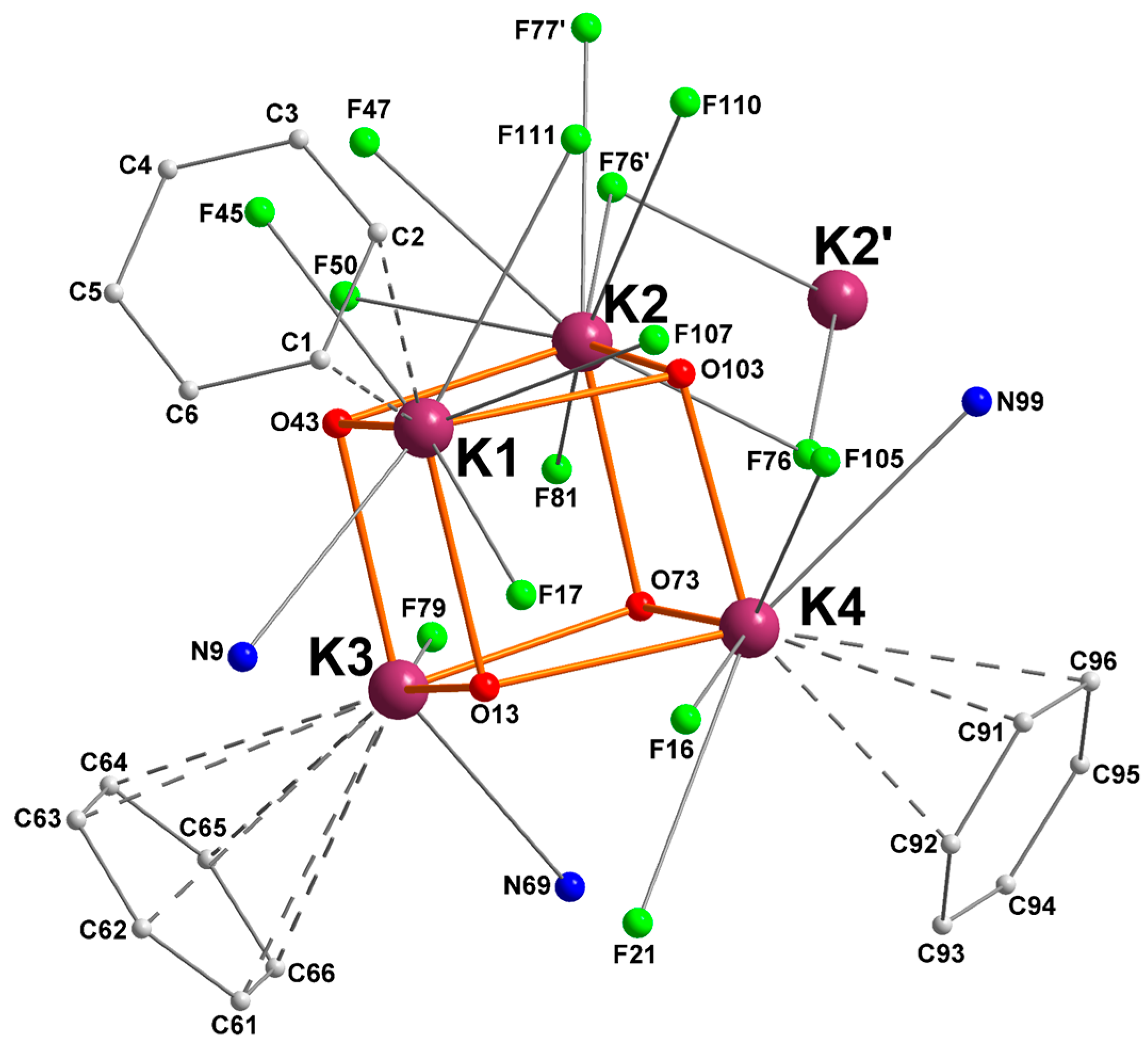
The compound [{RO1}K] crystallized as the distorted cubane [{RO1}K]4 ([1]4), a multinuclear structure typical of potassium alkoxides (Figure 1) [6,18,28,29]. Two K4O4 cuboid motifs are associated through bridging K···F interactions to generate a centrosymmetric macromolecular edifice containing eight potassium ions. The distances to the bridging fluorine atoms K2–F76′ (2.963(1) Å), K2–F76 (3.171(1) Å), and K2–F77′ (3.279(2) Å) are well below the sum of van der Waals radii for potassium (2.75 Å) and fluorine (1.47 Å), testifying to substantial interactions. They are also below the accepted distance for significant K–F interactions (ca. 3.40 Å) [30].
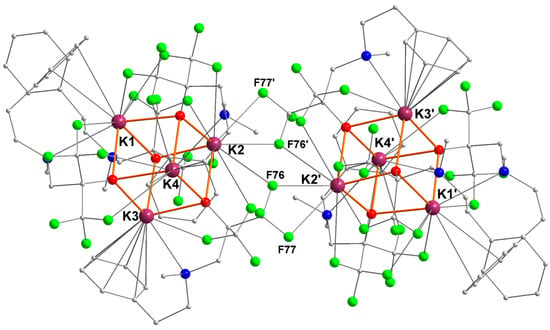
Figure 1.
Representation of the molecular solid-state structure of the potassium–arene complex [{RO1}K]4 ([1]4). Color code: purple, K; green, F; blue, N; red, O; grey, C. H atoms omitted for clarity.
A simplified view of the coordination pattern in [1]4 is depicted in Figure 2. In each of the two identical cubanes, each potassium atom is coordinated by three oxygen atoms in μ3-positions, with characteristic dK–O bond distances in the range 2.611(2)–2.825(2) Å.
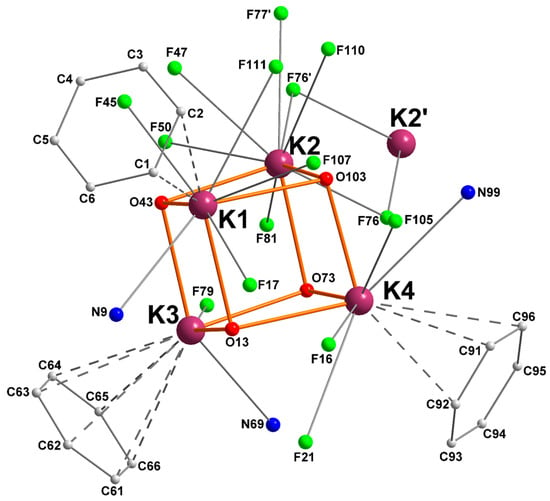
Figure 2.
Simplified representation of the molecular solid-state structure of complex [{RO1}K]4 ([1]4). Color code: purple, K; green, F; blue, N; red, O; grey, C. Hydrogen atoms are omitted for clarity. Only the heteroatoms and aryl substituents interacting with potassium are depicted.
The nitrogen atoms N9, N69, and N99 are bound to K1, K3, and K4, respectively; there are not any nitrogen atoms coordinated to K2. In addition, each metal ion is stabilized by multiple K+···F contacts: K1, K2, K3, and K4 are respectively involved in four, seven, one, and three such interactions. They range from very strong (dK–F = 2.806(1) Å for K3) to mild (dK–F = 3.324(2) Å for K4) [31]. Another prominent feature of this complex is the presence of K+···Cπ(arene) intramolecular interactions with three capping aromatic rings from the tethered side-arms of the ligands. Hence, K1, K3, and K4 show respectively η2, η6, and η3 π-interactions with the aromatic substituents. Such K+···Cπ(arene) contacts, all below 3.48 Å, are not uncommon for potassium [4,9,11,13]. A summary of relevant metric parameters for [1]4 is given in Table 1.

Table 1.
Key metric parameters in the potassium–arene complex [{RO1}K]4 ([1]4).
NMR spectroscopy did not provide information regarding the structure of [1]4 in solution. Its 1H NMR spectrum in [D6]benzene features broad resonances. In the 19F NMR spectrum, a unique, sharp singlet is detected at −76.34 ppm, indicating that all CF3 groups are equivalent on the NMR time-scale; there was no indication for the persistence of K+···F interactions in solution. 1H DOSY NMR measurements proved erratic, hence provided limited help in assessing the nuclearity of the complex in solution; they were, however, consistent with the existence of a multinuclear species.
2.2. Potassium–Alkyne Complex [{RO2}K]4 ([2]4)
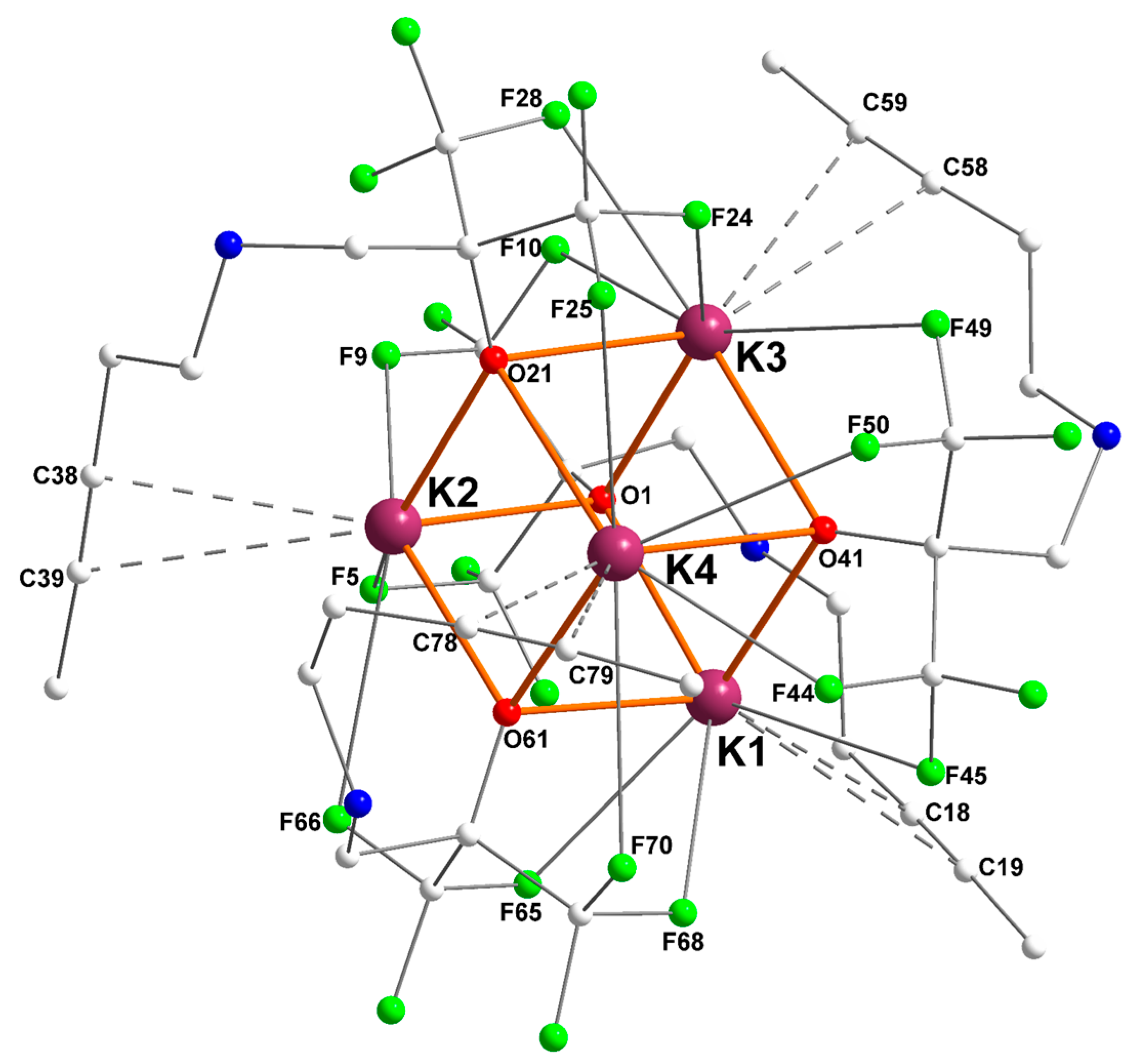
The potassium fluoroalkoxide [{RO2}K] bearing a dangling alkynyl side-arm recrystallized from pentane as the tetranuclear [{RO2}K]4 ([2]4) showing also a K4O4 cuboid arrangement (Figure 3). Besides the presence of multiple K+···F interactions (three or four per potassium), one of its main characteristic is the presence of η2-K+···Cπ(alkyne) interactions, in the range 3.131(3)–3.495(3) Å. Remarkably, none of the nitrogen atoms of the ligand backbones coordinates onto a potassium center (dK–N > 3.832(2) Å, and generally over 4.5 Å), thus highlighting the key contributions of K+···F and K+···Cπ(alkyne) secondary interactions in this complex. Of interest, [2]4 is a rare example of non-acetylide potassium–alkyne complex, the sole other occurrence being [{(C5Me4)2SiMe2C≡CPh}K·THF]∞ [23]. However, the K+···Cπ(alkyne) interatomic distances in [2]4 (in the range 3.131(3)–3.495(3) Å, see Table 2) are, for most of them, much shorter than in this latter compound (3.406 and 3.470 Å). On the other hand, they are much longer than in K+···Cπ(acetylide) compounds, where it often approximates 2.95–3.10 Å [21,22,31].
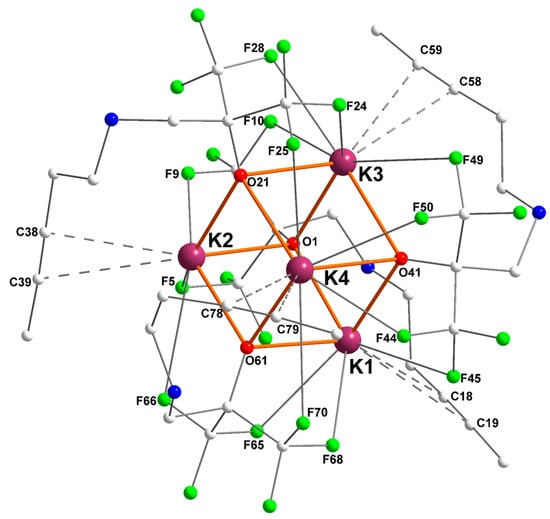
Figure 3.
Representation of the molecular solid-state structure of the potassium–alkyne complex [{RO2}K]4 ([2]4). Color code: purple, K; green, F; blue, N; red, O; grey, C. H atoms omitted for clarity.
Table 2.
Key metric parameters in the potassium–alkyne complex [{RO2}K]4 ([2]4).
Relevant metric parameters for [2]4 are collated in Table 2. Examination of the Ki···Cπ(alkyne) distances shows large discrepancies, with K4–C78 and K1–C18 being as short as 3.131(3) and 3.172(2) Å, respectively, whereas K3–C59 reaches 3.495(3) Å. One should note that for each potassium, the distance to the “internal” Cπ(alkyne) atom is systematically much shorter than that to the “external” one (internal and external Cπ(alkyne) atoms are respectively in γ and δ positions to the nitrogen atom), hence indicating a dissymmetric binding mode for the alkyne. As seen for [1]4, the intensity of K+···F interactions also varies largely in [2]4, in the range 2.682(1)–3.324(1) Å. The C≡C bond lengths in [2]4, in the region 1.153(5)–1.7179(4) Å, are unexceptional and are typical of non-coordinated (–CX2)–C≡C–CH3 fragments.
The solution NMR data (recorded in [D6]benzene) for [2]4 did not inform us about the nuclearity of the complex in solution. A sharp singlet is observed at −77.32 ppm in the 19F NMR spectrum, and the resonances at 78.19 (C≡C–CH3) and 76.94 (C≡C–CH3) ppm in the 13C{1H} NMR spectrum did not provide useful information about the potential coordination/dissociation of the alkyne in solution.
2.3. Potassium–Alkene Complex [{RO3}K]4 ([3]4)
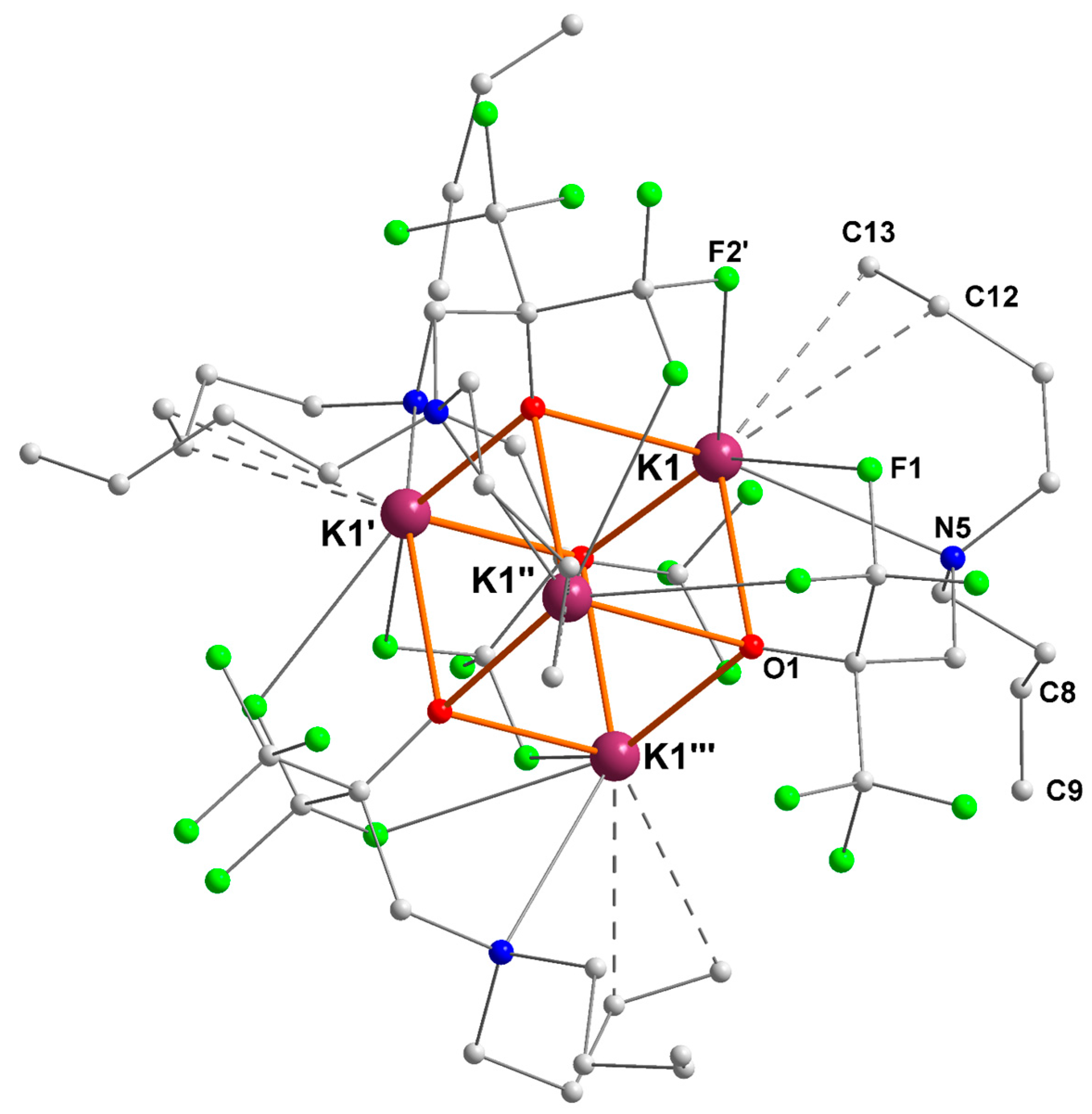
The potassium–alkene complex [{RO3}K] also crystallized as the tetranuclear cubane [{RO3}K]4 ([3]4) in the tetragonal space group P-421c (Figure 4). The four potassium centers are therefore symmetrically equivalent. Each exhibits η2-coordination of an olefin and two K+···F intramolecular interactions (K1–F1 = 3.062(2) Å, K1–F2′ = 2.928(2) Å). For each ligand, only one of the olefins is coordinated to potassium (K1–C12 = 3.192(4) and K1–C13 = 3.148(4) Å), whereas the second olefinic tether (corresponding to C8 and C9) is remote from the metal ion. The two K+–Cπ(alkene) distances in [3]4 are very comparable. They are in the range of those measured in [Sn{(Me3Si)CHCH=CH(SiMe3)}3]−[K·THF]+ (3.065(8) and 3.164(8) Å) [24], in the fulleride [KC60(THF)5]·2 THF (3.204(1) and 3.356(1) Å) [27], in [Zn{(Me3Si)CHCH=CH(SiMe3)}3]−[K]+ (2.942(3)–3.283(3) Å) [25], or in [Zn(CH2SiMe3)(TMP)(CH=CH2)]−[K·PTMEDA]+ (2.985(4) and 3.167(3) Å) [26], although the K+–olefin interaction was much more dissymmetric in these complexes. The main metric parameters for [3]4 are summarized in Table 3.
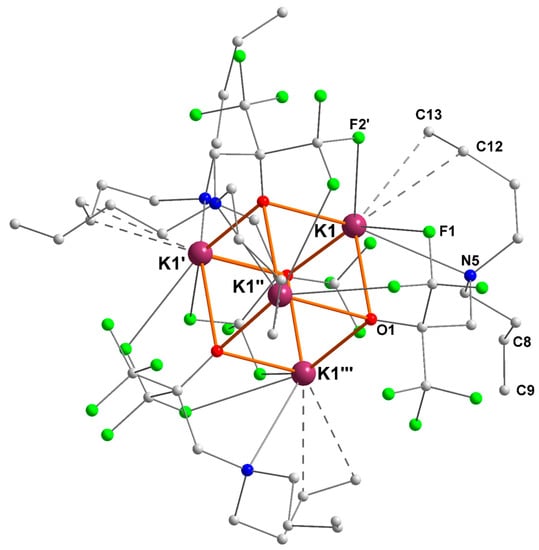
Figure 4.
Representation of the molecular solid-state structure of the potassium–alkene complex [{RO3}K]4 ([3]4). Color code: purple, K; green, F; blue, N; red, O; grey, C. H atoms omitted for clarity.
Table 3.
Key metric parameters in the potassium–alkene complex [{RO3}K]4 ([3]4).
The 19F NMR spectrum of [3]4 displays a sharp singlet at −75.86 ppm for all CF3 groups. The 1H and especially 13C{1H} spectra (δ13C = 137.70 and 116.01 ppm for the Cπ(alkene) atoms) did not show differences between the two types of olefins, coordinated and dissociated; this suggests that they either exchange very fast on the NMR time-scale, or that the tetranuclear arrangement is disrupted in [D6]benzene.
Since one of the two tethered olefins in the ligand {RO3}− is not directly involved in the coordination sphere of K+, we prepared a related proteo-ligand {RO5}H having only one dangling olefinic group, and where the other one is replaced by an isopropyl group. This new ligand led to the clean preparation of a compound of composition [{RO5}K]n according to NMR spectroscopy and elemental analysis; however, all attempts to grow X-ray-quality crystals proved unsuccessful, and we could not obtain useful information in the solid state.
2.4. Potassium complex [{RO4}K]4 ([4]4)
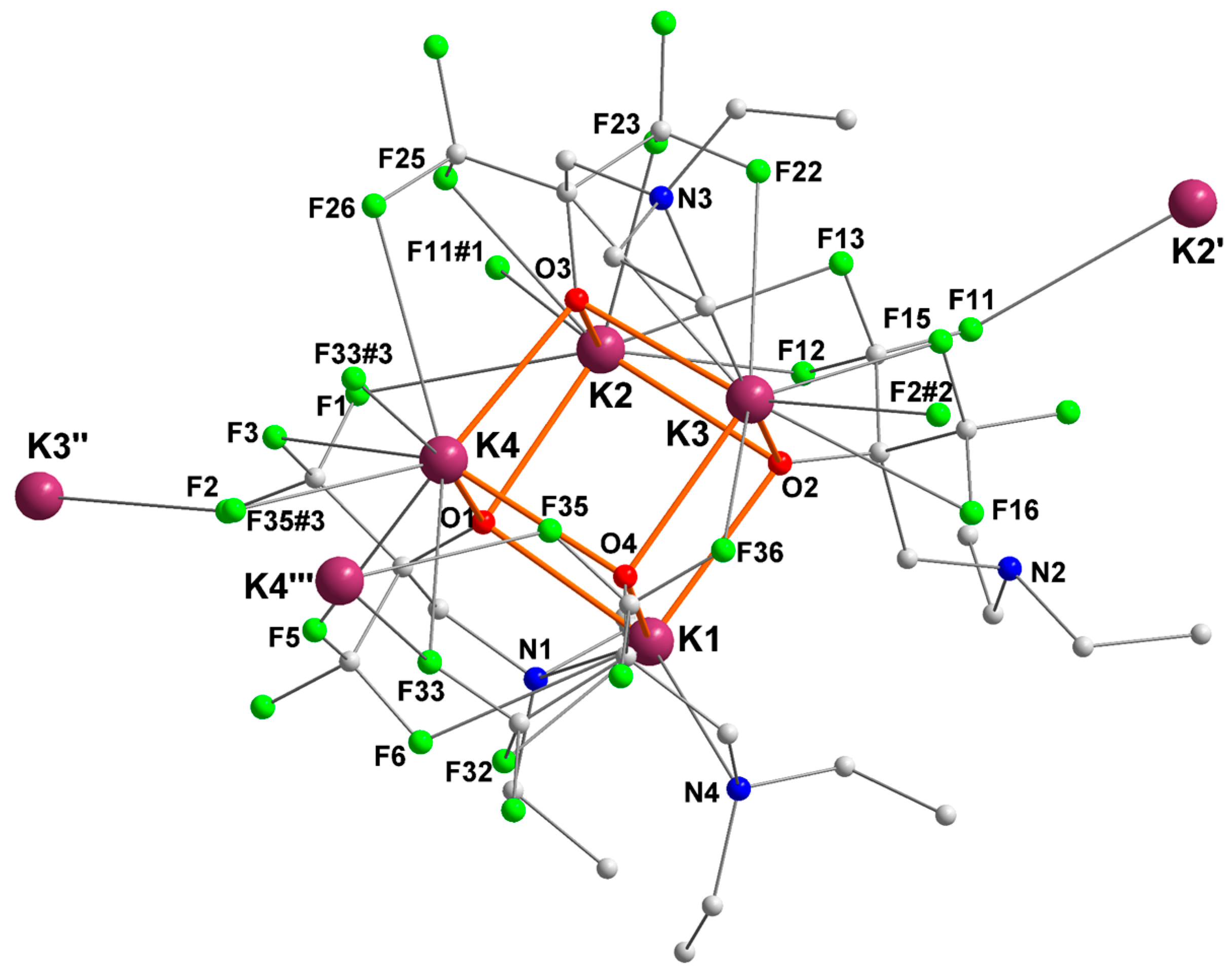
The potassium complex {RO4}K, where the ancillary ligand is devoid of π ligand, crystallized as the tetranuclear [{RO4}K]4 ([4]4) with a distorted K4O4 cuboid core (Figure 5). Each potassium is involved in several intermolecular (e.g., K2–F11#2, K2–F33#3, K3–F32#1, K4–F35#4, K2′–F11, K3″–F2, K4′′′–F35) and intramolecular K+···F interactions, leading to the formation of infinite two-dimensional layer coordination polymers. In addition to the array of K+···F contacts, all potassium centers are coordinated by three μ3-bridging oxygen atoms. K1 and K3 are also coordinated by a nitrogen atom (N4 and N3, respectively), but K2 and K4 are not. As a result, and to compensate for an otherwise overwhelming electron deficiency, K2 and K4 exhibit six K+···F interactions each. This is more than for K1 and K3, which respectively feature two and five interactions.
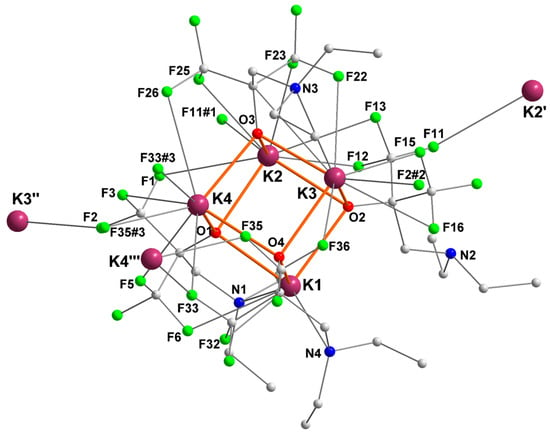
Figure 5.
Representation of the molecular solid-state structure of the potassium complex [{RO4}K]4 ([4]4). Only the main component of disordered ethyl groups is depicted. Color code: purple, K; green, F; blue, N; red, O; grey, C. H atoms omitted for clarity.
Table 4 displays the key metric parameters in [4]4. All K+–O bond lengths are in the same range, 2.586(2)–2.767(2) Å. The K+–N bond is weaker for K3 (K3–N3 = 3.162(2) Å) than for K1 (K1–N4 = 2.974(2) Å), which explains the greater number of K+···F contacts for the former.
Table 4.
Key metric parameters in the potassium complex [{RO4}K]4 ([4]4).
There is a unique sharp singlet at −76.38 ppm for all CF3 groups in the 19F NMR spectrum of [4]4, and its 1H NMR spectrum features only three resonances at 2.70 (s), 2.56 (q), and 0.82 (t) ppm. We could not obtain reliable information as to the nuclearity of the complex in solution.
3. Discussion
Compared to the polymeric [4]4—where electron depletion at the potassium centers is compensated solely by a large number of K+···F intramolecular and intermolecular interactions, resulting in the formation of two-dimensional networks—the presence of π ligands in [1]4 (arene), [2]4 (alkyne), and [3]4 (alkene) profoundly influences the coordination pattern of these compounds. A comparison of the structural and metric parameters for these complexes shows that as the number of K+···Cπ interactions increases, one generally observes a lowering of the number or the strength of K+···F contacts. This is perhaps best epitomized in the structure of the arene complex [1]4, where the four potassium centers display different coordination environments.
Beyond structural considerations, this work shows that, other than the well-known K+···Cπ(arene) interaction, alkenes and alkynes efficiently provide stabilization to potassium alkoxides. The potassium–alkyne complex described here is the only one of this type. This is in line with the recent account of the utilization of π ligands in alkaline-earth chemistry [19,20]. In an attempt to extend the range of potential π ligands for s-block metals, we have also prepared a ligand possessing a dangling allene moiety. However, we have so far been unable to grow X-ray-quality crystals for the resulting potassium complex. One should note that independently of the mode of coordination (η2, η3, or η6) of the arene in [1]4, the K+···Cπ(arene) distances are considerably longer than the distances to the coordinated alkyne and alkene in complexes [2]2 and [3]2. Density-functional theory (DFT) computations would be very useful to probe the respective intensities of the interactions between the π ligands and the potassium ions in these complexes, but they are precluded owing to the structural complexity of these polynuclear species, and because we have no reliable information about their structures in solution. For the same reason, bond valence sum analysis, which can be a convenient way to analyze the bonding pattern for a given complex [32], was also rendered prohibitively complicated.
We found no indication by NMR spectroscopy, especially 13C{1H} NMR, of any degree of covalence in the interaction between K+ and the three different types of π ligands. Instead, this interaction is thought to be purely electrostatic, as seen for alkaline earths [19], and it occurs without any detectable polarization of the carbon–carbon unsaturated bonds [33].
The present results constitute further support in favor of Ruhlandt-Senge’s statement that secondary interactions are a key tool to satisfy coordinative demands of electropositive elements, and eventually yield stable and unusual molecular compounds [1]. The interactions K+···Cπ and K+···F described here complement other non-covalent interactions reported before, such as agostic β-Si–H···K+ distortions seen in [KN(SiMe2H)2]∞ [15]. These (and other related) potassium fluoroalkoxides are convenient synthetic precursors for the introduction of the ligands onto other metals, such as alkaline earths or lanthanides, via salt metathesis reactions. We are continuing our efforts in this field, and are seeking to combine these types of interactions to yield stable alkali and alkaline-earth compounds. One route we are currently investigating is the use of enantiomerically pure chiral fluoroalkoxides to direct the formation of specific architectures.
4. Materials and Methods
4.1. General Protocols
All manipulations were performed under inert atmosphere using standard Schlenk techniques or in a dry, solvent-free glove-box (Jacomex; O2 < 1 ppm, H2O < 5 ppm). HN(SiMe3)2 (abcr; Karlsruhe, Germany) and HN(SiMe2H)2 (abcr) were dried over CaH2 and distilled prior to use. The compounds [KN(SiMe3)2] and [K(N(SiMe2H)2] were prepared following literature protocols [15]. The proteo-ligands {RO1}H–{RO3}H were obtained as described earlier [16,20]. The new {RO4}H was obtained following the same protocols, using HNEt2 as starting material; see the Supplementary Materials (Sections S1–S9) for detail. 2,2-Bis(trifluoromethyl)oxirane was purchased from Synquest Laboratories (Alachua, FL, USA) and used as received. Solvents (THF, Et2O, CH2Cl2, pentane, and toluene) were purified and dried (water content all below 10 ppm) over alumina columns (MBraun SPS). THF was further distilled under argon from sodium mirror/benzophenone ketyl prior to use. All deuterated solvents (Eurisotop, Saclay, France) were stored in sealed ampoules over activated 3 Å molecular sieves and were thoroughly degassed by several freeze-thaw-vacuum cycles.
NMR spectra were recorded on Bruker AM-400 and AM-500 spectrometers (Bruker BioSpin, Wissembourg, France) at the University of Rennes 1. All 1H and 13C{1H} chemicals shifts were determined using residual signals of the deuterated solvents and were calibrated vs. SiMe4. Assignment of the resonances was carried out using 1D (1H, 13C{1H}) and 2D (COSY, HMBC, HMQC) NMR experiments. Coupling constants are given in hertz. 19F{1H} chemical shifts were determined by external reference to an aqueous solution of NaBF4.
Elemental analyses performed on a Carlo Erba 1108 Elemental Analyzer at the London Metropolitan University by Stephen Boyer were the average of two independent measurements.
The November 2016 CSD database (CSDV37) was used for the searches of XRD structures.
4.2. Synthesis of Complex [{RO1}K]4 ([1]4)
KN(SiMe3)2 (0.06 g, 0.33 mmol) was added with a bent finger to a solution of {RO1}H (0.10 g, 0.33 mmol) in Et2O (10 mL). The reaction mixture was stirred at room temperature overnight. Volatiles were removed in vacuo to afford a sticky solid. Stripping with pentane (3 × 3 mL) afforded the title compound as a white solid (0.080 g, 69%). The compound was recrystallized from a concentrated pentane solution at −30 °C. 1H NMR (500.13 MHz, [D6]benzene, 298 K): δ 7.21–7.14 (m, 2H, m-C6H5), 7.12–7.08 (overlapping m, 3H, p-C6H5, and o-C6H5), 2.66–2.60 (br m, 2H, NCH2CH2), 2.56–2.52 (overlapping m, 4H, CH2C(CF3)2, and NCH2CH2), 2.18 (s, 3H, NCH3) ppm. 13C{1H} NMR (125.73 MHz, [D6]benzene, 298 K): δ 140.22, 129.06, 128.30, 126.69 (all C6H5), 127.62 (q, 1JC–F = 294.2 Hz, CF3), 81.24 (hept, 2JC–F = 22.6 Hz, C(CF3)2), 63.64 (NCH2CH2), 60.61 (CH2C(CF3)2), 45.33 (NCH3), 34.06 (NCH2CH2) ppm. 19F{1H} NMR (376.49 MHz, [D6]benzene, 298 K): δ −76.34 (s, 6F, CF3) ppm. Elemental analysis for C13H14F6KNO (353.35 g·mol−1): calc. C 44.2%, H 4.0%, N 4.0%; found C 44.3%, H 3.8%, N 3.9%.
4.3. Synthesis of Complex [{RO2}K]4 ([2]4)
KN(SiMe3)2 (0.08 g, 0.44 mmol) was added in solid portions with a bent finger to a solution of {RO2}H (0.13 g, 0.44 mmol) in Et2O (10 mL). The reaction mixture was stirred at room temperature overnight. Volatiles were removed under vacuum and the resulting oil was stripped with pentane (3 × 3 mL) to afford the title compound as a colorless solid. The compound was recrystallized from a concentrated pentane solution at −30 °C. Yield 50 mg (33%). 1H NMR (400.13 MHz, [D6]benzene, 298 K): δ 2.92 (hept, 1H, 3JH–H = 6.8 Hz, CH(CH3)2), 2.80 (t, 2H, 3JH–H = 6.5 Hz, NCH2CH2), 2.71 (s, 2H, CH2C(CF3)2), 2.39 (m, 2H, NCH2CH2), 1.60 (t, 3H, 2JH–H = 2.3 Hz, C≡C–CH3), 0.92 (d, 6H, 3JH–H = 6.5 Hz, CH(CH3)2) ppm. 13C{1H} NMR (100.63 MHz, [D6]benzene, 298 K): δ 127.81 (q, 1JC–F = 294.8 Hz, CF3), 81.97 (hept, 2JC–F = 22.1 Hz, C(CF3)2), 78.19 (C≡C–CH3), 76.94 (C≡C–CH3), 57.54 (CH2C(CF3)2), 51.91 (CH(CH3)2), 51.63 (NCH2CH2), 19.51 (NCH2CH2), 18.26 (CH(CH3)2), 3.06 (C≡C–CH3) ppm. 19F{1H} NMR (376.47 MHz, [D6]benzene, 298 K): −77.32 (s, 6F, CF3) ppm. Elemental analysis for C12H16F6KNO (343.35 g·mol−1): calc. C 42.0%, H 4.7%, N 4.1%; found C 42.0%, H 4.4%, N 4.1%.
4.4. Synthesis of Complex [{RO3}K]4 ([3]4)
KN(SiMe2H)2 (0.11 g, 0.65 mmol) was added in solid portions with a bent finger to a solution of {RO3}H (0.21 g, 0.67 mmol) in Et2O (10 mL). The reaction mixture was stirred at room temperature overnight. Volatiles were removed under vacuum and the resulting oil was stripped with pentane (3 × 4 mL) to afford the title compound as a colorless oil. In a matter of days, the oil crystallized and the title compound was isolated as off-white crystals. Yield (161 mg, 72%). 1H NMR (400.13 MHz, [D6]benzene, 298 K): δ 5.79 (ddt, 2H, 3JH–H (trans) = 17.4 Hz, 3JH–H (cis) = 9.9 Hz, 3JH–H = 6.7 Hz, CH=CH2), 5.13–5.01 (m, 4H, CH=CH2), 2.76 (s, 2H, CH2C(CF3)2), 2.62 (t, 4H, 3JH–H = 6.4 Hz, NCH2CH2), 2.06 (q, 3JH–H = 6.4 Hz, 4H, NCHCH2) ppm. 13C{1H} NMR (100.63 MHz, [D6]benzene, 298 K): δ 137.70 (CH=CH2), 127.78 (q, 1JC–F = 295.3 Hz, CF3), 116.01 (CH=CH2), 81.58 (hept, 2JC–F = 22.4 Hz, C(CF3)2), 61.03 (CH2C(CF3)2), 53.12 (NCH2CH2), 29.79 (NCH2CH2) ppm. 19F{1H} NMR (376.49 MHz, [D6]benzene, 298 K): δ −75.86 (s, 6F, CF3) ppm. Elemental analysis for C12H16F6KNO (343.35 g·mol−1): calc. C 42.0%, H 4.7%, N 4.1%; found C 42.1%, H 4.6%, N 4.2%.
4.5. Synthesis of Complex [{RO4}K]4 ([4]4)
KN(SiMe2H)2 (0.16 g, 0.80 mmol) was added with a bent finger to a solution of {RO4}H (0.20 mg, 0.80 mmol) in Et2O (10 mL). The reaction mixture was stirred overnight at room temperature. The volatiles were removed in vacuo to yield [{RO4}K]4 as a colorless solid (0.20 g, 85%). X-ray-quality crystals were obtained from a concentrated pentane solution at −30 °C. 1H NMR (400.16 MHz, [D6]benzene, 298 K): δ 2.70 (s, 2H, CH2C(CF3)2), 2.56 (q, 4H, 3JH–H = 6.9 Hz, NCH2CH3), 0.82 (t, 6H, 3JH–H = 7.0 Hz, NCH2CH3 ) ppm. 13C{1H}NMR (100.62 MHz, [D6]benzene, 298 K): δ 129.30 (q, 1JC–F = 295.3 Hz, CF3), 81.91 (hept, 2JCF = 22.8 Hz, C(CF3)2), 58.97 (CH2C(CF3)2), 47.36 (NCH2CH3), 10.01 (NCH2CH3) ppm. 19F{1H} NMR (376.49 MHz, [D6]benzene, 298 K): δ −76.38 (s, 6F, CF3) ppm. Elemental analysis for C8H12F6KNO (291.28 g·mol−1): calc. C 33.0%, H 4.1%, N 4.8%; found C 32.9%, H 4.0%, N 4.9%.
4.6. X-Ray Diffraction Crystallography
X-ray diffraction data were collected at 150 K using a Bruker APEX CCD diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) at the University of Rennes 1. A combination ω and Φ scans was carried out to obtain at least a unique data set. The crystal structures were solved by direct methods, and remaining atoms were located from difference Fourier synthesis followed by full-matrix least-squares based on F2 (programs SIR97 and SHELXL-97) [34,35]. Carbon-, oxygen-, and nitrogen-bound hydrogen atoms were placed at calculated positions and forced to ride on the attached atom. The hydrogen atom contributions were calculated, but not refined. All non-hydrogen atoms were refined with anisotropic displacement parameters. The locations of the largest peaks in the final difference Fourier map calculation as well as the magnitude of the residual electron densities were of no chemical significance. The crystallographic data for all compounds are available as CIF files from the Cambridge Crystallographic Database Centre (CCDC numbers 1530195–1530198). A summary of crystallographic data is given in Table 5.
Table 5.
Summary of crystallographic data for [1]4–[4]4.
Supplementary Materials
The following are available online at www.mdpi.com/2304-6740/5/1/13/s1, CIF and checkCIF files of [{ROx}K]4 ([1]4–[4]4), Sections S1–S9.
Acknowledgments
Sorin-Claudiu Roşca thanks the French Agence Nationale de la Recherche for a PhD grant (GreenLAkE, ANR-11-BS07-009-01).
Author Contributions
Sorin-Claudiu Roşca, Jean-François Carpentier and Yann Sarazin conceived and designed the experiments; Sorin-Claudiu Roşca and Hanieh Roueindeji performed the experiments; Vincent Dorcet and Thierry Roisnel carried out the X-ray crystal structure determinations and the interpretation of the crystal data; Sorin-Claudiu Roşca, Hanieh Roueindeji, Jean-François Carpentier and Yann Sarazin analyzed the data; Yann Sarazin wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Buchanan, W.D.; Allis, D.G.; Ruhlandt-Senge, K. Synthesis and stabilization—Advances in organoalkaline earth metal chemistry. Chem. Commun. 2010, 46, 4449–4465. [Google Scholar] [CrossRef] [PubMed]
- Samuels, J.A.; Lobkovsky, E.B.; Streib, W.E.; Folting, K.; Huffman, J.C.; Zwanziger, J.W.; Caulton, K.G. Organofluorine binding to sodium and thallium(I) in molecular fluoroalkoxide compounds. J. Am. Chem. Soc. 1993, 115, 5093–5104. [Google Scholar] [CrossRef]
- Samuels, J.A.; Folting, K.; Huffman, J.C.; Caulton, K.G. Structure/volatility correlation of sodium and zirconium fluoroalkoxides. Chem. Mater. 1996, 7, 929–935. [Google Scholar] [CrossRef]
- Buchanan, W.D.; Nagle, E.D.; Ruhlandt-Senge, K. π-Coordination as a structure determining principle: Structural characterization of [K(Odpp)]∞, and {[K2(Odpp)2H2O]2}∞. Main Group Chem. 2009, 8, 263–273. [Google Scholar] [CrossRef]
- Buchanan, W.D.; Ruhlandt-Senge, K. M···F Interactions and heterobimetallics: Furthering the understanding of heterobimetallic stabilization. Chem. Eur. J. 2013, 19, 10708–10715. [Google Scholar] [CrossRef] [PubMed]
- Lum, J.S.; Tahsini, L.; Golen, J.A.; Moore, C.; Rheingold, A.L.; Doerrer, L.H. K···F/O Interactions bridge copper(I) fluorinated alkoxide complexes and facilitate dioxygen activation. Chem. Eur. J. 2013, 19, 6374–6384. [Google Scholar] [CrossRef] [PubMed]
- Weinert, C.S.; Fanwick, P.E.; Rothwell, I.P. Synthesis of group 1 metal 2,6-Diphenylphenoxide complexes [M(OC6H3Ph2-2,6)] (M = Li, Na, K, Rb, Cs) and structures of the solvent-free complexes [Rb(OC6H3Ph2-2,6)]x and [Cs(OC6H3Ph2-2,6)]x: One-dimensional extended arrays of metal aryloxides. Inorg. Chem. 2003, 42, 6089–6094. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, D.A. Cation-π interactions in chemistry and biology: A new view of benzene, Phe, Tyr, and Trp. Science 1996, 271, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.C.; Dougherty, D.A. The cation–π interaction. Chem. Rev. 1997, 97, 1303–1324. [Google Scholar] [CrossRef] [PubMed]
- Salonen, L.M.; Ellermann, M.; Diederich, F. Aromatic rings in chemical and biological recognition: Energetics and structures. Angew. Chem. Int. Ed. 2011, 50, 4808–4842. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.M.; Collins, M.S.; Johnson, D.W. Ion–π interactions in ligand design for anions and main group cations. Acc. Chem. Res. 2013, 46, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Mahadevi, A.S.; Sastry, G.N. Cation–π interaction: Its role and relevance in chemistry, biology, and material science. Chem. Rev. 2013, 113, 2100–2138. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, D.A. The Cation–π interaction. Acc. Chem. Res. 2013, 46, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C.R.; Lin, S.; Jacobsen, E.N. The cation–π Interaction in small-molecule catalysis. Angew. Chem. Int. Ed. 2016, 55, 12596–12624. [Google Scholar] [CrossRef] [PubMed]
- Sarazin, Y.; Roşca, D.; Poirier, V.; Roisnel, T.; Silvestru, A.; Maron, L.; Carpentier, J.-F. Bis(dimethylsilyl)-amide complexes of the alkaline-earth metals stabilized by β-Si–H agostic interactions: Synthesis, characterization, and catalytic activity. Organometallics 2010, 29, 6569–6577. [Google Scholar] [CrossRef]
- Sarazin, Y.; Liu, B.; Roisnel, T.; Maron, L.; Carpentier, J.-F. Discrete, solvent-free alkaline-earth metal cations: Metal···Fluorine interactions and ROP catalytic activity. J. Am. Chem. Soc. 2011, 133, 9069–9087. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Roisnel, T.; Carpentier, J.-F.; Sarazin, Y. When Bigger Is Better: Intermolecular hydrofunctionalizations of activated alkenes catalyzed by heteroleptic alkaline earth complexes. Angew. Chem. Int. Ed. 2012, 51, 4943–4946. [Google Scholar] [CrossRef] [PubMed]
- Roşca, S.-C.; Roisnel, T.; Dorcet, V.; Carpentier, J.-F.; Sarazin, Y. Potassium and well-defined neutral and cationic calcium fluoroalkoxide complexes: Structural features and reactivity. Organometallics 2014, 33, 5630–5642. [Google Scholar] [CrossRef]
- Roşca, S.-C.; Dinoi, C.; Caytan, E.; Dorcet, V.; Etienne, M.; Carpentier, J.-F.; Sarazin, Y. Alkaline earth-olefin complexes with secondary interactions. Chem. Eur. J. 2016, 22, 6505–6509. [Google Scholar] [CrossRef] [PubMed]
- Roşca, S.-C.; Caytan, E.; Dorcet, V.; Roisnel, T.; Carpentier, J.-F.; Sarazin, Y. π Ligands in alkaline earth complexes. Organometallics 2017. under revision. [Google Scholar]
- Varga, V.; Hiller, J.; Polášek, M.; Thewalt, U.; Mach, K. Synthesis and structure of Titanium(III) Tweezer complexes with embedded alkali metal ions: [(C5HMe4)2Ti(η1-C≡C–SiMe3)2]−M+ (M = Li, Na, K, and Cs). J. Organomet. Chem. 1996, 515, 57–64. [Google Scholar] [CrossRef]
- Berben, L.A.; Long, J.R. Synthesis and alkali metal ion-binding properties of a Chromium(III) Triacetylide complex. J. Am. Chem. Soc. 2002, 124, 11588–11589. [Google Scholar] [CrossRef] [PubMed]
- Chadha, P.; Dutton, J.L.; Ragogna, P.J. Synthesis and reactivity of bis-alkynyl appended metallocenes of Ti, Fe, and Co. Can. J. Chem. 2010, 88, 1213–1221. [Google Scholar] [CrossRef]
- Layfield, R.A.; García, F.; Hannauer, J.; Humphrey, S.M. Ansa-Tris(allyl) Complexes of alkali metals: Tripodal analogues of cyclopentadienyl and ansa-metallocene ligands. Chem. Commun. 2007. [Google Scholar] [CrossRef] [PubMed]
- Gren, C.K.; Hanusa, T.P.; Rheingold, A.L. Threefold cation–π bonding in trimethylsilylated allyl complexes. Organometallics 2007, 26, 1643–1649. [Google Scholar] [CrossRef]
- Kennedy, A.R.; Klett, J.; Mulvey, R.E.; Wright, D.S. Synergic sedation of sensitive anions: Alkali-mediated zincation of cyclic ethers and ethene. Science 2009, 326, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Kromer, A.; Wedig, U.; Roduner, E.; Jansen, M.; Amsharov, K.Y. Counterintuitive anisotropy of electron transport properties in KC60(THF)5·2 THF Fulleride. Angew. Chem. Int. Ed. 2013, 52, 12610–12614. [Google Scholar] [CrossRef] [PubMed]
- Boyle, T.J.; Andrews, N.L.; Rodriguez, M.A.; Campana, C.; Yiu, T. Structural variations of potassium aryloxides. Inorg. Chem. 2003, 42, 5357–5366. [Google Scholar] [CrossRef] [PubMed]
- Veith, M.; Belot, C.; Huch, V.; Guyard, L.; Knorr, M.; Khatyr, A.; Wickleder, C. Syntheses, crystal structures, and physico-chemical studies of sodium and potassium alcoholates bearing thienyl substituents and their derived luminescent Samarium(III) alkoxides. Z. Anorg. Allg. Chem. 2010, 636, 2262–2275. [Google Scholar] [CrossRef]
- Plenio, H. The coordination chemistry of the CF unit in fluorocarbons. Chem. Rev. 1997, 97, 3363–3384. [Google Scholar] [CrossRef] [PubMed]
- Schiefer, M.; Hatop, H.; Roesky, H.W.; Schmidt, H.-G.; Noltemeyer, M. Organoaluminates with three terminal phenylethynyl groups and their interactions with alkali metal cations. Organometallics 2002, 21, 1300–1303. [Google Scholar] [CrossRef]
- Brown, I.D. Recent developments in the methods and applications of the bond valence model. Chem. Rev. 2009, 109, 6858–6919. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, J.-F.; Maryin, V.P.; Luci, J.; Jordan, R.F. Solution structures and dynamic properties of chelated d0 Metal Olefin Complexes {η5: η1-C5R4SiMe2NtBu}Ti(OCMe2CH2CH2CHCH2)+ (R = H, Me): Models for the {η5: η1-C5R4SiMe2NtBu}Ti(R′)(olefin)+ intermediates in “constrained geometry” catalysts. J. Am. Chem. Soc. 2001, 123, 898–909. [Google Scholar] [CrossRef] [PubMed]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R. SIR97: A new tool for crystal structure determination and refinement. Appl. Crystallogr. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXL-97, Program for Refinement of Crystal Structures; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).