




Hydrazine Oxidation in Aqueous Solutions I: N4H6 Decomposition


Abstract
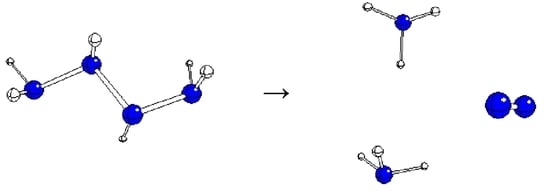
:
1. Introduction
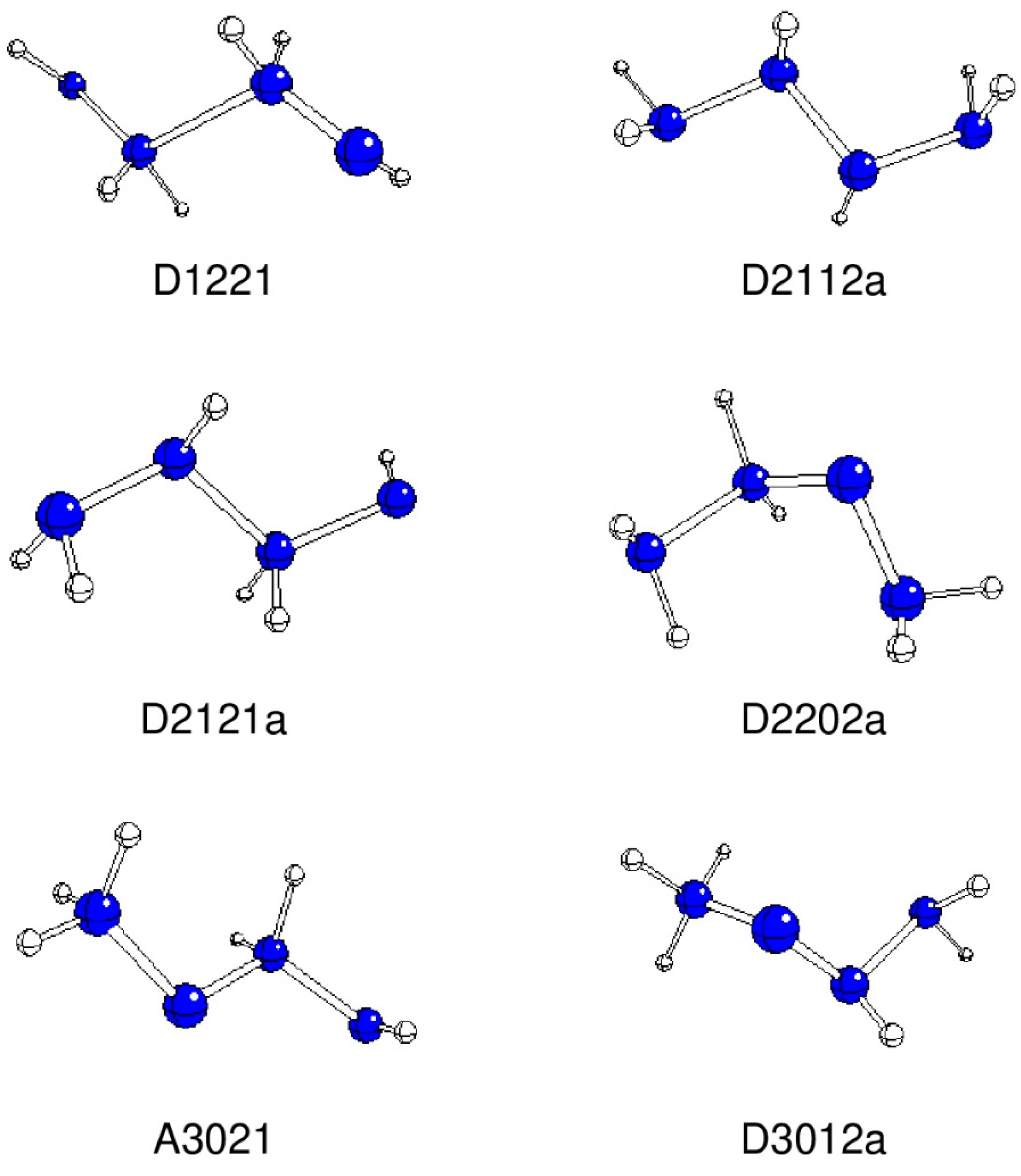
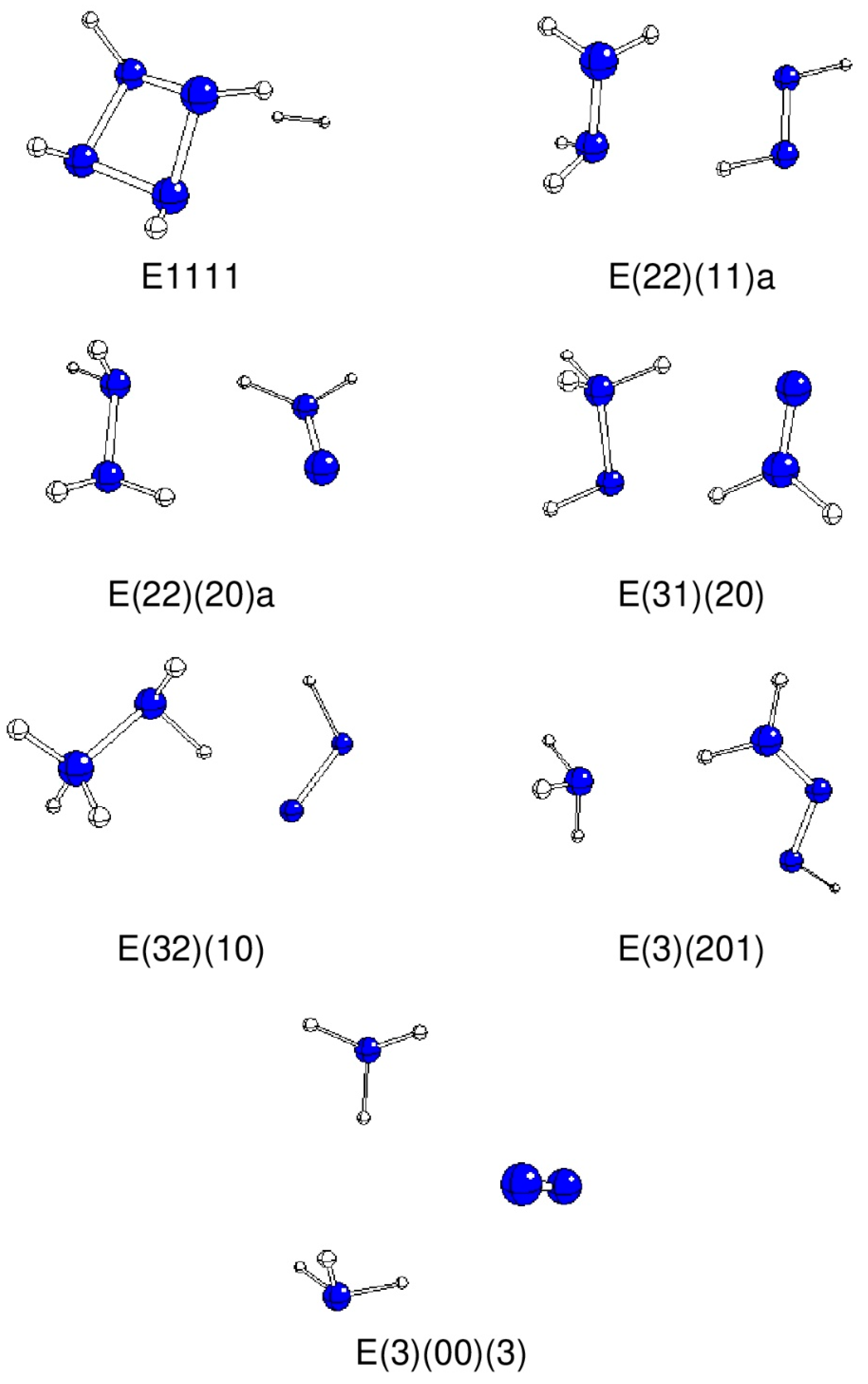
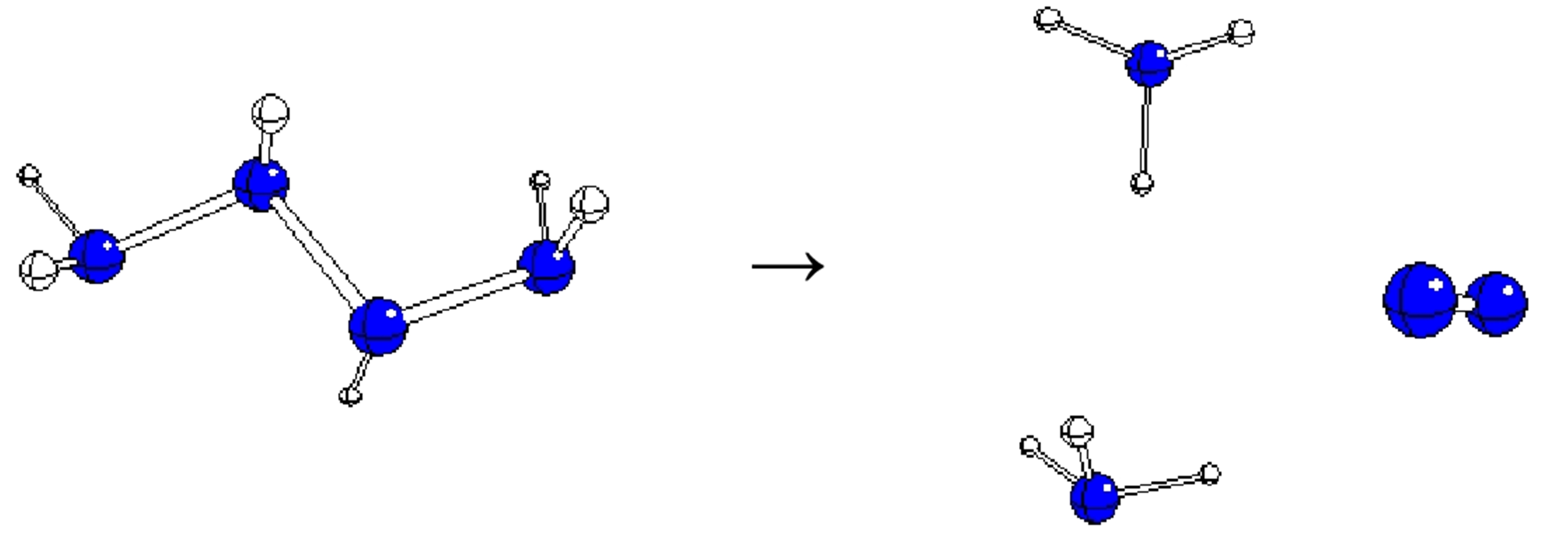
2. Results and Discussion
3. Method
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lauko, L.; Hudec, R.; Lenghartova, K.; Manova, A.; Cacho, F.; Beinrohr, E. Simple Electrochemical Determination of Hydrazine in Water. Pol. J. Environ. Stud. 2015, 24, 1659–1666. [Google Scholar] [CrossRef] [PubMed]
- Higginson, W.C.E.; Sutton, D. The Oxidation of Hydrazine in Aqueous Solution. Part II. The Use of 15N as a Tracer in the Oxidation of Hydrazine. J. Chem. Soc. 1953, 1402–1406. [Google Scholar] [CrossRef]
- Cahn, J.W.; Powell, R.E. Oxidation of Hydrazine in Solution. J. Am. Chem. Soc. 1954, 76, 2568–2572. [Google Scholar] [CrossRef]
- Petek, M.; Bruckenstein, S. An Isotopic Labeling Investigation of the Mechanism of the Electrooxidation of Hydrazine at Platinum. An Electrochemical Mass Spectrometric Study. Electroanal. Chem. Interrac. Electrochem. 1973, 47, 329–333. [Google Scholar] [CrossRef]
- Rice, F.O.; Sherber, F. The Hydrazino Radical and Tetrazane. J. Am. Chem. Soc. 1955, 77, 291–293. [Google Scholar] [CrossRef]
- Karp, S.; Meites, L. The Voltammetric Characteristics and Mechanism of Electrooxidation of Hydrazine. J. Am. Chem. Soc. 1962, 84, 906–912. [Google Scholar] [CrossRef]
- Ball, D.W. Tetrazane: Hartree-Fock, Gaussian-2 and -3, and Complete Basis Set Predictions of Some Thermochemical Properties of N4H6. J. Phys. Chem. A 2001, 105, 465–470. [Google Scholar] [CrossRef]
- Dana, A.G.; Moore, K.B., III; Jasper, A.W.; Green, W.H. Large Intermediates in Hydrazine Decomposition: A Theoretical Study of the N3H5 and N4H6 Potential Energy Surfaces. J. Phys. Chem. A 2019, 123, 4679–4692. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990; ISBN 9780198558651. [Google Scholar]
- Scuseria, G.E.; Janssen, C.L.; Schaefer, H.F., III. An efficient reformulation of the closed-shell coupled cluster single and double excitation (CCSD) equations. J. Chem. Phys. 1988, 89, 7382–7387. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Frisch, G.W.; Trucks, M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Biegler-König, F.; Schönbohm, J.; Bayles, D. AIM2000—A Program to Analyze and Visualize Atoms in Molecules. J. Comput. Chem. 2001, 22, 545–559. [Google Scholar]
- Ugliengo, P. MOLDRAW: A Program to Display and Manipulate Molecular and Crystal Structures, University Torino, Torino. 2012. Available online: https://www.moldraw.software.informer.com (accessed on 9 September 2019).

{kind=link}
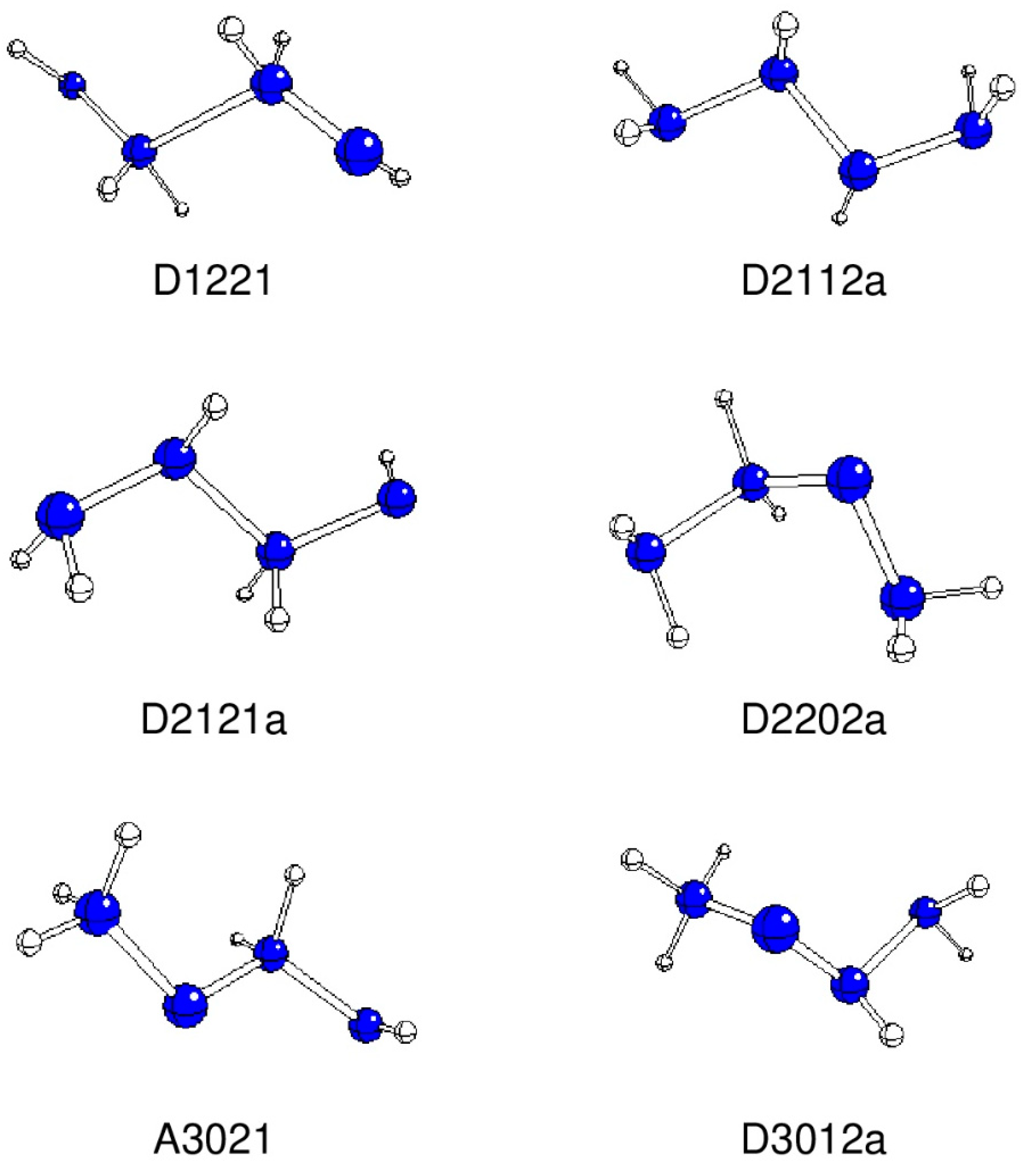{kind=link}
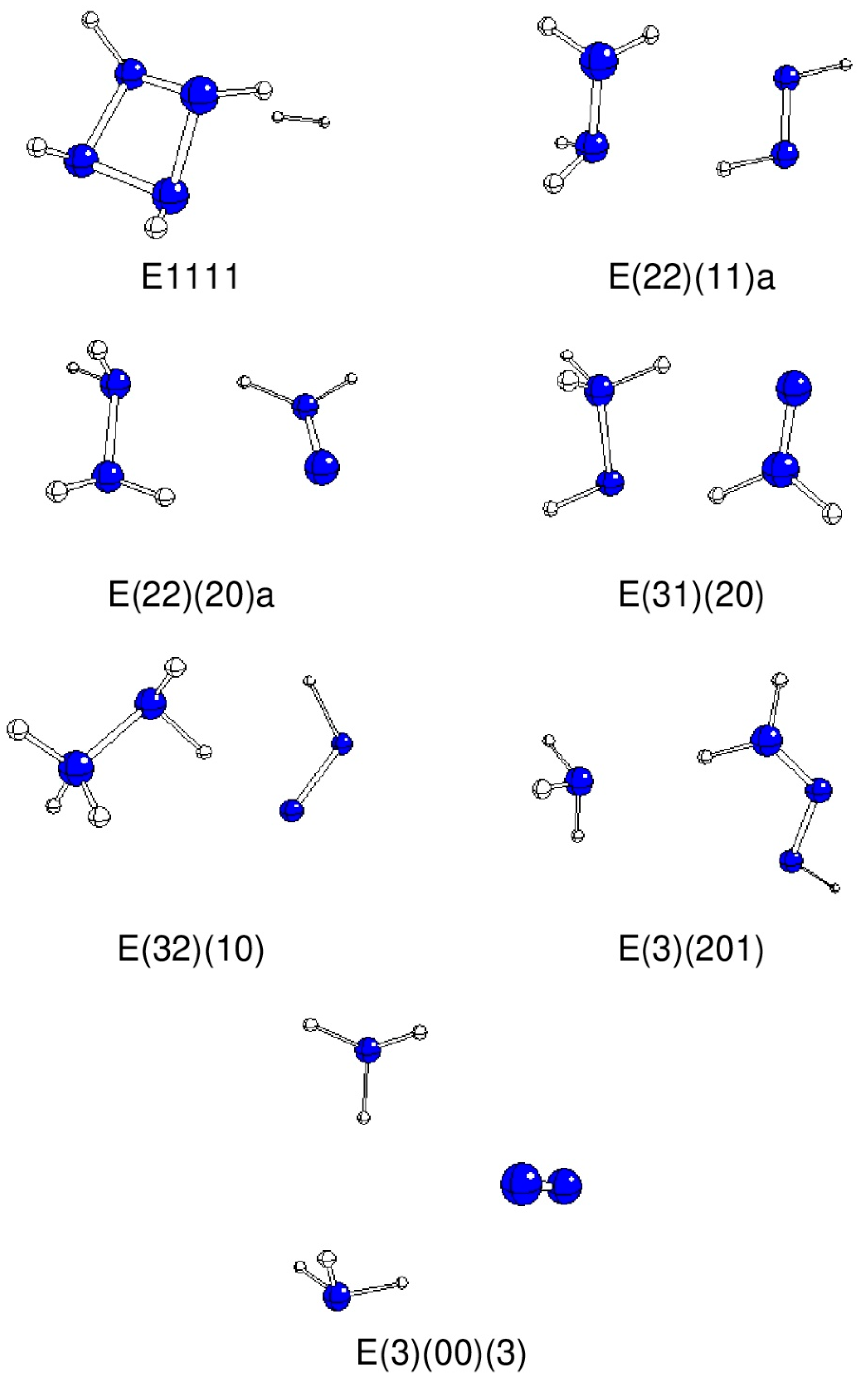{kind=link}
| Reaction | ΔEr (kJ/mol) | Ea (kJ/mol |
|---|---|---|
| N2H4 + H2N=N → NH2-NH-NH-NH2 | −103.6 | 50.6 |
| N2H4 + H2N=N → NH2NH2N=NH2 | 29.0 | 55.4 |
| NH2-NH-NH-NH2 → NH2NH=N + NH3 | 7.5 | 178.7 |
| NH2-NH-NH-NH2 → NH2-N=NH + NH3 | −102.5 | 214.1 |
| NH2-NH2-N=NH2 → NH2-N=NH + NH3 | −245.1 | 38.7 |
| NH2-NH-NH-NH2 → NH2-NH-NH2=NH | 151.1 | 158.6 |
| NH2-NH-NH2=NH → NH2-NH2-N=NH2 | −18.5 | 74.4 |
| N2H3• + N2H3• → NH2-NH-NH-NH2 | −152.9 | 0.2 |
| NH2-NH=NH• + NH2• → NH2-NH-NH-NH2 | 208.9 | 0.2 |
| NH=NH2-NH• + NH2• → NH2-NH-NH2=NH | 682.7 | 0.2 |
| NH2-N=NH2• + NH2• → NH2-NH2-N=NH2 | 37.9 | 2.8 |
| Starting | Optimized | Θ1234 [o] | G298 [Hartree] | ΔG298 [kJ/mol] | Remarks |
|---|---|---|---|---|---|
| A2112 | D2112a | 168.3 | −222.09177 | 0.00 | |
| A2121 | D2121a | −161.4 | −222.04654 | 118.75 | |
| A2211 | E(22)(11)a | −33.7 | −222.10760 | −41.56 | H2N-NH2 + HN=NH |
| A2202 | A2202 | −179.9 | −222.04618 | 119.71 | |
| A2220 | E(22)(20)a | 146.5 | −222.07817 | 35.7 | H2N-NH2 + H2N=N |
| A1221 | D1221 | −168.5 | −222.00357 | 231.58 | |
| A3210 | E(32)(10) | 14.3 | −222.04629 | 119.42 | H3N-NH2 + HN=N |
| A3201 | E(22)(11)b | −142.8 | −222.10755 | −41.43 | H2N-NH2 + HN=NH, 1→3 H rearrangement |
| A3201 | E(3)(201) | −26.5 | −222.14890 | −150.04 | NH3 + H2N-N=NH |
| A3111 | D2112b | 75.6 | −222.09311 | −3.52 | 1→4 H rearrangement |
| A3120 | E(31)(20) | −21.4 | −222.03229 | 156.16 | H3N-NH + H2N=N |
| A3102 | E(3)(102)a | −177.1 | −222.14926 | −150.94 | NH3 + HN=N-NH2 |
| A3012 | D3012a | 88.8 | −222.04832 | 114.07 | |
| A3021 | A3021 | 176.8 | −221.99866 | 244.46 | |
| A3003 | E(3)(00)(3) | 60.4 | −222.26295 | −449.44 | 2NH3 + N2 |
| B2112 | D2112c | 72.0 | −222.09665 | −12.80 | |
| B2121 | D2121b | −65.0 | −222.04530 | 122.02 | |
| B2121 | D2121c | −44.8 | −222.04982 | 110.13 | |
| B2211 | E(22)(11)a | −33.7 | −222.10760 | −41.56 | H2N-NH2 + HN=NH |
| B2202 | D2202a | 73.7 | −222.05056 | 108.21 | |
| B2220 | E(22)(20)b | −32.9 | −222.07820 | 35.64 | H2N-NH2 + H2N=N |
| B1221 | F12)(21 | 75.8 | −222.09760 | −15.30 | N2-N3 fission, N1-N4 bonding |
| B1221 | F1)(22)(1 | −33.3 | −222.10756 | −41.45 | H2N-NH2 + HN=NH, N1-N4 bonding |
| B3210 | E(22)(11)c | −34.0 | −222.10754 | −41.41 | H2N-NH2 + HN=NH, 1→4 H rearrangement |
| B3201 | D2202b | 80.1 | −222.04758 | 116.02 | 1→4 H rearrangement |
| B3201 | E(3)(201) | −26.5 | −222.14892 | −150.04 | NH3 + H2N=N-NH |
| B3111 | D2112b | 75.6 | −222.09311 | −3.52 | 1→4 H rearrangement |
| B3120 | D2121d | 68.8 | −222.04651 | 118.82 | 1→4 H rearrangement |
| B3102 | E(3)(102)b | −20.6 | −222.14678 | −144.42 | NH3 + HN=N-NH2 |
| B3012 | D3012b | −59.7 | −222.04910 | 112.02 | |
| B3021 | D2022 | −73.8 | −222.05056 | 108.21 | 1→4 H rearrangement |
| B3003 | E(3)(00)(3) | 60.4 | −222.26235 | −449.44 | 2 NH3 + N2 |
| C2211 | E(22)(11)d | 12.3 | −222.10759 | −41.45 | H2N-NH2 + HN=NH |
| C2202 | E(22)(02) | 29.6 | −222.07822 | 35.58 | H2N-NH2 + N=NH2 |
| C2121 | E1111 | 23.1 | −221.99631 | 250.64 | Cyclo-N4H4 + H2 |
| Structure | N1-N2 | N2-N3 | N3-N4 | N1-H | N2-H | N3-H | N4-H |
|---|---|---|---|---|---|---|---|
| D1221 | 1.341 | 1.840 | 1.338 | 1.017 | 1.019 1.016 | 1.015 1.021 | 1.018 |
| D2112a | 1.423 | 1.467 | 1.431 | 1.012 1.018 | 1.014 | 1.016 | 1.011 1.014 |
| D2112b | 1.432 | 1.419 | 1.440 | 1.012 1.015 | 1.018 | 1.013 | 1.011 1.015 |
| D2112c | 1.424 | 1.428 | 1.437 | 1.013 1.017 | 1.016 | 1.014 | 1.011 1.015 |
| D2121a | 1.413 | 1.480 | 1.412 | 1.011 1.017 | 1.015 | 1.020 1.021 | 1.018 |
| D2121b | 1.423 | 1.467 | 1.417 | 1.010 1.013 | 1.018 | 1.017 1.020 | 1.019 |
| D2121c | 1.413 | 1.504 | 1.409 | 1.013 1.024 | 1.017 | 1.017 1.020 | 1.020 |
| D2121d | 1.423 | 1.467 | 1.415 | 1.010 1.013 | 1.018 | 1.016 1.021 | 1.018 |
| A2202 | 1.427 | 1.454 | 1.443 | 1.016(2×) | 1.021(2×) | - | 1.013(2×) |
| D2202a | 1.459 | 1.422 | 1.446 | 1.017(2×) | 1.016 1.022 | - | 1.012 1.013 |
| D2202b | 1.464 | 1.418 | 1.446 | 1.016 1.018 | 1.017 1.021 | - | 1.012 1.014 |
| D2022 | 1.459 | 1.421 | 1.447 | 1.017(2×) | - | 1.016 1.022 | 1.012 1.013 |
| A3021 | 1.463 | 1.452 | 1.433 | 1.016 1.024(2×) | - | 1.020 1.025 | 1.019 |
| D3012a | 1.463 | 1.418 | 1.463 | 1.016 1.021(2×) | - | 1.013 | 1.015 1.017 |
| D3012b | 1.493 | 1.395 | 1.485 | 1.014 1.021(2×) | - | 1.021 | 1.014 1.017 |
| System | N1-N2 | N2-N3 | N3-N4 | N1-H | N2-H | N3-H | N4-H |
|---|---|---|---|---|---|---|---|
| E1111 (a) | 1.476 | 1.481 | 1.481 | 1.023 | 1.017 | 1.017 | 1.017 |
| E(22)(11)a | 1.446 | 3.118 | 1.245 | 1.012 1.014 | 1.011 1.014 | 1.030 | 1.027 |
| E(22)(11)b | 1.446 | 3.465 | 1.245 | 1.011 1.014 | 1.012 1.014 | 1.030 | 1.027 |
| E(22)(11)c | 1.445 | 3.292 | 1.245 | 1.011 1.014 | 1.012 1.014 | 1.027 | 1.030 |
| E(22)(11)d | 1.446 | 3.116 | 1.245 | 1.012 1.014 | 1.011 1.014 | 1.030 | 1.027 |
| E(22)(20)a | 1.446 | 3.271 | 1.225 | 1.011 1.014 | 1.013 1.014 | 1.028 1.034 | - |
| E(22)(20)b | 1.446 | 2.971 | 1.225 | 1.013 1.014 | 1.011 1.014 | 1.028 1.033 | - |
| E(22)(02) | 1.447 | 3.276 | 1.225 | 1.011 1.014 | 1.013 1.014 | - | 1.028 1.033 |
| E(31)(20) | 1.468 | 2.750 | 1.230 | 1.018(2×) 1.029 | 1.016 | 1.029 1.057 | - |
| E(32)(10) | 1.445 | 3.035 | 1.242 | 1.018(2×) 1.021 | 1.015 1.079 | 1.076 | - |
| E(3)(201) | 3.088 | 1.350 | 1.249 | 1.013(3×) | 1.006 1.022 | - | 1.019 |
| E(3)(102)a | 3.117 | 1.243 | 1.365 | 1.013(3×) | 1.026 | - | 1.008 1.014 |
| E(3)(102)b | 3.760 | 1.246 | 1.356 | 1.013(2×) 1.014 | 1.032 | - | 1.007 1.024 |
| E(3)(00)(3) | 3.636 | 1.096 | 3.711 | 1.014(3×) | - | - | 1.013(3×) |
| F(11)(22) | 1.245 | 3.291 | 1.446 | 1.030 | 1.027 | 1.012 1.014 | 1.011 1.014 |
| F12)(21 (b) | 1.430 | 3.017 | 1.424 | 1.012 | 1.012 1.018 | 1.012 1.017 | 1.017 |
| Structure | N1-N2 | N2-N3 | N3-N4 | N1-H | N2-H | N3-H | N4-H |
|---|---|---|---|---|---|---|---|
| D1221 | 0.3705 | 0.1281 | 0.3734 | 0.3450 | 0.3483 0.3528 | 0.3467 0.3534 | 0.3448 |
| D2112a | 0.3156 | 0.2911 | 0.3092 | 0.3459 0.3515 | 0.3574 | 0.3551 | 0.3492 0.3518 |
| D2112b | 0.3092 | 0.3237 | 0.3036 | 0.3494 0.3508 | 0.3527 | 0.3561 | 0.3484 0.3514 |
| D2112c | 0.3149 | 0.3165 | 0.3050 | 0.3472 0.3513 | 0.3542 | 0.3552 | 0.3484 0.3515 |
| D2121a | 0.3212 | 0.2827 | 0.3080 | 0.3458 0.3516 | 0.3544 | 0.3520 0.3527 | 0.3415 |
| D2121b | 0.3149 | 0.2920 | 0.3074 | 0.3509 0.3517 | 0.3524 | 0.3521 0.3551 | 0.3404 |
| D2121c | 0.3224 | 0.2668 | 0.3133 | 0.3406 0.3505 | 0.3542 | 0.3508 0.3556 | 0.3404 |
| D2121d | 0.3149 | 0.2928 | 0.3086 | 0.3503 0.3521 | 0.3523 | 0.3509 0.3559 | 0.3417 |
| A2202 | 0.3148 | 0.2864 | 0.2965 | 0.3478(2×) | 0.3514(2×) | - | 0.3504 0.3503 |
| D2202a | 0.2928 | 0.3098 | 0.2951 | 0.3469 0.3474 | 0.3499 0.3553 | - | 0.3506(2×) |
| D2202b | 0.2898 | 0.3107 | 0.2956 | 0.3447 0.3478 | 0.3507 0.3549 | - | 0.3501 0.3510 |
| D2022 | 0.2949 | 0.3102 | 0.2930 | 0.3505 0.3507 | - | 0.3498 0.3552 | 0.3470 0.3474 |
| A3021 | 0.2759 | 0.2964 | 0.2956 | 0.3431 0.3437 0.3480 | - | 0.3499 0.3542 | 0.3397 |
| D3012a | 0.2761 | 0.3196 | 0.2874 | 0.3454 0.3473 0.3500 | - | 0.3564 | 0.3463 0.3476 |
| D3012b | 0.2562 | 0.3362 | 0.2736 | 0.3446 0.3450 0.3501 | - | 0.3488 | 0.3455 0.3497 |
| System | N1-N2 | N2-N3 | N3-N4 | N1-H | N2-H | N3-H | N4-H |
|---|---|---|---|---|---|---|---|
| E1111 (a) | 0.2858 | 0.2828 | 0.2824 | 0.3506 | 0.3545 | 0.3566 | 0.3546 |
| E(22)(11)a | 0.2953 | - | 0.4863 | 0.3502 0.3529 | 0.3500 0.3529 | 0.3463 | 0.3483 |
| E(22)(11)b | 0.2953 | - | 0.4826 | 0.3500 0.3529 | 0.3502 0.3529 | 0.3462 | 0.3482 |
| E(22)(11)c | 0.2954 | - | 0.4863 | 0.3500 0.3529 | 0.3503 0.3529 | 0.3482 | 0.3462 |
| E(22)(11)d | 0.2953 | - | 0.4863 | 0.3502 0.3529 | 0.3500 0.3529 | 0.3463 | 0.3482 |
| E(22)(20)a | 0.2947 | - | 0.4970 | 0.3499 0.3529 | 0.3501 0.3518 | 0.3367 0.3422 | - |
| E(22)(20)b | 0.2945 | - | 0.4967 | 0.3501 0.3517 | 0.3499 0.3529 | 0.3367 0.3423 | - |
| E(22)(02) | 0.2945 | - | 0.4967 | 0.3500 0.3529 | 0.3501 0.3517 | - | 0.3367 0.3423 |
| E(31)(20) | 0.2696 | - | 0.4927 | 0.3382 0.3486 0.3493 | 0.3426 | 0.3123 0.3412 | - |
| E(32)(10) | 0.2929 | - | 0.4831 | 0.3471 0.3453 0.3474 | 0.2909 0.3485 | 0.3032 | - |
| E(3)(201) | - | 0.3794 | 0.4825 | 0.3434 0.3435 0.3436 | 0.3531 0.3368 | - | 0.3503 |
| E(3)(102)a | - | 0.4891 | 0.3669 | 0.3435(2×) 0.3436 | 0.3448 | - | 0.3457 0.3521 |
| E(3)(102)b | - | 0.4833 | 0.3719 | 0.3432 0.3435 0.3436 | 0.3375 | - | 0.3345 0.3523 |
| E(3)(00)(3) | - | 0.7140 | - | 0.3432 0.3433 0.3433 | - | - | 0.3435 0.3437 0.3442 |
| F(11)(22) | 0.4863 | - | 0.2954 | 0.3463 | 0.3482 | 0.3502 0.3529 | 0.3500 0.3529 |
| F12)(21 (b) | 0.3096 | - | 0.3150 | 0.3573 | 0.3451 0.3515 | 0.3470 0.3515 | 0.3522 |
| Structure | N1-N2 | N2-N3 | N3-N4 | N1-H | N2-H | N3-H | N4-H |
|---|---|---|---|---|---|---|---|
| D1221 | 0.230 | 0.107 | 0.231 | 0.048 | 0.015 0.017 | 0.015 0.016 | 0.047 |
| D2112a | 0.003 | 0.149 | 0.024 | 0.045 0.050 | 0.041 | 0.036 | 0.046 0.051 |
| D2112b | 0.040 | 0.039 | 0.012 | 0.046 0.051 | 0.043 | 0.051 | 0.047 0.051 |
| D2112c | 0.027 | 0.046 | 0.015 | 0.044 0.047 | 0.047 | 0.050 | 0.046 0.050 |
| D2121a | 0.027 | 0.123 | 0.198 | 0.048 0.051 | 0.046 | 0.007 0.009 | 0.073 |
| D2121b | 0.035 | 0.070 | 0.182 | 0.045 0.049 | 0.034 | 0.011 0.015 | 0.074 |
| D2121c | 0.026 | 0.074 | 0.192 | 0.039 0.048 | 0.038 | 0.013(2×) | 0.071 |
| D2121d | 0.025 | 0.069 | 0.178 | 0.045 0.050 | 0.033 | 0.012 0.013 | 0.073 |
| A2202 | 0.060 | 0.302 | 0.089 | 0.036(2×) | 0.008(2×) | - | 0.055(2×) |
| D2202a | 0.045 | 0.288 | 0.084 | 0.034 0.035 | 0.012 0.013 | - | 0.053(2×) |
| D2202b | 0.086 | 0.301 | 0.087 | 0.039 0.041 | 0.010 0.082 | - | 0.052 0.053 |
| D2022 | 0.084 | 0.288 | 0.046 | 0.052 0.053 | - | 0.012 0.013 | 0.034 0.035 |
| A3021 | 0.268 | 0.222 | 0.169 | 0.106 0.108 0.005 | - | 0.006 0.009 | 0.079 |
| D3012a | 0.267 | 0.124 | 0.064 | 0.006 0.007 0.008 | - | 0.049 | 0.044 0.048 |
| D3012b | 0.248 | 0.113 | 0.123 | 0.004 0.005(2×) | - | 0.051 | 0.038(2×) |
| System | N1-N2 | N2-N3 | N3-N4 | N1-H | N2-H | N3-H | N4-H |
|---|---|---|---|---|---|---|---|
| E1111 (a) | 0.103 | 0.108 | 0.108 | 0.029 | 0.030 | 0.027 | 0.030 |
| E(22)(11)a | 0.008 | - | 0.189 | 0.047 0.049 | 0.046 0.050 | 0.004 | 0.004 |
| E(22)(11)b | 0.008 | - | 0.189 | 0.046 0.050 | 0.047 0.049 | 0.004 | 0.004 |
| E(22)(11)c | 0.008 | - | 0.189 | 0.046 0.050 | 0.047 0.049 | 0.004 | 0.004 |
| E(22)(11)d | 0.008 | - | 0.189 | 0.047 0.049 | 0.046 0.049 | 0.004 | 0.004 |
| E(22)(20)a | 0.008 | - | 0.021 | 0.046 0.049 | 0.047(2×) | 0.035 0.038 | - |
| E(22)(20)b | 0.007 | - | 0.020 | 0.047(2×) | 0.046 0.049 | 0.035 0.038 | - |
| E(22)(02) | 0.007 | - | 0.020 | 0.046 0.049 | 0.047(2×) | - | 0.035 0.039 |
| E(31)(20) | 0.156 | - | 0.005 | 0.006 0.011 0.012 | 0.080 | 0.029 0.035 | - |
| E(32)(10) | 0.089 | - | 0.072 | 0.009 0.010(2×) | 0.027 0.045 | 0.005 | - |
| E(3)(201) | - | 0.138 | 0.229 | 0.033(3×) | 0.043 0.053 | - | 0.008 |
| E(3)(102)a | - | 0.218 | 0.118 | 0.326 0.327(2×) | 0.005 | - | 0.047 0.051 |
| E(3)(102)b | - | 0.238 | 0.133 | 0.324(2×) 0.329 | 0.001 | - | 0.041 0.052 |
| E(3)(00)(3) | - | 0.000 | - | 0.033 0.034(2×) | - | - | 0.033(3×) |
| F(11)(22) | 0.189 | - | 0.008 | 0.004 | 0.004 | 0.047 0.049 | 0.046 0.050 |
| F12)(21 (b) | 0.012 | - | 0.031 | 0.054 | 0.046 0.051 | 0.045 0.047 | 0.049 |
| Structure | N1 | N2 | N3 | N4 | H(N1) | H(N2) | H(N3) | H(N4) |
|---|---|---|---|---|---|---|---|---|
| D1221 | −0.657 | −0.488 | −0.485 | −0.649 | 0.342 | 0.452 0.455 | 0.444 0.457 | 0.342 |
| D2112a | −0.699 | −0.347 | −0.367 | −0.706 | 0.379 0.392 | 0.391 | 0.382 | 0.391 0.404 |
| D2112b | −0.691 | −0.357 | −0.354 | −0.726 | 0.378 0.394 | 0.372 | 0.395 | 0.387 0.398 |
| D2112c | −0.711 | −0.354 | −0.368 | −0.729 | 0.377 0.389 | 0.382 | 0.396 | 0.389 0.401 |
| D2121a | −0.700 | −0.341 | −0.398 | −0.787 | 0.394 0.413 | 0.417 | 0.452(2×) | 0.309 |
| D2121b | −0.704 | −0.361 | −0.394 | −0.811 | 0.400 0.416 | 0.405 | 0.470 0.560 | 0.302 |
| D2121c | −0.709 | −0.365 | −0.412 | −0.800 * | 0.396 0.407 * | 0.406 | 0.463 0.468 | 0.310 |
| D2121d | −0.707 | −0.361 | −0.395 | −0.809 | 0.402 0.420 | 0.408 | 0.458 0.471 | 0.304 |
| A2202 | −0.664 | −0.388 | −0.435 | −0.750 | 0.418(2×) | 0.450(2×) | - | 0.362(2×) |
| D2202a | −0.712 | −0.404 | −0.430 | −0.760 | 0.409 0.410 | 0.455 0.475 | - | 0.361 0.364 |
| D2202b | −0.705 | −0.397 | −0.432 | −0.737 | 0.407 0.411 | 0.459 0.465 | - | 0.357 0.367 |
| D2022 | −0.761 | −0.430 | −0.402 | −0.711 | 0.361 0.365 | - | 0.455 0.475 | 0.409 0.410 |
| A3021 | −0.730 | −0.368 | −0.388 | −0.824 | 0.460 0.461 0.496 | - | 0.403 0.423 | 0.286 |
| D3012a | −0.732 | −0.436 | −0.390 | −0.739 | 0.449(2×) 0.472 | - | 0.370 | 0.360 0.372 |
| D3012b | −0.762 | −0.417 | −0.384 | −0.754 * | 0.444 0.466 * 0.473 | - | 0.345 | 0.376 0.378 |
| System | N1 | N2 | N3 | N4 | H(N1) | H(N2) | H(N3) | H(N4) |
|---|---|---|---|---|---|---|---|---|
| E1111 | −0.345 | −0.367 * | −0.373 | −0.367 | 0.383 | 0.408 | 0.396 * | 0.403 |
| E(22)(11)a | −0.707 | −0.727 * | −0.358 | −0.348 * | 0.380 0.392 * | 0.385 0.393 | 0.409 * | 0.380 |
| E(22)(11)b | −0.727 * | −0.707 | −0.360 | −0.348 * | 0.384 0.393 | 0.380 0.388 * | 0.409 * | 0.380 |
| E(22)(11)c | −0.726 * | −0.706 | −0.349h | −0.360 | 0.384 0.393 | 0.380 0.387 * | 0.380 | 0.409h |
| E(22)(11)d | −0.706 | −0.726 * | −0.359 | −0.347 * | 0.380 0.388 * | 0.384 0.393 | 0.409 * | 0.380 |
| E(22)(20)a | −0.732 * | −0.714 | −0.519 | −0.271 * | 0.380 0.393 * | 0.387 0.395 | 0.417 0.460 * | - |
| E(22)(20)b | −0.713 | −0.732 * | −0.517 | −0.273 * | 0.380 0.393 * | 0.387 0.395 | 0.417 0.461 * | - |
| E(22)(02) | −0.714 | −0.732 * | −0.272 * | −0.517 | 0.380 0.393 * | 0.387 0.395 | - | 0.417 0.460 * |
| E(31)(20) | −0.751 | −0.831 * | −0.543 | −0.306 * | 0.447 0.452 0.491 * | 0.315 | 0.408 0.511 * | - |
| E(32)(10) | −0.718 | −0.731 | −0.426 | −0.530 * | 0.496 0.508(2×) | 0.408 0.501 * | 0.185 | - |
| E(3)(201) | −1.079 * | −0.734 | −0.035 | −0.436 | 0.394(3×) | 0.443 0.473 * | - | 0.388 |
| E(3)(102)a | −1.076 * | −0.454 | −0.033 | −0.686 | 0.394(3×) | 0.428 * | - | 0.429 0.445 |
| E(3)(102)b | −1.084 * | −0.396 | −0.030 | −0.739 | 0.394 0.395 0.396 | 0.352 | - | 0.445 0.470 * |
| E(3)(00)(3) | −1.077 * | 0.076 * | −0.049 | −1.059 | 0.382 0.382 * 0.384 | - | - | 0.373 0.380 * 0.386 |
| F(11)(22) | −0.359 | −0.347 * | −0.706 | −0.725 * | 0.409 * | 0.380 | 0.380 0.388 * | 0.393 0.394 |
| F12)(21 | −0.356 | −0.722 | −0.702 | −0.368 | 0.403 | 0.377 0.392 | 0.371 0.395 | 0.381 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breza, M.; Manova, A. Hydrazine Oxidation in Aqueous Solutions I: N4H6 Decomposition. Inorganics 2023, 11, 413. https://doi.org/10.3390/inorganics11100413
Breza M, Manova A. Hydrazine Oxidation in Aqueous Solutions I: N4H6 Decomposition. Inorganics. 2023; 11(10):413. https://doi.org/10.3390/inorganics11100413
Chicago/Turabian StyleBreza, Martin, and Alena Manova. 2023. "Hydrazine Oxidation in Aqueous Solutions I: N4H6 Decomposition" Inorganics 11, no. 10: 413. https://doi.org/10.3390/inorganics11100413
APA StyleBreza, M., & Manova, A. (2023). Hydrazine Oxidation in Aqueous Solutions I: N4H6 Decomposition. Inorganics, 11(10), 413. https://doi.org/10.3390/inorganics11100413