
Hydrogen Sulfide—Clues from Evolution and Implication for Neonatal Respiratory Diseases



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
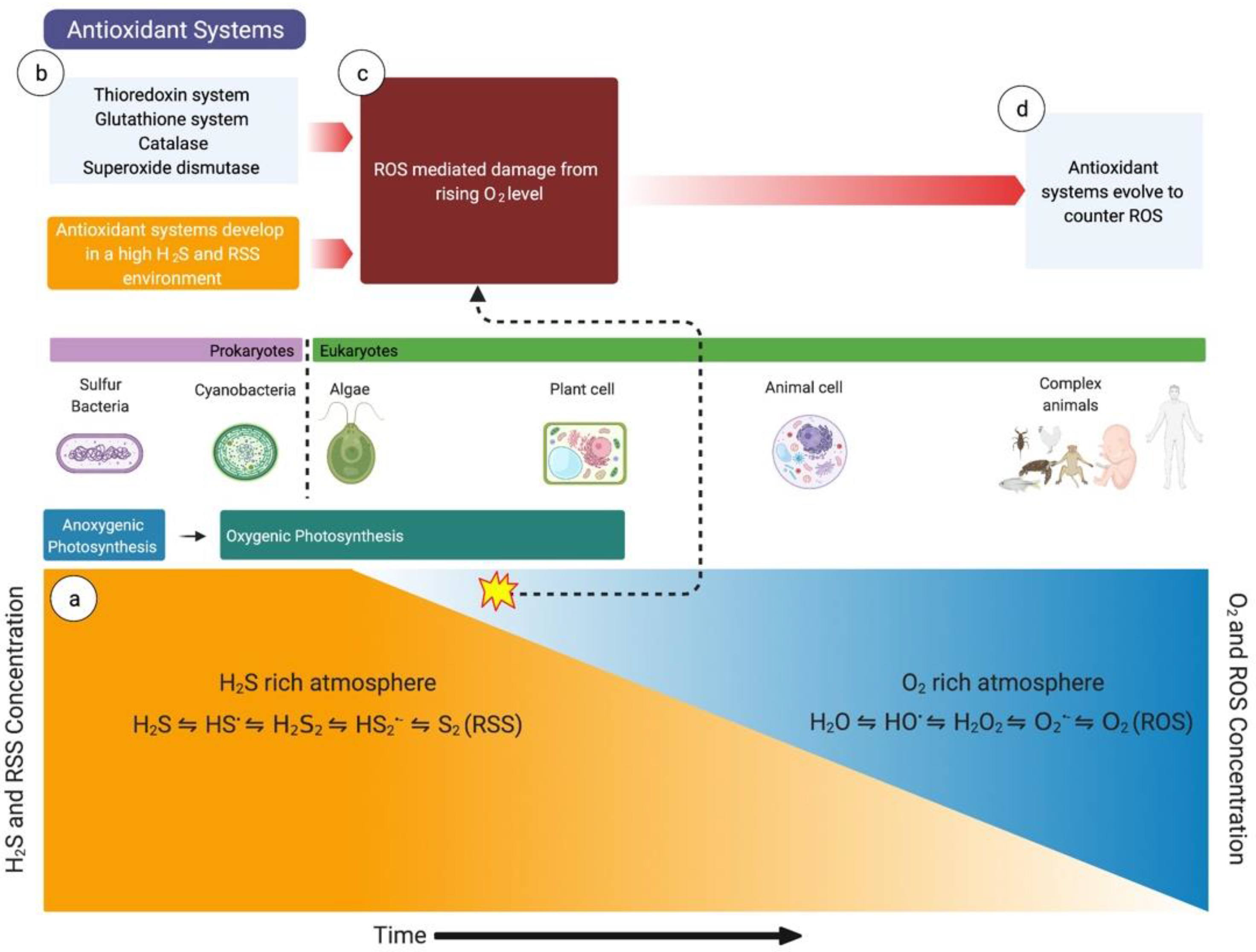
2. Evolution of Oxidant and Antioxidant Pathways and the Importance of H2S
3. Sulfur Homeostasis and Metabolism
3.1. H2S Biogenesis
3.2. H2S Metabolism
4. Mechanisms of H2S Signaling
4.1. Direct Antioxidant Effect
4.2. Interaction with Metalloproteins
4.3. Post-Translational Modification of Proteins
5. Current State of Antioxidant Therapy in Oxidative Neonatal Respiratory Disease
5.1. Treatment Modalities
5.2. Possible Reasons for the Antioxidant Therapy Failures
6. H2S in the Developing Lung and Neonatal Respiratory Diseases
7. Future Direction
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017. [Google Scholar] [CrossRef]
- Torres-Cuevas, I.; Parra-Llorca, A.; Sánchez-Illana, A.; Nuñez-Ramiro, A.; Kuligowski, J.; Cháfer-Pericás, C.; Cernada, M.; Escobar, J.; Vento, M. Oxygen and oxidative stress in the perinatal period. Redox Biol. 2017, 12, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.M.; Auten, R.L. Maturation of the antioxidant system and the effects on preterm birth. Semin. Fetal. Neonatal. Med. 2010, 15, 191–195. [Google Scholar] [CrossRef]
- Berkelhamer, S.K.; Farrow, K.N. Developmental Regulation of Antioxidant Enzymes and Their Impact on Neonatal Lung Disease. Antioxid. Redox Signal. 2014, 21, 1837–1848. [Google Scholar] [CrossRef]
- Ofman, G.; Tipple, T.E. Antioxidants & bronchopulmonary dysplasia: Beating the system or beating a dead horse? Free Radic. Biol. Med. 2019, 142, 138–145. [Google Scholar] [PubMed]
- Kimura, H. Production and Physiological Effects of Hydrogen Sulfide. Antioxid. Redox Signal. 2014, 20, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Madurga, A.; Golec, A.; Pozarska, A.; Ishii, I.; Mizikova, I.; Nardiello, C.; Vadász, I.; Herold, S.; Mayer, K.; Reichenberger, F.; et al. The H2S-generating enzymes cystathionine beta-synthase and cystathionine gamma-lyase play a role in vascular development during normal lung alveolarization. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L710–L724. [Google Scholar] [CrossRef]
- Madurga, A.; Mižíková, I.; Ruiz-Camp, J.; Vadász, I.; Herold, S.; Mayer, K.; Fehrenbach, H.; Seeger, W.; Morty, R.E. Systemic hydrogen sulfide administration partially restores normal alveolarization in an experimental animal model of bronchopulmonary dysplasia. Am. J. Physiol. Cell. Mol. Physiol. 2014, 306, L684–L697. [Google Scholar] [CrossRef]
- Bartman, C.M.; Schiliro, M.; Helan, M.; Prakash, Y.S.; Linden, D.; Pabelick, C. Hydrogen sulfide, oxygen, and calcium regulation in developing human airway smooth muscle. FASEB J. 2020, 34, 12991–13004. [Google Scholar] [CrossRef]
- Olson, K.R. Reactive oxygen species or reactive sulfur species: Why we should consider the latter. J. Exp. Biol. 2020, 223, jeb196352. [Google Scholar] [CrossRef]
- Muyzer, G.; Stams, A.J.M. The ecology and biotechnology of sulphate-reducing bacteria. Nat. Rev. Genet. 2008, 6, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Vigneron, A.; Cruaud, P.; Alsop, E.; De Rezende, J.R.; Head, I.M.; Tsesmetzis, N. Beyond the tip of the iceberg; a new view of the diversity of sulfite- and sulfate-reducing microorganisms. ISME J. 2018, 12, 2096–2099. [Google Scholar] [CrossRef]
- Olson, K.R.; Straub, K.D. The Role of Hydrogen Sulfide in Evolution and the Evolution of Hydrogen Sulfide in Metabolism and Signaling. Physiology 2016, 31, 60–72. [Google Scholar] [CrossRef]
- Barley, M.; Bekker, A.; Krapez, B. Late Archean to Early Paleoproterozoic global tectonics, environmental change and the rise of atmospheric oxygen. Earth Planet. Sci. Lett. 2005, 238, 156–171. [Google Scholar] [CrossRef]
- Holland, H.D. The oxygenation of the atmosphere and oceans. Philos. Trans. R Soc. Lond. B Biol. Sci. 2006, 361, 903–915. [Google Scholar] [CrossRef]
- Kolluru, G.K.; Shen, X.; Kevil, C.G. Reactive Sulfur Species: A New Redox Player in Cardiovascular Pathophysiology. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 874–884. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.M.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell Metab. 2016, 23, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Biological Production, Detection, and Fate of Hydrogen Peroxide. Antioxid. Redox Signal. 2018, 29, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Lau, N.; Pluth, M.D. Reactive sulfur species (RSS): Persulfides, polysulfides, potential, and problems. Curr. Opin. Chem. Biol. 2019, 49, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Giles, G.I.; Nasim, M.J.; Ali, W.; Jacob, C. The Reactive Sulfur Species Concept: 15 Years On. Antioxidants 2017, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, R.; Wu, L.; Yang, G. Hydrogen sulfide signaling in regulation of cell behaviors. Nitric Oxide 2020, 103, 9–19. [Google Scholar] [CrossRef]
- Kimura, H. Hydrogen Sulfide: From Brain to Gut. Antioxid. Redox Signal. 2010, 12, 1111–1123. [Google Scholar] [CrossRef]
- Giuffrè, A.; Tomé, C.S.; Fernandes, D.G.F.; Zuhra, K.; Vicente, J.B. Hydrogen Sulfide Metabolism and Signaling in the Tumor Microenvironment. Clin. Biol. Mol. Asp. Covid-19 2020, 1219, 335–353. [Google Scholar]
- de Cabo, R.; Diaz-Ruiz, A. A Central Role for the Gasotransmitter H2S in Aging. Cell Metab. 2020, 31, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Ling, K.; Xu, A.; Chen, Y.; Chen, X.; Li, Y.; Wang, W. Protective effect of a hydrogen sulfide donor on balloon injury-induced restenosis via the Nrf2/HIF-1α signaling pathway. Int. J. Mol. Med. 2019, 43, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Bazhanov, N.; Ansar, M.; Ivanciuc, T.; Garofalo, R.P.; Casola, A. Hydrogen Sulfide: A Novel Player in Airway Development, Pathophysiology of Respiratory Diseases, and Antiviral Defenses. Am. J. Respir. Cell Mol. Biol. 2017, 57, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Vitvitsky, V.; Kabil, O.; Banerjee, R. High Turnover Rates for Hydrogen Sulfide Allow for Rapid Regulation of Its Tissue Concentrations. Antioxid. Redox Signal. 2012, 17, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Furne, J.; Saeed, A.; Levitt, M.D. Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. Am. J. Physiol. Integr. Comp. Physiol. 2008, 295, R1479–R1485. [Google Scholar] [CrossRef]
- Yang, J.; Minkler, P.; Grove, D.; Wang, R.; Willard, B.; Dweik, R.; Hine, C. Non-enzymatic hydrogen sulfide production from cysteine in blood is catalyzed by iron and vitamin B6. Commun. Biol. 2019, 2, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Searcy, D.G.; Lee, S.H. Sulfur reduction by human erythrocytes. J. Exp. Zool. 1998, 282, 310–322. [Google Scholar] [CrossRef]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Regulators of the transsulfuration pathway. Br. J. Pharm. 2018, 176, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Teng, H.; Wu, B.; Zhao, K.; Yang, G.; Wu, L.; Wang, R. Oxygen-sensitive mitochondrial accumulation of cystathionine β-synthase mediated by Lon protease. Proc. Natl. Acad. Sci. USA 2013, 110, 12679–12684. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, N.; Ito, T.; Kitamura, H.; Nishino, T. Tissue and subcellular distribution of mercaptopyruvate sulfurtransferase in the rat: Confocal laser fluorescence and immunoelectron microscopic studies combined with biochemical analysis. Histochem. Cell Biol. 1998, 110, 243–250. [Google Scholar] [CrossRef]
- Zuhra, K.; Augsburger, F.; Majtan, T.; Szabo, C. Cystathionine-beta-Synthase: Molecular Regulation and Pharmacological Inhibition. Biomolecules 2020, 10, 697. [Google Scholar] [CrossRef]
- Mudd, S.H.; Finkelstein, J.D.; Irreverre, F.; Laster, L. Homocystinuria: An Enzymatic Defect. Science 1964, 143, 1443–1445. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Zou, C.G. Redox regulation and reaction mechanism of human cystathionine-beta-synthase: A PLP-dependent hemesensor protein. Arch. Biochem. Biophys. 2005, 433, 144–156. [Google Scholar] [CrossRef]
- Chiku, T.; Padovani, D.; Zhu, W.; Singh, S.; Vitvitsky, V.; Banerjee, R. H2S biogenesis by human cystathionine gamma-lyase leads to the novel sulfur metabolites lanthionine and homolanthionine and is responsive to the grade of hyperhomocysteinemia. J. Biol. Chem. 2009, 284, 11601–11612. [Google Scholar] [CrossRef]
- Mosharov, E.; Cranford, M.R.; Banerjee, R. The Quantitatively Important Relationship between Homocysteine Metabolism and Glutathione Synthesis by the Transsulfuration Pathway and Its Regulation by Redox Changes. Biochemistry 2000, 39, 13005–13011. [Google Scholar] [CrossRef]
- Akiyama, M.; Unoki, T.; Shinkai, Y.; Ishii, I.; Ida, T.; Akaike, T.; Yamamoto, M.; Kumagai, Y. Environmental Electrophile-Mediated Toxicity in Mice Lacking Nrf2, CSE, or Both. Environ. Heal. Perspect. 2019, 127, 067002. [Google Scholar] [CrossRef]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen Sulfide Protects Against Cellular Senescence via S-Sulfhydration of Keap1 and Activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Banerjee, R. Human polycomb 2 protein is a SUMO E3 ligase and alleviates substrate-induced inhibition of cystathionine beta-synthase sumoylation. PLoS ONE 2008, 3, e4032. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.K.; Yamada, K.; Chiku, T.; Koutmos, M.; Banerjee, R. Structure and Kinetic Analysis of H2S Production by Human Mercaptopyruvate Sulfurtransferase. J. Biol. Chem. 2013, 288, 20002–20013. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, N.; Yoshii, T.; Abe, Y.; Matsumura, T. Thioredoxin-dependent Enzymatic Activation of Mercaptopyruvate Sulfurtransferase. J. Biol. Chem. 2007, 282, 1561–1569. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, N.; Tanaka, M.; Yoshida, M.; Ogasawara, Y.; Togawa, T.; Ishii, K.; Kimura, H. 3-Mercaptopyruvate Sulfurtransferase Produces Hydrogen Sulfide and Bound Sulfane Sulfur in the Brain. Antioxid. Redox Signal. 2009, 11, 703–714. [Google Scholar] [CrossRef]
- Kabil, O.; Banerjee, R. Enzymology of H2S Biogenesis, Decay and Signaling. Antioxid. Redox Signal. 2014, 20, 770–782. [Google Scholar] [CrossRef]
- Bouillaud, F.; Blachier, F. Mitochondria and Sulfide: A Very Old Story of Poisoning, Feeding, and Signaling? Antioxid. Redox Signal. 2011, 15, 379–391. [Google Scholar] [CrossRef]
- Szczesny, B.; Marcatti, M.; Zatarain, J.R.; Druzhyna, N.; Wiktorowicz, J.E.; Nagy, P.; Hellmich, M.R.; Szabo, C. Inhibition of hydrogen sulfide biosynthesis sensitizes lung adenocarcinoma to chemotherapeutic drugs by inhibiting mitochondrial DNA repair and suppressing cellular bioenergetics. Sci. Rep. 2016, 6, 36125. [Google Scholar] [CrossRef]
- Merz, T.; Vogt, J.A.; Wachter, U.; Calzia, E.; Szabo, C.; Wang, R.; Radermacher, P.; McCook, O. Impact of hyperglycemia on cystathionine-γ-lyase expression during resuscitated murine septic shock. Intensiv. Care Med. Exp. 2017, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Vitvitsky, V.; Yadav, P.K.; Kurthen, A.; Banerjee, R. Sulfide Oxidation by a Noncanonical Pathway in Red Blood Cells Generates Thiosulfate and Polysulfides. J. Biol. Chem. 2015, 290, 8310–8320. [Google Scholar] [CrossRef] [PubMed]
- Bostelaar, T.; Vitvitsky, V.; Kumutima, J.; Lewis, B.E.; Yadav, P.K.; Brunold, T.C.; Filipovic, M.; Lehnert, N.; Stemmler, T.L.; Banerjee, R. Hydrogen Sulfide Oxidation by Myoglobin. J. Am. Chem. Soc. 2016, 138, 8476–8488. [Google Scholar] [CrossRef] [PubMed]
- Hancock, J.T.; Whiteman, M. Hydrogen sulfide signaling: Interactions with nitric oxide and reactive oxygen species. Ann. N. Y. Acad. Sci. 2015, 1365, 5–14. [Google Scholar] [CrossRef]
- Bełtowski, J. Synthesis, Metabolism, and Signaling Mechanisms of Hydrogen Sulfide: An Overview. Methods Mol. Biol. 2019, 2007, 1–8. [Google Scholar] [PubMed]
- Murphy, B.; Bhattacharya, R.; Mukherjee, P. Hydrogen sulfide signaling in mitochondria and disease. FASEB J. 2019, 33, 13098–13125. [Google Scholar] [CrossRef]
- Wedmann, R.; Bertlein, S.; Macinkovic, I.; Böltz, S.; Miljkovic, J.; Muñoz, L.E.; Herrmann, M.; Filipovic, M.R. Working with “H2S”: Facts and apparent artifacts. Nitric Oxide 2014, 41, 85–96. [Google Scholar] [CrossRef]
- Filipovic, M.R.; Miljkovic, J.; Allgäuer, A.; Chaurio, R.; Shubina, T.; Herrmann, M.; Ivanovic-Burmazovic, I. Biochemical insight into physiological effects of H₂S: Reaction with peroxynitrite and formation of a new nitric oxide donor, sulfinyl nitrite. Biochem. J. 2012, 441, 609–621. [Google Scholar] [CrossRef]
- Wang, C.; Wang, H.-Y.; Liu, Z.-W.; Fu, Y.; Zhao, B. Effect of endogenous hydrogen sulfide on oxidative stress in oleic acid-induced acute lung injury in rats. Chin. Med. J. 2011, 124, 3476–3480. [Google Scholar] [PubMed]
- Xie, Z.-Z.; Liu, Y.; Bian, J.-S. Hydrogen Sulfide and Cellular Redox Homeostasis. Oxidative Med. Cell. Longev. 2016, 2016, 1–12. [Google Scholar] [CrossRef]
- Nordberg, J.; Arnér, E.S. Reactive oxygen species, antioxidants, and the mammalian thioredoxin. Free Radic. Biol. Med. 2001, 31, 1287–1312. [Google Scholar] [CrossRef]
- Nicholson, C.K.; Lambert, J.P.; Molkentin, J.D.; Sadoshima, J.; Calvert, J.W. Thioredoxin 1 Is Essential for Sodium Sulfide–Mediated Cardioprotection in the Setting of Heart Failure. Arter. Thromb. Vasc. Biol. 2013, 33, 744–751. [Google Scholar] [CrossRef]
- Xiao, L.; Dong, J.-H.; Jing-Hui, D.; Xue, H.-M.; Guo, Q.; Teng, X.; Wu, Y.-M. Hydrogen Sulfide Improves Endothelial Dysfunction via Downregulating BMP4/COX-2 Pathway in Rats with Hypertension. Oxidative Med. Cell. Longev. 2016, 2016, 1–10. [Google Scholar] [CrossRef]
- Al-Magableh, M.R.; Kemp-Harper, B.K.; Ng, H.H.; Miller, A.A.; Hart, J.L. Hydrogen sulfide protects endothelial nitric oxide function under conditions of acute oxidative stress in vitro. Naunyn. Schmiedebergs Arch. Pharmacol. 2013, 387, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Predmore, B.L.; Lefer, D.J.; Gojon, G. Hydrogen Sulfide in Biochemistry and Medicine. Antioxid. Redox Signal. 2012, 17, 119–140. [Google Scholar] [CrossRef]
- Haouzi, P.; Klingerman, C.M. Fate of intracellular H2S/HS− and metallo-proteins. Respir. Physiol. Neurobiol. 2013, 188, 229–230. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yoshikawa, S.; Shimada, A. Reaction mechanism of cytochrome c oxidase. Chem. Rev. 2015, 115, 1936–1989. [Google Scholar] [CrossRef] [PubMed]
- Módis, K.; Panopoulos, P.; Coletta, C.; Papapetropoulos, A.; Szabo, C. Hydrogen sulfide-mediated stimulation of mitochondrial electron transport involves inhibition of the mitochondrial phosphodiesterase 2A, elevation of cAMP and activation of protein kinase A. Biochem. Pharm. 2013, 86, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Vitvitsky, V.; Miljkovic, J.L.; Bostelaar, T.; Adhikari, B.; Yadav, P.K.; Steiger, A.K.; Torregrossa, R.; Pluth, M.D.; Whiteman, M.; Banerjee, R.; et al. Cytochrome c Reduction by H2S Potentiates Sulfide Signaling. ACS Chem. Biol. 2018, 13, 2300–2307. [Google Scholar] [CrossRef]
- Zhou, Z.; Martin, E.; Sharina, I.; Esposito, I.; Szabo, C.; Bucci, M.; Cirino, G.; Papapetropoulos, A. Regulation of soluble guanylyl cyclase redox state by hydrogen sulfide. Pharm. Res. 2016, 111, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Potor, L.; Mehes, G.; Hendrik, Z.; Jeney, V.; Pethő, D.; Vasas, A.; Pálinkás, Z.; Balogh, E.; Gyetvai, A.; Whiteman, M.; et al. Hydrogen Sulfide Abrogates Hemoglobin-Lipid Interaction in Atherosclerotic Lesion. Oxidative Med. Cell. Longev. 2018, 2018, 1–16. [Google Scholar] [CrossRef]
- Zhao, K.; Li, S.; Wu, L.; Lai, C.; Yang, G. Hydrogen Sulfide Represses Androgen Receptor Transactivation by Targeting at the Second Zinc Finger Module. J. Biol. Chem. 2014, 289, 20824–20835. [Google Scholar] [CrossRef]
- Bucci, M.; Papapetropoulos, A.; Vellecco, V.; Zhou, Z.; Pyriochou, A.; Roussos, C.; Roviezzo, F.; Brancaleone, V.; Cirino, G. Hydrogen Sulfide is an Endogenous Inhibitor of Phosphodiesterase Activity. Arter. Thromb. Vasc. Biol. 2010, 30, 1998–2004. [Google Scholar] [CrossRef]
- Yang, C.-T.; Devarie-Baez, N.O.; Hamsath, A.; Fu, X.-D.; Xian, M. S-Persulfidation: Chemistry, Chemical Biology, and Significance in Health and Disease. Antioxid. Redox Signal. 2020, 33, 1092–1114. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Liu, K.; He, J.; Tian, C.; Yu, X.; Yang, J. Direct Proteomic Mapping of Cysteine Persulfidation. Antioxid. Redox Signal. 2020, 33, 1061–1076. [Google Scholar] [CrossRef] [PubMed]
- Cuevasanta, E.; Lange, M.; Bonanata, J.; Coitiño, E.L.; Ferrer-Sueta, G.; Filipovic, M.R.; Alvarez, B. Reaction of Hydrogen Sulfide with Disulfide and Sulfenic Acid to Form the Strongly Nucleophilic Persulfide. J. Biol. Chem. 2015, 290, 26866–26880. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.-H.; Krokowski, D.; Guan, B.-J.; Bederman, I.R.; Majumder, M.; Parisien, M.; Diatchenko, L.; Kabil, O.; Willard, B.; Banerjee, R.; et al. Quantitative H2S-mediated protein sulfhydration reveals metabolic reprogramming during the integrated stress response. eLife 2015, 4, e10067. [Google Scholar] [CrossRef]
- Vandiver, M.S.; Paul, B.D.; Xu, R.; Karuppagounder, S.S.; Rao, F.; Snowman, A.M.; Ko, H.S.; Lee, Y.I.; Dawson, V.L.; Dawson, T.M.; et al. Sulfhydration mediates neuroprotective actions of parkin. Nat. Commun. 2013, 4, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Paul, B.D.; Gadalla, M.M.; Mustafa, A.K.; Sen, T.; Xu, R.; Kim, S.; Snyder, S.H. Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol. Cell. 2012, 45, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Chakraborty, P.K.; Xiong, X.; Dwivedi, S.K.; Mustafi, S.B.; Leigh, N.R.; Ramchandran, R.; Mukherjee, P.; Bhattacharya, R. Cystathionine β-synthase regulates endothelial function via protein S-sulfhydration. FASEB J. 2016, 30, 441–456. [Google Scholar] [CrossRef]
- Zhao, S.; Song, T.; Gu, Y.; Zhang, Y.; Cao, S.; Miao, Q.; Zhang, X.; Chen, H.; Gao, Y.; Zhang, L.; et al. Hydrogen sulfide alleviates liver injury via S-sulfhydrated-Keap1/Nrf2/LRP1 pathway. Hepatology 2020, 73, 282–302. [Google Scholar] [CrossRef]
- Davidson, L.M.; Berkelhamer, S.K. Bronchopulmonary Dysplasia: Chronic Lung Disease of Infancy and Long-Term Pulmonary Outcomes. J. Clin. Med. 2017, 6, 4. [Google Scholar] [CrossRef]
- Stoll, B.J.; Hansen, N.I.; Bell, E.F.; Shankaran, S.; Laptook, A.R.; Walsh, M.C.; Hale, E.C.; Newman, N.S.; Schibler, K.; Carlo, W.A.; et al. Neonatal Outcomes of Extremely Preterm Infants From the NICHD Neonatal Research Network. Pediatrics 2010, 126, 443–456. [Google Scholar] [CrossRef]
- Filbrun, A.G.; Popova, A.P.; Linn, M.J.; McIntosh, N.A.; Hershenson, M.B. Longitudinal measures of lung function in infants with bronchopulmonary dysplasia. Pediatr. Pulmonol. 2010, 46, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, A.; Martin, R.J. Vulnerability of the developing airway. Respir. Physiol. Neurobiol. 2019, 270, 103263. [Google Scholar] [CrossRef] [PubMed]
- Vento, M.; Moro, M.; Escrig, R.; Arruza, L.; Villar, G.; Izquierdo, I.; Roberts, L.J.; Arduini, A.; Escobar, J.J.; Sastre, J.; et al. Preterm Resuscitation With Low Oxygen Causes Less Oxidative Stress, Inflammation, and Chronic Lung Disease. Pediatrics 2009, 124, e439–e449. [Google Scholar] [CrossRef] [PubMed]
- Huizing, M.J.; Cavallaro, G.; Moonen, R.M.; González-Luis, G.E.; Mosca, F.; Vento, M.; Villamor, E. Is the C242T Polymorphism of the CYBA Gene Linked with Oxidative Stress-Associated Complications of Prematurity? Antioxid. Redox Signal. 2017, 27, 1432–1438. [Google Scholar] [CrossRef] [PubMed]
- Parinandi, N.L.; Kleinberg, M.A.; Usatyuk, P.V.; Cummings, R.J.; Pennathur, A.; Cardounel, A.J.; Zweier, J.L.; Garcia, J.G.N.; Natarajan, V. Hyperoxia-induced NAD(P)H oxidase activation and regulation by MAP kinases in human lung endothelial cells. Am. J. Physiol. Cell. Mol. Physiol. 2003, 284, L26–L38. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.A. DNA damage and cell cycle checkpoints in hyperoxic lung injury: Braking to facilitate repair. Am. J. Physiol. Cell. Mol. Physiol. 2001, 281, L291–L305. [Google Scholar] [CrossRef] [PubMed]
- Tyson, J.E.; Wright, L.L.; Oh, W.; Kennedy, K.A.; Mele, L.; Ehrenkranz, R.A.; Stoll, B.J.; Lemons, J.A.; Stevenson, D.K.; Bauer, C.R.; et al. Vitamin A Supplementation for Extremely-Low-Birth-Weight Infants. N. Engl. J. Med. 1999, 340, 1962–1968. [Google Scholar] [CrossRef]
- Ahola, T.; Lapatto, R.; Raivio, K.O.; Selander, B.; Stigson, L.; Jónsson, B.; Jonsbo, F.; Esberg, G.; Stövring, S.; Kjartansson, S.; et al. N-acetylcysteine does not prevent bronchopulmonary dysplasia in immature infants: A randomized controlled trial. J. Pediatr. 2003, 143, 713–719. [Google Scholar] [CrossRef]
- Davis, J.M.; Rosenfeld, W.N.; Richter, S.E.; Parad, R.; Gewolb, I.H.; Spitzer, A.R.; Carlo, W.A.; Couser, R.J.; Price, A.; Flaster, E.; et al. Safety and Pharmacokinetics of Multiple Doses of Recombinant Human CuZn Superoxide Dismutase Administered Intratracheally to Premature Neonates With Respiratory Distress Syndrome. Pediatrics 1997, 100, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Stey, C.; Steurer, J.; Bachmann, S.; Medici, T.; Tramèr, M. The effect of oral N-acetylcysteine in chronic bronchitis: A quantitative systematic review. Eur. Respir. J. 2000, 16, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Tse, H.N.; Tseng, C.Z.S. Update on the pathological processes, molecular biology, and clinical utility of N-acetylcysteine in chronic obstructive pulmonary disease. Int. J. Chronic. Obs. Pulm. Dis. 2014, 9, 825. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chessex, P.; Watson, C.; Kaczala, G.W.; Rouleau, T.; Lavoie, M.-È.; Friel, J.; Lavoie, J.-C. Determinants of oxidant stress in extremely low birth weight premature infants. Free Radic. Biol. Med. 2010, 49, 1380–1386. [Google Scholar] [CrossRef]
- Joung, K.E.; Kim, H.-S.; Lee, J.; Shim, G.H.; Choi, C.W.; Kim, E.-K.; Kim, B.I.; Choi, J.-H. Correlation of urinary inflammatory and oxidative stress markers in very low birth weight infants with subsequent development of bronchopulmonary dysplasia. Free Radic. Res. 2011, 45, 1024–1032. [Google Scholar] [CrossRef]
- Perrone, S.; Tataranno, M.L.; Negro, S.; Longini, M.; Marzocchi, B.; Proietti, F.; Iacoponi, F.; Capitani, S.; Buonocore, G. Early identification of the risk for free radical-related diseases in preterm newborns. Early Hum. Dev. 2010, 86, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Watterberg, K.L.; Demers, L.M.; Scott, S.M.; Murphy, S. Chorioamnionitis and early lung inflammation in infants in whom bronchopulmonary dysplasia develops. Pediatrics 1996, 97, 210–215. [Google Scholar] [PubMed]
- Roberts, D.; Brown, J.; Medley, N.; Dalziel, S.R. Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst. Rev. 2017, 3, CD004454. [Google Scholar] [CrossRef] [PubMed]
- Tindell, R.; Tipple, T. Selenium: Implications for outcomes in extremely preterm infants. J. Perinatol. 2018, 38, 197–202. [Google Scholar] [CrossRef]
- Darlow, B.A.; Austin, N. Selenium supplementation to prevent short-term morbidity in preterm neonates. Cochrane Database Syst. Rev. 2003, CD003312. [Google Scholar] [CrossRef]
- Amata, E.; Pittalà, V.; Marrazzo, A.; Parenti, C.; Prezzavento, O.; Arena, E.; Nabavi, S.M.; Salerno, L. Role of the Nrf2/HO-1 axis in bronchopulmonary dysplasia and hyperoxic lung injuries. Clin. Sci. 2017, 131, 1701–1712. [Google Scholar] [CrossRef]
- Olson, K.R.; Whitfield, N.L.; Bearden, S.E.; Leger, J.S.; Nilson, E.; Gao, Y.; Madden, J.A. Hypoxic pulmonary vasodilation: A paradigm shift with a hydrogen sulfide mechanism. Am. J. Physiol. Integr. Comp. Physiol. 2010, 298, R51–R60. [Google Scholar] [CrossRef]
- Liu, T.; Mukosera, G.T.; Blood, A.B. The role of gasotransmitters in neonatal physiology. Nitric Oxide 2020, 95, 29–44. [Google Scholar] [CrossRef]
- Olson, K.R. Hydrogen sulfide as an oxygen sensor. Antioxid. Redox Signal. 2015, 22, 377–397. [Google Scholar] [CrossRef]
- Madden, J.A.; Ahlf, S.B.; Dantuma, M.W.; Olson, K.R.; Roerig, D.L. Precursors and inhibitors of hydrogen sulfide synthesis affect acute hypoxic pulmonary vasoconstriction in the intact lung. J. Appl. Physiol. 2012, 112, 411–418. [Google Scholar] [CrossRef]
- Dyson, R.M.; Palliser, H.K.; Latter, J.L.; Chwatko, G.; Glowacki, R.; Wright, I.M.R. A Role for H2S in the Microcirculation of Newborns: The Major Metabolite of H2S (Thiosulphate) is Increased in Preterm Infants. PLoS ONE 2014, 9, e105085. [Google Scholar] [CrossRef]
- Hilgendorff, A.; Reiss, I.; Ehrhardt, H.; Eickelberg, O.; Alvira, C.M. Chronic Lung Disease in the Preterm Infant: Lessons Learned From Animal Models. Am. J. Respir. Cell Mol. Biol. 2013, 50, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Prabhudesai, S.; Zhang, Y.; Rao, G.; Thirugnanam, K.; Hossen, M.N.; Dwivedi, S.K.D.; Ramchandran, R.; Mukherjee, P.; Bhattacharya, R. Cystathione beta-synthase regulates HIF-1alpha stability through persulfidation of PHD2. Sci. Adv. 2020, 6, eaaz8534. [Google Scholar] [CrossRef] [PubMed]
- Floen, M.J.; Forred, B.J.; Bloom, E.J.; Vitiello, P.F. Thioredoxin-1 redox signaling regulates cell survival in response to hyperoxia. Free Radic. Biol. Med. 2014, 75, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Forred, B.J.; Daugaard, D.R.; Titus, B.K.; Wood, R.R.; Floen, M.J.; Booze, M.L.; Vitiello, P.F. Detoxification of Mitochondrial Oxidants and Apoptotic Signaling Are Facilitated by Thioredoxin-2 and Peroxiredoxin-3 during Hyperoxic Injury. PLoS ONE 2017, 12, e0168777. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wall, S.B.; Ren, C.; Velten, M.; Hill, C.L.; Locy, M.L.; Rogers, L.K.; Tipple, T.E. Thioredoxin Reductase Inhibition Attenuates Neonatal Hyperoxic Lung Injury and Enhances Nuclear Factor E2–Related Factor 2 Activation. Am. J. Respir. Cell Mol. Biol. 2016, 55, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Tipple, T.E. The Thioredoxin System in Neonatal Lung Disease. Antioxid. Redox Signal. 2014, 21, 1916–1925. [Google Scholar] [CrossRef]
- Faller, S.; Ryter, S.W.; Choi, A.M.K.; Loop, T.; Schmidt, R.; Hoetzel, A. Inhaled Hydrogen Sulfide Protects against Ventilator-induced Lung Injury. Anesthesiology 2010, 113, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Spassov, S.; Pfeifer, D.; Strosing, K.; Ryter, S.; Hummel, M.; Faller, S.; Hoetzel, A. Genetic Targets of Hydrogen Sulfide in Ventilator-Induced Lung Injury—A Microarray Study. PLoS ONE 2014, 9, e102401. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Sun, J.; Fei, A.; Gao, C.; Pan, S.; Wu, Z. Hydrogen sulfide treatment alleviated ventilator-induced lung injury through regulation of autophagy and endoplasmic reticulum stress. Int. J. Biol. Sci. 2019, 15, 2872–2884. [Google Scholar] [CrossRef] [PubMed]
- Guan, R.; Wang, J.; Li, D.; Li, Z.; Liu, H.; Ding, M.; Cai, Z.; Liang, X.; Yang, Q.; Long, Z.; et al. Hydrogen sulfide inhibits cigarette smoke-induced inflammation and injury in alveolar epithelial cells by suppressing PHD2/HIF-1α/MAPK signaling pathway. Int. Immunopharmacol. 2020, 81, 105979. [Google Scholar] [CrossRef]
- Lin, F.; Liao, C.; Sun, Y.; Zhang, J.; Lu, W.; Bai, Y.; Liao, Y.; Li, M.; Ni, X.; Hou, Y.; et al. Hydrogen Sulfide Inhibits Cigarette Smoke-Induced Endoplasmic Reticulum Stress and Apoptosis in Bronchial Epithelial Cells. Front. Pharm. 2017, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, P.; Yang, G.; Cao, Q.; Wang, R. The Inhibitory Role of Hydrogen Sulfide in Airway Hyperresponsiveness and Inflammation in a Mouse Model of Asthma. Am. J. Pathol. 2013, 182, 1188–1195. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Wu, R.; Geng, B.; Qi, Y.-F.; Wang, P.-P.; Yao, W.-Z.; Tang, C.-S. Endogenous hydrogen sulfide reduces airway inflammation and remodeling in a rat model of asthma. Cytokine 2009, 45, 117–123. [Google Scholar] [CrossRef]
- Mendes, J.A.; Ribeiro, M.C.; Filho, G.J.R.; Rocha, T.; Muscará, M.N.; Costa, S.K.; Ferreira, H.H. Hydrogen sulfide inhibits apoptosis and protects the bronchial epithelium in an allergic inflammation mice model. Int. Immunopharmacol. 2019, 73, 435–441. [Google Scholar] [CrossRef]
- Saito, J.; Zhang, Q.; Hui, C.; Macedo, P.; Gibeon, D.; Menzies-Gow, A.; Bhavsar, P.K.; Chung, K.F. Sputum hydrogen sulfide as a novel biomarker of obstructive neutrophilic asthma. J. Allergy Clin. Immunol. 2013, 131, 232–234.e3. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Wang, Y.; Lu, Y.-Q.; Yan, M.; Jiang, Y.-H.; Zhao, D.-Y. Correlation between serum H2S and pulmonary function in children with bronchial asthma. Mol. Med. Rep. 2012, 6, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Chen, Y.; Yao, W. Correlation between levels of exhaled hydrogen sulfide and airway inflammatory phenotype in patients with chronic persistent asthma. Respirology 2014, 19, 1165–1169. [Google Scholar] [CrossRef]
- Ivanciuc, T.; Sbrana, E.; Ansar, M.; Bazhanov, N.; Szabo, C.; Casola, A.; Garofalo, R.P. Hydrogen Sulfide Is an Antiviral and Antiinflammatory Endogenous Gasotransmitter in the Airways. Role in Respiratory Syncytial Virus Infection. Am. J. Respir. Cell Mol. Biol. 2016, 55, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Oliver, E.; Ma, Y.; Escaffre, O.; Ivanciuc, T.; Komaravelli, N.; Kelley, J.P.; Coletta, C.; Szabo, C.; Rockx, B.; Garofalo, R.P.; et al. Role of Hydrogen Sulfide in Paramyxovirus Infections. J. Virol. 2015, 89, 5557–5568. [Google Scholar]
- Yang, G. H2S as a potential defense against COVID-19? Am. J. Physiol. Physiol. 2020, 319, C244–C249. [Google Scholar] [CrossRef] [PubMed]
- Renieris, G.; Katrini, K.; Damoulari, C.; Akinosoglou, K.; Psarrakis, C.; Kyriakopoulou, M.; Dimopoulos, G.; Lada, M.; Koufargyris, P.; Giamarellos-Bourboulis, E.J. Serum Hydrogen Sulfide and Outcome Association in Pneumonia by the SARS-CoV-2 Coronavirus. Shock 2020, 54, 633–637. [Google Scholar] [CrossRef]
- Citi, V.; Martelli, A.; Brancaleone, V.; Brogi, S.; Gojon, G.; Montanaro, R.; Morales, G.; Testai, L.; Calderone, V. Anti-inflammatory and antiviral roles of hydrogen sulfide: Rationale for considering H 2 S donors in COVID-19 therapy. Br. J. Pharm. 2020, 177, 4931–4941. [Google Scholar] [CrossRef] [PubMed]
- Zlotkin, S.H.; Anderson, G.H. The Development of Cystathionase Activity during the First Year of Life. Pediatr. Res. 1982, 16, 65–68. [Google Scholar] [CrossRef]
- Viña, J.; Vento, M.; García-Sala, F.; Puertes, I.R.; Gascó, E.; Sastre, J.; Asensi, M.; Pallardó, F.V. L-cysteine and glutathione metabolism are impaired in premature infants due to cystathionase deficiency. Am. J. Clin. Nutr. 1995, 61, 1067–1069. [Google Scholar] [CrossRef]
- Vadivel, A.; Alphonse, R.S.; Ionescu, L.; Machado, D.S.; O’Reilly, M.; Eaton, F.; Haromy, A.; Michelakis, E.D.; Thébaud, B. Exogenous Hydrogen Sulfide (H2S) Protects Alveolar Growth in Experimental O2-Induced Neonatal Lung Injury. PLoS ONE 2014, 9, e90965. [Google Scholar] [CrossRef]
- Schiliro, M.; Bartman, C.M.; Pabelick, C. Understanding hydrogen sulfide signaling in neonatal airway disease. Expert Rev. Respir. Med. 2021, 15, 351–372. [Google Scholar] [CrossRef]
- Ardini-Poleske, M.E.; Clark, R.F.; Ansong, C.; Carson, J.P.; Corley, R.A.; Deutsch, G.H.; Hagood, J.S.; Kaminski, N.; Mariani, T.J.; Potter, S.S.; et al. LungMAP: The Molecular Atlas of Lung Development Program. Am. J. Physiol. Cell. Mol. Physiol. 2017, 313, L733–L740. [Google Scholar] [CrossRef]
- Renga, B.; Cipriani, S.; Carino, A.; Simonetti, M.; Zampella, A.; Fiorucci, S. Reversal of Endothelial Dysfunction by GPBAR1 Agonism in Portal Hypertension Involves a AKT/FOXOA1 Dependent Regulation of H2S Generation and Endothelin-1. PLoS ONE 2015, 10, e0141082. [Google Scholar] [CrossRef] [PubMed]
- Sen, N. Functional and Molecular Insights of Hydrogen Sulfide Signaling and Protein Sulfhydration. J. Mol. Biol. 2017, 429, 543–561. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Hu, Q.; Zhu, D. An Update on Hydrogen Sulfide and Nitric Oxide Interactions in the Cardiovascular System. Oxidative Med. Cell. Longev. 2018, 2018, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Rawlins, E.L.; Perl, A.K. The a“MAZE”ing world of lung-specific transgenic mice. Am. J. Respir. Cell Mol. Biol. 2012, 46, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-Y.; Jedlicka, A.E.; Gladwell, W.; Marzec, J.; McCaw, Z.R.; Bienstock, R.J.; Kleeberger, S.R. Association of Nrf2 polymorphism haplotypes with acute lung injury phenotypes in inbred strains of mice. Antioxid. Redox Signal. 2015, 22, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Leary, S.; Das, P.; Ponnalagu, D.; Singh, H.; Bhandari, V. Genetic Strain and Sex Differences in a Hyperoxia-Induced Mouse Model of Varying Severity of Bronchopulmonary Dysplasia. Am. J. Pathol. 2019, 189, 999–1014. [Google Scholar] [CrossRef]
- Ronchi, J.A.; Figueira, T.R.; Ravagnani, F.G.; Oliveira, H.C.; Vercesi, A.E.; Castilho, R.F. A spontaneous mutation in the nicotinamide nucleotide transhydrogenase gene of C57BL/6J mice results in mitochondrial redox abnormalities. Free Radic. Biol. Med. 2013, 63, 446–456. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ganguly, A.; Ofman, G.; Vitiello, P.F. Hydrogen Sulfide—Clues from Evolution and Implication for Neonatal Respiratory Diseases. Children 2021, 8, 213. https://doi.org/10.3390/children8030213
Ganguly A, Ofman G, Vitiello PF. Hydrogen Sulfide—Clues from Evolution and Implication for Neonatal Respiratory Diseases. Children. 2021; 8(3):213. https://doi.org/10.3390/children8030213
Chicago/Turabian StyleGanguly, Abhrajit, Gaston Ofman, and Peter F Vitiello. 2021. "Hydrogen Sulfide—Clues from Evolution and Implication for Neonatal Respiratory Diseases" Children 8, no. 3: 213. https://doi.org/10.3390/children8030213
APA StyleGanguly, A., Ofman, G., & Vitiello, P. F. (2021). Hydrogen Sulfide—Clues from Evolution and Implication for Neonatal Respiratory Diseases. Children, 8(3), 213. https://doi.org/10.3390/children8030213