Abstract
Background/Objectives: Cardiac amyloidosis (CA) is an underdiagnosed and potentially life-threatening infiltrative cardiomyopathy characterized by the extracellular deposition of misfolded amyloid fibrils in cardiac tissue. It is most commonly associated with light-chain (AL) amyloidosis and transthyretin (ATTR) amyloidosis, either hereditary or wild-type. The disease often presents with non-specific symptoms, leading to delayed diagnosis and treatment. This study aims to provide a comprehensive overview of the pathophysiology, diagnostic strategies, and current therapeutic approaches for cardiac amyloidosis, with a focus on improving early detection and clinical outcomes. Methods: A narrative review was conducted using databases such as PubMed and Scopus, covering the period from September 2016 to March 2025. Keywords such as “cardiac amyloidosis”, “cardiac amyloidosis from transthyretin”, “cardiomyopathy”, “transthyretin”, “immunoglobulin light-chain amyloidosis”, and “familial amyloidosis” were used. Relevant clinical trials and guideline-based management recommendations were also included. Results: This review highlights that non-invasive imaging modalities and serum biomarker analyses are key to reducing diagnostic delays. New therapeutic developments, including gene-editing technologies and RNA-based therapies, show promise in early trials. Multidisciplinary management and increased awareness are crucial for timely diagnosis and treatment optimization. Conclusions: The early recognition of cardiac amyloidosis remains a major clinical challenge. Advances in non-invasive diagnostics and emerging disease-modifying therapies are transforming the prognosis of affected patients. Continued research and heightened clinical suspicion are essential to improve outcomes in this complex and heterogeneous disease.
1. Introduction
Amyloidosis is a disorder characterized by the extracellular accumulation of fibrils composed of low-molecular-weight subunits derived from various serum proteins. This condition can affect multiple organs, with cardiac, renal, hepatic, and autonomic nervous system involvements being the primary contributors to morbidity and mortality. Several factors increase the risk of developing amyloidosis, including advanced age, male sex, African ancestry, chronic or infectious diseases, and a family history of the condition, as certain types of amyloidosis are hereditary [1]. Moreover, the transthyretin (TTR) Ile122 variant—strongly associated with cardiac amyloidosis (CA) in individuals of African descent—has an allele frequency of 66/3376 (2.0%) among African-Americans across the U.S., suggesting it is a common, often unrecognized contributor to cardiac disease in this population [2].
CA is characterized by the extracellular buildup of misfolded proteins within the heart, marked by a unique histological feature: green birefringence under cross-polarized light following Congo red staining. CA involves the extracellular accumulation of fibrillar and insoluble protein aggregates within the myocardium, leading to cardiac dysfunction [3]. Although over 30 types of amyloidogenic proteins have been identified [4], five of them affect the heart, including immunoglobulin heavy and light chain (AL), transthyretin (TTR), amyloid A, and apolipoprotein A1. Among these, the AL and ATTR types (wild-type [ATTRwt] and hereditary/variant [ATTRv]) account for 95% of all CA cases [5]. The pathogenesis of CA starts with the misfolding of precursor proteins, which form insoluble fibrils that deposit throughout the myocardial interstitium and perivascular spaces, ultimately disrupting the normal tissue architecture and compliance [6,7]. As these deposits accumulate, the heart develops a restrictive physiology characterized by impaired ventricular filling and, over time, systolic dysfunction [8]. Despite its clinical significance, CA is often underdiagnosed: the systematic screening of older adults with heart failure and preserved ejection fraction (HFpEF) has revealed CA in up to 16% of these patients, underscoring a significant diagnostic gap in this population [9].
The infiltrative process in the heart results in progressive myocardial dysfunction, often accompanied by the impairment of the cardiac conduction system. Amyloid cardiomyopathy is increasingly recognized as a significant yet frequently underdiagnosed cause of heart failure and cardiac arrhythmias, particularly in older adults. In parallel with mechanical impairment, amyloid fibrils infiltrate the cardiac conduction system—including the sinoatrial and atrioventricular nodes and the His–Purkinje network—creating areas of conduction block and promoting reentrant circuits [9]. As a result, atrial fibrillation (AF) arises in up to 73% of patients with transthyretin amyloidosis, especially those of an advanced age, a higher disease stage, and enlarged left atrial volumes [10]. The combination of a poor rate control tolerance and a markedly elevated stroke risk makes AF management challenging; current evidence supports early rhythm-control strategies and indicates that novel oral anticoagulants provide a thromboembolic protection comparable to warfarin with fewer major bleedings, while timely AF ablation may reduce both mortality and heart-failure hospitalizations [10,11]. Conduction blocks and ventricular tachyarrhythmias also occur frequently, often necessitating permanent pacing for symptomatic bradyarrhythmias or advanced atrioventricular blocks [12]. Although implantable cardioverter-defibrillators (ICDs) are used for sudden cardiac death (SCD) prevention, their impact on the overall survival in CA remains uncertain, as a high mortality persists despite device therapy [13].
Although traditionally regarded as a rare condition, emerging evidence indicates that cardiac amyloidosis is often overlooked as a contributing factor to common cardiac diseases and syndromes. Recent advancements in cardiac imaging, diagnostic techniques, and therapeutic approaches have significantly enhanced the identification and management of this condition [14].
Advancements in cardiac imaging and increased physician awareness have significantly improved the diagnosis of cardiac amyloidosis over the past decade. Cardiac involvement in amyloidosis is associated with a poor prognosis, making early detection essential. A timely diagnosis is critical for implementing appropriate treatment strategies that can potentially alter the disease’s natural course and improve patient outcomes [15]. This review highlights that non-invasive imaging modalities and serum biomarker analyses are key to reducing diagnostic delays. New therapeutic developments, including gene-editing technologies and RNA-based therapies, show promise in early trials. Multidisciplinary management and increased awareness are crucial for timely diagnosis and treatment optimization.
2. Materials and Methods
This work was designed as a narrative review, with its primary focus being cardiac amyloidosis. A comprehensive literature search was independently performed by two researchers using the PubMed and Scopus databases from September 2016 to March 2025. The search strategy included a range of relevant keyword combinations, such as “cardiac amyloidosis”, “cardiac amyloidosis from transthyretin”, “cardiomyopathy”, “transthyretin”, “immunoglobulin light-chain amyloidosis”, and “familial amyloidosis”.
Review articles and meta-analyses were included. Articles were excluded based on the following criteria: non-English language publications, case reports, editorials, or studies lacking relevance to the review topic. After applying the exclusion criteria, a total of 37 studies were included in the final review.
3. Clinical and Multimodal Imaging Features of Cardiac Amyloidosis
The diagnostic assessment of a patient with suspected cardiac amyloidosis begins with a comprehensive clinical evaluation—a detailed history and physical examination—to identify and assess the key cardiac and extracardiac manifestations of AL and ATTR amyloidosis, as summarized in Table 1 [16].

Table 1.
Clinical manifestations of AL and ATTR amyloidosis (in order of frequency).
The diagnosis of cardiac amyloidosis requires a high level of clinical suspicion but is often delayed due to limited disease awareness and the diverse range of presenting symptoms. Many patients remain asymptomatic until the disease has significantly progressed, and even at that stage, symptoms are often non-specific, further complicating early detection [17].
The key cardiac findings are summarized in Table 2. One of the hallmark electrocardiographic features raising the suspicion of cardiac amyloidosis is disproportionately low QRS voltages relative to the left ventricular (LV) wall thickness. Echocardiography serves as the initial imaging modality for suspected cardiac amyloidosis and can reveal various abnormalities, including LV hypertrophy with or without right ventricular (RV) free wall hypertrophy, bi-atrial dilation, myocardial granular sparkling on non-harmonic imaging, thickened mitral and tricuspid valve leaflets, atrial septal thickening, low-flow/low-gradient aortic stenosis, diastolic dysfunction of varying severity, and abnormal left ventricular global longitudinal strain, typically exhibiting an apical sparing pattern [17,18].
Table 2.
Main findings in cardiac amyloidosis.
Further tissue characterization using cardiovascular magnetic resonance (CMR) imaging may reveal a diffuse subendocardial or transmural late gadolinium enhancement, along with an increased extracellular volume as assessed by T1 mapping, providing additional diagnostic insight [19].
4. Diagnostic Criteria for Cardiac Amyloidosis
Although over thirty proteins are known to aggregate as amyloid in vivo, only nine of these amyloidogenic proteins accumulate in the myocardium and lead to clinically significant cardiac disease.
However, certain forms of cardiac amyloidosis, such as AApoAI, AApoAII, AApoAIV, Aβ2M, AFib, and AGel, are extremely rare. Additionally, cardiac amyloidosis secondary to chronic inflammatory or infectious diseases (AA) has become much less common [20,21,22]. Currently, over 98% of diagnosed cases of cardiac amyloidosis are attributed to fibrils composed of either monoclonal immunoglobulin light chains (AL) or transthyretin (ATTR), which can occur in hereditary (ATTRv) or acquired (ATTRwt) forms [22]. Table 1 outlines the key characteristics of each type of cardiac amyloidosis.
Cardiac amyloidosis is confirmed by the presence of amyloid fibrils within cardiac tissue. Diagnostic criteria can be categorized into invasive and non-invasive methods. The invasive criteria, which require a histological confirmation of amyloid through a biopsy, apply to all CA subtypes. In contrast, the non-invasive criteria—characterized by typical echo/CMR findings combined with Grade 2–3 uptake on 99mTc-PYP/DPD/HMDP scintigraphy and the exclusion of a monoclonal protein—are validated exclusively for ATTR amyloidosis [21].
4.1. Non-Invasive Diagnostic Criteria
Cardiac ATTR amyloidosis can be diagnosed without histological confirmation if typical echocardiographic or cardiac magnetic resonance (CMR) findings are present, and scintigraphy with 99mTc-Pyrophosphate (PYP), 99mTc-DPD, or 99mTc-HMDP shows a Grade 2 or 3 myocardial radiotracer uptake. Additionally, clonal dyscrasia must be ruled out through the following tests: serum-free light chain (FLC) assay, serum protein electrophoresis with immunofixation (SPIE), and urine protein electrophoresis with immunofixation (UPIE). The combined use of SPIE, UPIE, and serum FLC quantification achieves a 99% sensitivity for identifying abnormal pro-amyloidotic precursors in AL amyloidosis. Importantly, serum and urine electrophoresis should always include immunofixation to enhance sensitivity for detecting monoclonal proteins [23,24]. The initial diagnostic steps involve nuclear scintigraphy using 99mTc-PYP, 99mTc-DPD, or 99mTc-HMDP to assess myocardial uptake, alongside a monoclonal protein assessment through serum protein electrophoresis with immunofixation (SPIE), urine protein electrophoresis with immunofixation (UPIE), and the quantification of serum-free light chains (FLCs) [25,26]. The interpretation of results involves identifying AL amyloidosis in cases with positive monoclonal proteins, necessitating a hematologic evaluation and confirmatory biopsy, while the absence of monoclonal proteins with Grade 2/3 uptake on scintigraphy suggests ATTR amyloidosis, requiring genetic testing to differentiate between wild-type (ATTRwt) and variant (ATTRv) forms. If results remain inconclusive, an endomyocardial or extracardiac biopsy should be performed for a definitive diagnosis and amyloid typing. This structured diagnostic approach ensures the accurate identification of cardiac amyloidosis subtypes, facilitating the timely initiation of targeted treatment strategies [27], based on the results of scintigraphy and monoclonal protein assessments.
4.2. Invasive Diagnostic Criteria
Cardiac amyloidosis is confirmed through an endomyocardial biopsy that reveals amyloid deposits via Congo red staining, irrespective of the extent of the left ventricular (LV) wall thickness (Figure 1). A significant number of patients with CA require tissue sampling for diagnosis, and histological typing can provide prognostic information and guide treatment [28]. Treatments are available for many types of amyloid but are specific to each type; thus, the confirmation and typing of amyloid are crucial before starting therapy [29]. Patients with AL amyloidosis are particularly susceptible to the cardiovascular toxicity associated with AL-directed chemotherapy (daratumumab-CyBorD) [30].
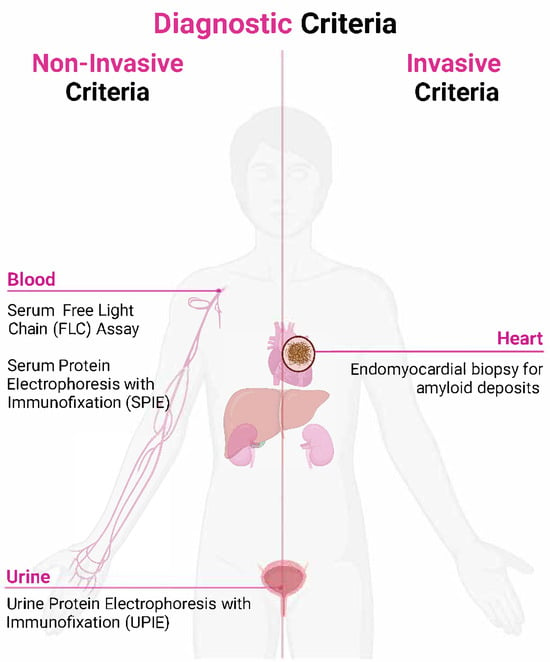
Figure 1.
Diagnostic criteria for cardiac amyloidosis.
Upon the detection of amyloid deposits, precise fibril typing is essential to guide therapy: a laser-capture microdissection coupled with mass spectrometry-based proteomics is now considered the gold standard for amyloid typing, offering unparalleled sensitivity and specificity [31]. While immunohistochemistry and immunoelectron microscopy remain valuable, more widely available methods for routine classification in specialized centers is common [23]. Once amyloid is detected, the fibril protein must be classified. While mass spectrometry remains the gold standard for amyloid typing, immunohistochemistry and immunoelectron microscopy are commonly used in specialized centers for routine identification [23].
A diagnosis can also be established if amyloid deposits are identified in an extracardiac biopsy, provided there are characteristic features of cardiac amyloidosis observed on echocardiography—without another explanation for an increased LV wall thickness or distinctive findings in cardiac magnetic resonance (CMR) imaging [24,32,33].
A recent multicenter study proposed an echocardiographic scoring system to aid in diagnosing AL or ATTR amyloidosis in cases of increased LV wall thickness. Although not yet externally validated, a score of ≥ 8 points, combined with an LV wall thickness ≥ 12 mm and amyloid deposits confirmed by an extracardiac biopsy, may also be considered diagnostic for cardiac amyloidosis [32,33,34].
4.3. Challenges in Interpretation
Low-level monoclonal proteins or mild abnormalities in the kappa–lambda FLC ratio can occur in patients with chronic kidney disease (CKD) or monoclonal gammopathy of undetermined significance (MGUS). In CKD, a reduced glomerular filtration rate (GFR) leads to the decreased renal clearance of polyclonal FLCs, causing serum levels to rise. The FLC ratio varies based on the assay used. For the Freelite Assay (Binding Site), the FLC ratio increases with a declining GFR. A range of 0.37–3.1 is considered normal for patients with CKD. For the N Latex Assay (Siemens), the FLC ratio decreases as the GFR declines, but no reference range has been proposed for CKD. For patients with moderate CKD (eGFR < 45 mL/min/1.73 m2 using the CKD-EPI formula), a FLC ratio up to 2.0 (or 3.1 in dialysis) can typically be regarded as normal in the context of normal SPIE/UPIE results [24,25].
If FLC findings are unclear or abnormal, a consultation with a hematologist is recommended for further evaluation. In the absence of a detectable monoclonal protein and an abnormal serum FLC ratio, the specificity of Grade 2 or 3 bone scintigraphy for diagnosing cardiac ATTR amyloidosis in suspected cases is estimated to be nearly 100%. However, it is crucial to perform scintigraphy with single-photon emission computed tomography (SPECT) to ensure that the observed uptake corresponds to the myocardium and not the cardiac chambers [35]. It is important to note that rare conditions may also cause a positive cardiac uptake on scintigraphy. These alternative causes should always be considered when interpreting the results to avoid misdiagnosis [36]. Once cardiac ATTR amyloidosis is confirmed, genetic counseling and testing should be conducted to identify TTR mutations, enabling a differentiation between wild-type ATTR (ATTRwt) and hereditary ATTR (ATTRv). Genetic testing is recommended even in elderly patients, as a notable proportion may carry TTR mutations [37,38,39].
The diagnostic process for cardiac amyloidosis involves two key phases:
- Suspicion phase: identifying clinical and imaging features that raise suspicion for cardiac amyloidosis.
- Definitive diagnosis phase: confirming the presence of amyloid deposits and accurately typing the amyloid fibrils to guide targeted treatments [40].
Cardiac amyloidosis is frequently associated with extracardiac manifestations that, in conjunction with characteristic cardiac imaging findings, can significantly raise clinical suspicion. These manifestations, referred to as “red flags”, serve as critical indicators for the early recognition of the disease (Figure 2).
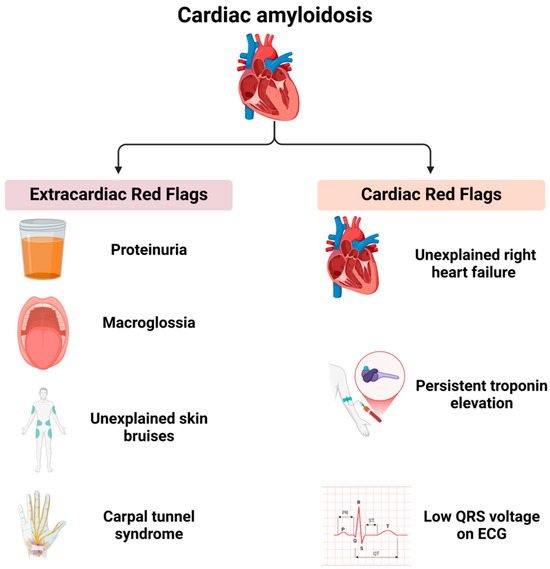
Figure 2.
Red flags in cardiac amyloidosis.
Extracardiac red flags include proteinuria (even if mild), macroglossia (enlarged tongue), unexplained skin bruising, and carpal tunnel syndrome. Cardiac red flags comprise heart failure with disproportionately elevated N-terminal pro-B-type natriuretic peptide (NT-proBNP) levels relative to echocardiographic findings, unexplained right heart failure despite normal ventricular and valvular function, idiopathic pericardial effusion, persistent troponin elevation, a low QRS voltage on an electrocardiogram (ECG) disproportionate to the left ventricular (LV) wall thickness, and an early-onset conduction system disease. The recognition of these extracardiac and cardiac red flags should prompt a further evaluation for cardiac amyloidosis, particularly when imaging findings are suggestive of the condition [41,42,43,44]. Beyond specific red flags, certain clinical scenarios warrant a heightened suspicion for cardiac amyloidosis. Cardiac disease occurring in the context of a systemic condition, such as plasma cell dyscrasia, nephrotic syndrome, peripheral neuropathy, or chronic systemic inflammatory conditions, should prompt a thorough diagnostic workup for amyloidosis when accompanied by compatible cardiac imaging findings. An increased left ventricular wall thickness in a non-dilated left ventricle is a hallmark feature of cardiac amyloidosis and necessitates further investigation, particularly in elderly patients presenting with heart failure with preserved ejection fraction (HFpEF), hypertrophic cardiomyopathy, or severe aortic stenosis. Patients undergoing a transcatheter aortic valve replacement (TAVR) should be evaluated for cardiac amyloidosis, given that transthyretin (ATTR) amyloidosis has been identified in 7–19% of such cases [44]. Non-invasive diagnostic methods facilitate early identification, and individuals with an increased wall thickness presenting with heart failure, aortic stenosis, or red flag symptoms should be assessed for cardiac amyloidosis, particularly those over the age of 65 [45].
About 5% of patients with a history of bilateral carpal tunnel syndrome are also later found to have cardiac amyloidosis [46].
The early differentiation of AL versus ATTR amyloidosis is essential. Since most cases of cardiac amyloidosis are attributed to AL or ATTR subtypes, the diagnostic strategy primarily focuses on distinguishing between these forms. Begin with serum and urine immunofixation plus free light-chain quantification to detect a monoclonal protein, and perform bone scintigraphy (99mTc-PYP/DPD/HMDP) for myocardial uptake. A positive monoclonal protein directs the patient to a hematology and biopsy confirmation for AL amyloidosis. In contrast, an isolated Grade 2–3 tracer uptake without a monoclonal protein confirms ATTR amyloidosis and prompts genetic testing (ATTRv vs. ATTRwt). If either pathway remains equivocal, proceed to an endomyocardial or extracardiac biopsy with amyloid typing. Table 3 summarizes these four diagnostic scenarios.
Table 3.
Scenarios that can guide the diagnostic pathway of cardiac amyloidosis.
5. Discussion
Cardiac amyloidosis (CA) is a complex and progressive infiltrative cardiomyopathy caused by the extracellular deposition of amyloid fibrils within myocardial tissue, leading to restrictive physiology and progressive heart failure. The early recognition and differentiation of subtypes are critical for guiding treatment and improving outcomes in this frequently underdiagnosed condition.
The main subtypes of cardiac amyloidosis include immunoglobulin light-chain (AL) amyloidosis and transthyretin (ATTR) amyloidosis. AL amyloidosis arises from a clonal plasma cell disorder producing misfolded light chains that deposit systemically, often associated with multiple myeloma. In contrast, ATTR amyloidosis results from misfolded transthyretin proteins, which can be inherited (variant, ATTRv) or acquired (wild-type, ATTRwt) forms [1,14]. While AL amyloidosis progresses rapidly and is potentially fatal without prompt therapy, ATTR amyloidosis often has a more indolent course, particularly the wild-type form.
Clinically, CA may present with non-specific symptoms, such as exertional dyspnea, fatigue, edema, and signs of heart failure with preserved ejection fraction (HFpEF), contributing to diagnostic delays [47,48]. Arrhythmias, particularly atrial fibrillation and conduction abnormalities, are also common. Extracardiac manifestations—including neuropathy, carpal tunnel syndrome, and nephrotic-range proteinuria—may offer important diagnostic clues.
A systematic review and meta-analysis by Antonopoulos et al. (2022) [49] confirmed that ATTR-CA is the most common form of cardiac amyloidosis, although AL-CA may account for up to 18% of cases. The average age at diagnosis is typically between 74 and 90 years, with a clear male predominance (50–100%). ATTR-CA is more prevalent than previously believed, particularly among patients with HFpEF, severe aortic stenosis (AS), or hypertrophic cardiomyopathy [49].
The diagnostic approach to CA is multimodal. Echocardiography (ECHO) is generally the first-line tool, but findings such as an increased wall thickness, elevated left ventricular mass index (LVMI), and diastolic dysfunction are often non-specific and may overlap with hypertensive heart disease or hypertrophic cardiomyopathy [14,50]. Jaiswal et al. (2022) [51] demonstrated that subtle echocardiographic differences in wall thickness and diastolic parameters may help screen AS patients for CA. Still, optimal thresholds for these markers require further validation [51].
Advanced cardiac imaging has substantially improved diagnostic accuracy. Cardiac magnetic resonance imaging (CMR) with late gadolinium enhancement enables the visualization of amyloid infiltration and tissue characterization. Tissue mapping techniques—such as T1 mapping and extracellular volume (ECV) quantification—have prognostic value. Studies found that elevated native T1 times, a higher ECV, and lower myocardial-to-skeletal T2 ratios correlate with worse outcomes, reinforcing the importance of CMR in risk stratification [52].
Positron emission tomography (PET), particularly using tracers like florbetapir, is a promising modality in CA diagnosis. A systematic review by Kim et al. (2020) [53] highlighted the utility of PET, especially when paired with a semi-quantitative analysis, in differentiating between AL and ATTR amyloidosis. These results support the use of PET alongside CMR and bone scintigraphy to enhance diagnostic precision [53].
An invasive tissue biopsy remains the gold standard for diagnosis, with Congo red staining and immunohistochemical subtyping performed on samples from the abdominal fat pad, rectal mucosa, or endomyocardial tissue. While a fat pad biopsy is less invasive, it may have a reduced sensitivity in ATTR cases, necessitating a myocardial biopsy in selected patients [54,55]. Fine-needle fat aspiration is integrated into CA diagnosis, reliably documenting amyloid deposits and indicating cardiac involvement when combined with imaging [56]. The sensitivity is 85.1% and specificity is 97.1% under optimal conditions [40]. The abdominal fat pad excisional biopsy (FPEB) and fat fine-needle biopsy identify and classify CA, offering simplicity, low costs, and minimal complications [56]. In AL-CA patients, the abdominal fat fine-needle aspiration shows a 100% specificity and an 84% sensitivity, though a lower sensitivity for ATTR (45% for ATTRv and 15% for ATTRwt) [56]. Fine-needle aspiration provides better material for amyloid typing than FPEB. FPEB’s sensitivity for AL amyloidosis varies from 50% (<700 mm3) to 100% (>700 mm3) [56,57]. The initial surgical sample should be 1400 mm3 to allow division for immunofluorescence, mass spectrometry, and electron microscopy studies.
The importance of subtype differentiation is underscored by Xin et al. (2019) [58], who demonstrated the distinct prognostic implications of AL vs. ATTR cardiac amyloidosis. AL amyloidosis requires urgent chemotherapy targeting the plasma cell clone, while ATTR amyloidosis may be managed with transthyretin-stabilizing agents. Tafamidis and patisiran, for example, have demonstrated survival benefits in ATTR-CA, particularly in early stages of the disease [1,49]. Diflunisal and inotersen are also being considered as therapeutic options in clinical evaluations; however, their substantial out-of-pocket and drug acquisition costs continue to be significant barriers to their widespread adoption.
Heart failure management in CA is complex. Diuretics are used cautiously for volume overload, but beta-blockers and ACE inhibitors may not be well tolerated due to low-output states. Anticoagulation is indicated in patients with atrial fibrillation or left atrial dysfunction to prevent thromboembolic events [59]. Patients with bradyarrhythmias or a conduction disease may require pacemakers or defibrillators. For those with obstructive symptoms, septal alcohol ablation has been explored, although the restrictive physiology complicates procedural success [60]. Electrophysiological studies and catheter ablation may provide symptomatic relief in selected cases [61,62].
In patients with severe AS undergoing a transcatheter aortic valve replacement (TAVR), concomitant ATTR-CA is found in up to 13.3% of cases [63]. These individuals face higher mortality and cardiovascular hospitalization rates compared to AS patients without amyloidosis. Recent studies noted that the left ventricular wall thickness (LVWT) independently predicts mortality in AS-CA patients, regardless of ejection fraction or surgical interventions. The authors advocate for randomized controlled trials to evaluate the benefit of combining an amyloid-specific therapy with standard valve treatments [64,65].
Despite these advancements, CA remains underdiagnosed. Heightened clinical suspicion in high-risk populations, improved access to advanced imaging, and multidisciplinary collaboration between cardiologists, hematologists, and imaging specialists are essential. Moreover, future research should focus on large-scale epidemiological studies to define the global burden of ATTR and AL cardiac amyloidosis and assess the efficacy of emerging treatments tailored to genetic variants. Table 4 shows the most recent articles about the diagnosis and management of cardiac amyloidosis.
Table 4.
Systematic reviews and meta-analyses on the diagnosis of cardiac amyloidosis.
In parallel with advances in diagnostic techniques, the therapeutic landscape for cardiac amyloidosis is evolving rapidly, driven by innovative approaches that go beyond traditional stabilizers. Transthyretin (TTR) silencers now include subcutaneous small interfering RNA (siRNA) agents, such as patisiran, which in the APOLLO-B trial demonstrated sustained reductions in NT-proBNP and hospitalization rates among ATTR cardiomyopathy patients [94], and vutrisiran, whose HELIOS-B study corroborated these benefits with an improved dosing profile and high tolerability [95]. Antisense oligonucleotides—exemplified by inotersen and its successor eplontersen (AKCEA-TTR-LRx)—have also shown a robust TTR knockdown, with eplontersen offering monthly dosing and a favorable safety signal [96]. On the stabilization front, acoramidis (AG10) received approval after phase 3 ATTRibute-CM results confirmed a marked symptomatic improvement and waveform normalization, complementing the longstanding tafamidis platform [97].
Perhaps the most transformative prospect lies in in vivo gene editing. Early first-in-human studies of NTLA-2001—a CRISPR/Cas9-based therapy—have achieved up to a 90% serum TTR reduction after a single infusion, underscoring the potential for a one-time curative approach [98]. Real-world anecdotes—such as the case of a septuagenarian treated by lipid–nanoparticle-delivered CRISPR machinery—highlight both the promise and delivery challenges of this modality. As these modalities mature, head-to-head and combination trials—especially those assessing long-term durability, off-target effects, and cardiac remodeling—will be crucial.
Collectively, these emerging therapies offer a spectrum of options—from lifelong stabilization and periodic silencing to potentially curative gene editing—that promise to transform cardiac amyloidosis from a relentlessly progressive disease into a manageable, and perhaps even reversible, condition. Continued multidisciplinary collaboration, patient access initiatives, and cost-effectiveness analyses will be essential to ensure the equitable delivery of these cutting-edge treatments.
To provide clinicians with an at-a-glance, evidence-based action plan for the subtype-specific management of cardiac amyloidosis, we have summarized the major society recommendations in Table 5. Disease-modifying therapies include transthyretin stabilizers (tafamidis; Class I, Level B) and RNA silencers (patisiran/inotersen; Class IIa, Level B) for ATTR amyloidosis [25,99], as well as proteasome-inhibitor-based chemotherapy (±daratumumab; Class I, Level B) and autologous stem-cell transplant (Class IIa, Level B) for AL amyloidosis [100,101]. Supportive care measures—diuretics (Class I, Level C), withholding β-blockers/ACE inhibitors in hypotensive restrictive physiology (Class IIb, Level C) and anticoagulation for atrial fibrillation or atrial dysfunction (Class I, Level C)—are drawn from the ESC heart failure and atrial fibrillation guidelines [73,102,103]. Finally, device and procedural interventions, such as TAVR for severe AS with concomitant CA (Class IIa, Level B) and ICD placement for the secondary prevention of VT/VF in AL amyloidosis (Class IIa, Level C), are recommended by the ACC/AHA and ACC expert consensus documents [99,104].
Table 5.
Guideline-based management recommendations.
6. Limitations of the Study
Several limitations of this narrative review merit consideration. First, by design, our review was not conducted as a fully systematic or meta-analytic study; although we searched two major databases (PubMed and Scopus) over a defined interval, we did not apply formal risk-of-bias assessments or GRADE evidence grading, and we may have omitted relevant studies published outside the search window or in non-English journals. Second, the heterogeneity of the included studies—in terms of study populations, diagnostic criteria, imaging protocols, and therapeutic regimens—precludes a quantitative synthesis and limits the generalizability of our conclusions to specific patient subgroups or practice settings. Third, emerging therapies and diagnostic tools in cardiac amyloidosis are evolving rapidly; some of the most recent clinical trials and consensus statements may not yet be fully reflected, and cost-effectiveness considerations (particularly for high-cost agents such as tafamidis, inotersen, and diflunisal) were beyond the scope of our review. Finally, as with all narrative overviews, selection bias and publication bias—favoring positive or high-profile studies—may influence the balance of the evidence presented. Despite these limitations, our review aims to provide a comprehensive clinical perspective on current diagnostic advances and emerging therapies in cardiac amyloidosis, highlighting areas in need of further rigorous investigation.
7. Future Directions
Despite rapid advances in both diagnostic modalities and targeted treatments, several critical questions remain unanswered. First, robust, prospective risk-stratification models that integrate imaging metrics (ECV, strain patterns), biomarkers (NT-proBNP, troponin), genotypes (ATTRv vs. ATTRwt), and clinical features are lacking; developing and validating such multidimensional scores will be essential to identify patients most likely to benefit from early intervention. Second, with an expanding armamentarium—from TTR stabilizers (tafamidis and acoramidis) and RNA silencers (patisiran, vutrisiran, inotersen, and eplontersen) to in vivo gene editors—head-to-head or adaptive trials are urgently needed to define the optimal sequencing or combination of these agents, to clarify whether certain subgroups (for example, early-stage vs. advanced-stage ATTR-CA) derive greater incremental benefits from one class over another, and to assess long-term safety and durability. Third, real-world effectiveness and cost-effectiveness analyses—particularly in health systems with constrained resources—are scarce, yet crucial for informing policy, reimbursements, and equitable access. Finally, the field would benefit from large, multicenter registries and biobanks that can support translational research into novel biomarkers and mechanisms of disease progression. Addressing these gaps through collaborative, interdisciplinary efforts will be pivotal in transforming cardiac amyloidosis from an often-fatal disorder into a condition that can be reliably detected early, stratified by risk, and treated with precision.
8. Conclusions
Cardiac amyloidosis presents significant diagnostic and therapeutic challenges due to its heterogeneous presentations and underlying etiologies. Innovations in imaging modalities, molecular therapies, and interdisciplinary care models are reshaping the management paradigm and offering renewed hope for affected patients.
Author Contributions
Conceptualization, D.E.M., and A.C.M.; methodology, D.E.M., D.C., and O.A.; software, D.C.; validation, D.E.M., A.C.M., and A.C.S.; formal analysis, A.C.S., and M.A.; investigation, O.A., and T.C.; resources, S.R.D.; data curation, O.A.; writing—original draft preparation, D.E.M.; writing—review and editing, A.C.M., O.A., and S.R.D.; visualization, M.A.; supervision, S.R.D., D.C., and T.C.; project administration, A.C.M.; funding acquisition, A.C.M. All authors have read and agreed to the published version of the manuscript.
Funding
We would like to acknowledge “Victor Babes” University of Medicine and Pharmacy for their support in covering the publication costs for this research paper.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Rubin, J.; Maurer, M.S. Cardiac Amyloidosis: Overlooked, Underappreciated, and Treatable. Annu. Rev. Med. 2020, 71, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, D.R.; Pastore, R.; Pool, S.; Malendowicz, S.; Kane, I.; Shivji, A.; Embury, S.H.; Ballas, S.K.; Buxbaum, J.N. Revised Transthyretin Ile 122 Allele Frequency in African-Americans. Hum. Genet. 1996, 98, 236–238. [Google Scholar] [CrossRef]
- Sipe, J.D.; Cohen, A.S. Review: History of the Amyloid Fibril. J. Struct. Biol. 2000, 130, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid Nomenclature 2018: Recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid 2018, 25, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Falk, R.H.; Alexander, K.M.; Liao, R.; Dorbala, S. AL (Light-Chain) Cardiac Amyloidosis. JACC 2016, 68, 1323–1341. [Google Scholar] [CrossRef]
- Mohammed, S.F.; Mirzoyev, S.A.; Edwards, W.D.; Dogan, A.; Grogan, D.R.; Dunlay, S.M.; Roger, V.L.; Gertz, M.A.; Dispenzieri, A.; Zeldenrust, S.R.; et al. Left Ventricular Amyloid Deposition in Patients with Heart Failure and Preserved Ejection Fraction. JACC Heart Fail. 2014, 2, 113–122. [Google Scholar] [CrossRef]
- Castaño, A.; Narotsky, D.L.; Hamid, N.; Khalique, O.K.; Morgenstern, R.; DeLuca, A.; Rubin, J.; Chiuzan, C.; Nazif, T.; Vahl, T.; et al. Unveiling Transthyretin Cardiac Amyloidosis and Its Predictors among Elderly Patients with Severe Aortic Stenosis Undergoing Transcatheter Aortic Valve Replacement. Eur. Heart J. 2017, 38, 2879–2887. [Google Scholar] [CrossRef]
- Pollak, A.; Falk, R.H. Left Ventricular Systolic Dysfunction Precipitated by Verapamil in Cardiac Amyloidosis. Chest 1993, 104, 618–620. [Google Scholar] [CrossRef]
- Laptseva, N.; Rossi, V.A.; Sudano, I.; Schwotzer, R.; Ruschitzka, F.; Flammer, A.J.; Duru, F. Arrhythmic Manifestations of Cardiac Amyloidosis: Challenges in Risk Stratification and Clinical Management. J. Clin. Med. 2023, 12, 2581. [Google Scholar] [CrossRef]
- Donnellan, E.; Wazni, O.M.; Hanna, M.; Elshazly, M.B.; Puri, R.; Saliba, W.; Kanj, M.; Vakamudi, S.; Patel, D.R.; Baranowski, B.; et al. Atrial Fibrillation in Transthyretin Cardiac Amyloidosis: Predictors, Prevalence, and Efficacy of Rhythm Control Strategies. JACC Clin. Electrophysiol. 2020, 6, 1118–1127. [Google Scholar] [CrossRef]
- Mitrani, L.R.; De Los Santos, J.; Driggin, E.; Kogan, R.; Helmke, S.; Goldsmith, J.; Biviano, A.B.; Maurer, M.S. Anticoagulation with Warfarin Compared to Novel Oral Anticoagulants for Atrial Fibrillation in Adults with Transthyretin Cardiac Amyloidosis: Comparison of Thromboembolic Events and Major Bleeding. Amyloid 2021, 28, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Hartnett, J.; Jaber, W.; Maurer, M.; Sperry, B.; Hanna, M.; Collier, P.; Patel, D.R.; Wazni, O.M.; Donnellan, E. Electrophysiological Manifestations of Cardiac Amyloidosis: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncology 2021, 3, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Liżewska-Springer, A.; Sławiński, G.; Lewicka, E. Arrhythmic Sudden Cardiac Death and the Role of Implantable Cardioverter-Defibrillator in Patients with Cardiac Amyloidosis—A Narrative Literature Review. J. Clin. Med. 2021, 10, 1858. [Google Scholar] [CrossRef] [PubMed]
- Belfeki, N.; Ghriss, N.; Monchi, M.; Moini, C. State of the Art of Cardiac Amyloidosis. Biomedicines 2023, 11, 1045. [Google Scholar] [CrossRef]
- Maurer, M.S.; Dunnmon, P.; Fontana, M.; Quarta, C.C.; Prasad, K.; Witteles, R.M.; Rapezzi, C.; Signorovitch, J.; Lousada, I.; Merlini, G. Proposed Cardiac End Points for Clinical Trials in Immunoglobulin Light Chain Amyloidosis: Report From the Amyloidosis Forum Cardiac Working Group. Circ. Heart Fail. 2022, 15, e009038. [Google Scholar] [CrossRef]
- aus dem Siepen, F.; Hansen, T. Diagnosing AL and ATTR Amyloid Cardiomyopathy: A Multidisciplinary Approach. J. Clin. Med. 2024, 13, 5873. [Google Scholar] [CrossRef]
- Selvanayagam, J.B.; Hawkins, P.N.; Paul, B.; Myerson, S.G.; Neubauer, S. Evaluation and Management of the Cardiac Amyloidosis. JACC 2007, 50, 2101–2110. [Google Scholar] [CrossRef]
- Martinez-Naharro, A.; Hawkins, P.N.; Fontana, M. Cardiac Amyloidosis. Clin. Med. 2018, 18, s30–s35. [Google Scholar] [CrossRef]
- Pan, J.A.; Kerwin, M.J.; Salerno, M. Native T1 Mapping, Extracellular Volume Mapping, and Late Gadolinium Enhancement in Cardiac Amyloidosis: A Meta-Analysis. JACC Cardiovasc. Imaging 2020, 13, 1299–1310. [Google Scholar] [CrossRef]
- Rapezzi, C.; Elliott, P.; Damy, T.; Nativi-Nicolau, J.; Berk, J.L.; Velazquez, E.J.; Boman, K.; Gundapaneni, B.; Patterson, T.A.; Schwartz, J.H.; et al. Efficacy of Tafamidis in Patients with Hereditary and Wild-Type Transthyretin Amyloid Cardiomyopathy. JACC Heart Fail. 2021, 9, 115–123. [Google Scholar] [CrossRef]
- Gertz, M.A.; Dispenzieri, A.; Sher, T. Pathophysiology and Treatment of Cardiac Amyloidosis. Nat. Rev. Cardiol. 2015, 12, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, A.; Porcari, A.; Patel, R.K.; Razvi, Y.; Sinigiani, G.; Martinez-Naharro, A.; Venneri, L.; Moon, J.; Rauf, M.U.; Lachmann, H.; et al. Rare Forms of Cardiac Amyloidosis: Diagnostic Clues and Phenotype in Apo AI and AIV Amyloidosis. Circ. Cardiovasc. Imaging 2023, 16, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Briasoulis, A.; Bampatsias, D.; Papamichail, A.; Kuno, T.; Skoularigis, J.; Xanthopoulos, A.; Triposkiadis, F. Invasive and Non-Invasive Diagnostic Pathways in the Diagnosis of Cardiac Amyloidosis. J. Cardiovasc. Dev. Dis. 2023, 10, 256. [Google Scholar] [CrossRef] [PubMed]
- Vergaro, G.; Aimo, A.; Barison, A.; Genovesi, D.; Buda, G.; Passino, C.; Emdin, M. Keys to Early Diagnosis of Cardiac Amyloidosis: Red Flags from Clinical, Laboratory and Imaging Findings. Eur. J. Prev. Cardiol. 2020, 27, 1806–1815. [Google Scholar] [CrossRef]
- Garcia-Pavia, P.; Rapezzi, C.; Adler, Y.; Arad, M.; Basso, C.; Brucato, A.; Burazor, I.; Caforio, A.L.P.; Damy, T.; Eriksson, U.; et al. Diagnosis and Treatment of Cardiac Amyloidosis: A Position Statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2021, 42, 1554–1568. [Google Scholar] [CrossRef]
- Moore, P.T.; Burrage, M.K.; Mackenzie, E.; Law, W.P.; Korczyk, D.; Mollee, P. The Utility of 99mTc-DPD Scintigraphy in the Diagnosis of Cardiac Amyloidosis: An Australian Experience. Heart Lung Circ. 2017, 26, 1183–1190. [Google Scholar] [CrossRef]
- Ioannou, A.; Patel, R.K.; Razvi, Y.; Porcari, A.; Knight, D.; Martinez-Naharro, A.; Kotecha, T.; Venneri, L.; Chacko, L.; Brown, J.; et al. Multi-Imaging Characterization of Cardiac Phenotype in Different Types of Amyloidosis. Cardiovasc. Imaging 2023, 16, 464–477. [Google Scholar] [CrossRef]
- Gonzalez-Lopez, E.; McPhail, E.D.; Salas-Anton, C.; Dominguez, F.; Gertz, M.A.; Dispenzieri, A.; Dasari, S.; Milani, P.; Verga, L.; Grogan, M.; et al. Histological Typing in Patients with Cardiac Amyloidosis. JACC 2024, 83, 1085–1099. [Google Scholar] [CrossRef]
- Leung, N.; Nasr, S.H.; Sethi, S. How I Treat Amyloidosis: The Importance of Accurate Diagnosis and Amyloid Typing. Blood 2012, 120, 3206–3213. [Google Scholar] [CrossRef]
- Morfino, P.; Aimo, A.; Castiglione, V.; Chianca, M.; Vergaro, G.; Cipolla, C.M.; Fedele, A.; Emdin, M.; Fabiani, I.; Cardinale, D. Cardiovascular Toxicity from Therapies for Light Chain Amyloidosis. Front. Cardiovasc. Med. 2023, 10, 1212983. [Google Scholar] [CrossRef]
- Hill, M.M.; Dasari, S.; Mollee, P.; Merlini, G.; Costello, C.E.; Hazenberg, B.P.C.; Grogan, M.; Dispenzieri, A.; Gertz, M.A.; Kourelis, T.; et al. The Clinical Impact of Proteomics in Amyloid Typing. Mayo Clin. Proc. 2021, 96, 1122–1127. [Google Scholar] [CrossRef] [PubMed]
- Brownrigg, J.; Lorenzini, M.; Lumley, M.; Elliott, P. Diagnostic Performance of Imaging Investigations in Detecting and Differentiating Cardiac Amyloidosis: A Systematic Review and Meta-Analysis. ESC Heart Fail. 2019, 6, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Dicorato, M.M.; Basile, P.; Muscogiuri, G.; Carella, M.C.; Naccarati, M.L.; Dentamaro, I.; Guglielmo, M.; Baggiano, A.; Mushtaq, S.; Fusini, L.; et al. Novel Insights into Non-Invasive Diagnostic Techniques for Cardiac Amyloidosis: A Critical Review. Diagnostics 2024, 14, 2249. [Google Scholar] [CrossRef]
- Cotella, J.; Randazzo, M.; Maurer, M.S.; Helmke, S.; Scherrer-Crosbie, M.; Soltani, M.; Goyal, A.; Zareba, K.; Cheng, R.; Kirkpatrick, J.N.; et al. Limitations of Apical Sparing Pattern in Cardiac Amyloidosis: A Multicentre Echocardiographic Study. Eur. Heart J.—Cardiovasc. Imaging 2024, 25, 754–761. [Google Scholar] [CrossRef]
- Paeng, J.C.; Choi, J.Y. Nuclear Imaging for Cardiac Amyloidosis: Bone Scan, SPECT/CT, and Amyloid-Targeting PET. Nucl. Med. Mol. Imaging 2021, 55, 61–70. [Google Scholar] [CrossRef]
- Auer, B.; Kijewski, M.F.; Dorbala, S. Quantitative ATTR-Cardiac Amyloidosis SPECT/CT Imaging: The Time Is Now! J. Nucl. Cardiol. 2023, 30, 1246–1249. [Google Scholar] [CrossRef]
- Oerlemans, M.I.F.J.; Rutten, K.H.G.; Minnema, M.C.; Raymakers, R.A.P.; Asselbergs, F.W.; de Jonge, N. Cardiac Amyloidosis: The Need for Early Diagnosis. Neth. Heart J. 2019, 27, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Ruberg, F.L.; Berk, J.L. Transthyretin (TTR) Cardiac Amyloidosis. Circulation 2012, 126, 1286–1300. [Google Scholar] [CrossRef]
- Siddiqi, O.K.; Ruberg, F.L. Cardiac Amyloidosis: An Update on Pathophysiology, Diagnosis, and Treatment. Trends Cardiovasc. Med. 2018, 28, 10–21. [Google Scholar] [CrossRef]
- Kyriakou, P.; Mouselimis, D.; Tsarouchas, A.; Rigopoulos, A.; Bakogiannis, C.; Noutsias, M.; Vassilikos, V. Diagnosis of Cardiac Amyloidosis: A Systematic Review on the Role of Imaging and Biomarkers. BMC Cardiovasc. Disord 2018, 18, 221. [Google Scholar] [CrossRef]
- Maloberti, A.; Ciampi, C.; Politi, F.; Fabbri, S.; Musca, F.; Giannattasio, C. Cardiac Amyloidosis Red Flags: What All the Cardiologist Have to Know. Int. J. Cardiol. Cardiovasc. Risk Prev. 2024, 21, 200271. [Google Scholar] [CrossRef] [PubMed]
- Debonnaire, P.; Claeys, M.; Smet, M.D.; Trenson, S.; Lycke, M.; Demeester, C.; Droogenbroeck, J.V.; Vriese, A.S.D.; Verhoeven, K.; Vantomme, N.; et al. Trends in Diagnosis, Referral, Red Flag Onset, Patient Profiles and Natural Outcome of de Novo Cardiac Amyloidosis and Their Multidisciplinary Implications. Acta Cardiol. 2022, 77, 791–804. [Google Scholar] [CrossRef]
- Blanco-López, E.; Martínez-del Río, J.; López-Calles, A.; Negreira-Caamaño, M.; Águila-Gordo, D.; Soto-Martín, P.; Soto-Pérez, M.M.; Cubides-Novoa, A.F.; Gonzalez-Barderas, M.; Sánchez-Pérez, I.; et al. Cardiac Amyloidosis and Red Flags: Natural History and Its Impact in Morbimortality. Med. Clínica (Engl. Ed.) 2025, 164, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.S.; Farzaneh-Far, A.; Mamas, M.A. Red Flags in Cardiac Amyloidosis. Eur. J. Prev. Cardiol. 2020, 27, 1804–1805. [Google Scholar] [CrossRef] [PubMed]
- Papathanasiou, M.; Carpinteiro, A.; Rischpler, C.; Hagenacker, T.; Rassaf, T.; Luedike, P. Diagnosing Cardiac Amyloidosis in Every-Day Practice: A Practical Guide for the Cardiologist. IJC Heart Vasc. 2020, 28, 100519. [Google Scholar] [CrossRef]
- Westin, O.; Fosbøl, E.L.; Maurer, M.S.; Leicht, B.P.; Hasbak, P.; Mylin, A.K.; Rørvig, S.; Lindkær, T.H.; Johannesen, H.H.; Gustafsson, F. Screening for Cardiac Amyloidosis 5 to 15 Years After Surgery for Bilateral Carpal Tunnel Syndrome. J. Am. Coll. Cardiol. 2022, 80, 967–977. [Google Scholar] [CrossRef]
- Yamamoto, H.; Yokochi, T. Transthyretin Cardiac Amyloidosis: An Update on Diagnosis and Treatment. ESC Heart Fail. 2019, 6, 1128–1139. [Google Scholar] [CrossRef]
- Driggin, E.; Maurer, M.S. The Quintessential Form of Diastolic Heart Failure in Older Adults: Wild Type Transthyretin Cardiac Amyloidosis. Clin. Cardiol. 2020, 43, 171–178. [Google Scholar] [CrossRef]
- Antonopoulos, A.S.; Panagiotopoulos, I.; Kouroutzoglou, A.; Koutsis, G.; Toskas, P.; Lazaros, G.; Toutouzas, K.; Tousoulis, D.; Tsioufis, K.; Vlachopoulos, C. Prevalence and Clinical Outcomes of Transthyretin Amyloidosis: A Systematic Review and Meta-Analysis. Eur. J. Heart Fail. 2022, 24, 1677–1696. [Google Scholar] [CrossRef]
- Novosad, O.; Rudiuk, T.; Shevchuk, L.; Kundina, V.; Schmidt, A.G. Secondary Systemic AL-Amyloidosis Associated with Multiple Myeloma: Clinical Case and Literature Review. Res. Res. Square 2022. [Google Scholar] [CrossRef]
- Jaiswal, V.; Ang, S.P.; Chia, J.E.; Abdelazem, E.M.; Jaiswal, A.; Biswas, M.; Gimelli, A.; Parwani, P.; Siller-Matula, J.M.; Mamas, M.A. Echocardiographic Predictors of Presence of Cardiac Amyloidosis in Aortic Stenosis. Eur. Heart J. Cardiovasc. Imaging 2022, 23, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Llerena-Velastegui, J.; Zumbana-Podaneva, K. Advances in the Diagnosis and Management of Cardiac Amyloidosis: A Literature Review. Cardiol. Res. 2024, 15, 211–222. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, Y.S.; Kim, S.-J. Diagnostic Performance of PET for Detection of Cardiac Amyloidosis: A Systematic Review and Meta-Analysis. J. Cardiol. 2020, 76, 618–625. [Google Scholar] [CrossRef]
- Sattar, Y.; Maya, T.R.; Zafrullah, F.; Patel, N.; Latchana, S. Diagnosis and Management of a Cardiac Amyloidosis Case Mimicking Hypertrophic Cardiomyopathy. Cureus 2018, 10, e3749. [Google Scholar] [CrossRef] [PubMed]
- Herzog, E.; Leibowitz, D.; Gatt, M.E.; Koren, T.; Pollak, A. Pathway for the Diagnosis and Management of Cardiac Amyloidosis. Crit. Pathw. Cardiol. 2023, 22, 114–119. [Google Scholar] [CrossRef]
- Musetti, V.; Greco, F.; Castiglione, V.; Aimo, A.; Palmieri, C.; Genovesi, D.; Giorgetti, A.; Emdin, M.; Vergaro, G.; McDonnell, L.A.; et al. Tissue Characterization in Cardiac Amyloidosis. Biomedicines 2022, 10, 3054. [Google Scholar] [CrossRef] [PubMed]
- Garcia, Y.; Collins, A.B.; Stone, J.R. Abdominal Fat Pad Excisional Biopsy for the Diagnosis and Typing of Systemic Amyloidosis. Hum. Pathol. 2018, 72, 71–79. [Google Scholar] [CrossRef]
- Xin, Y.; Hu, W.; Chen, X.; Hu, J.; Sun, Y.; Zhao, Y. Prognostic Impact of Light-Chain and Transthyretin-Related Categories in Cardiac Amyloidosis: A Systematic Review and Meta-Analysis. Hell. J. Cardiol. 2019, 60, 375–383. [Google Scholar] [CrossRef]
- Meier-Ewert, H.K.; Sanchorawala, V.; Berk, J.L.; Ruberg, F.L. Cardiac Amyloidosis: Evolving Approach to Diagnosis and Management. Curr. Treat. Options Cardiovasc. Med. 2011, 13, 528–542. [Google Scholar] [CrossRef]
- Fanta, L.E.; Ewer, S.M.; Gimelli, G.; Reilly, N. Alcohol Septal Ablation for Left Ventricular Outflow Tract Obstruction in Cardiac Amyloidosis: New Indication for an Established Therapy. Catheter. Cardiovasc. Interv. 2022, 100, 910–914. [Google Scholar] [CrossRef]
- Tan, N.Y.; MOHSIN, Y.; Hodge, D.O.; Lacy, M.Q.; Packer, D.L.; Dispenzieri, A.; Grogan, M.; Asirvatham, S.J.; Madhavan, M.; McLeod, C.J. Catheter Ablation for Atrial Arrhythmias in Patients with Cardiac Amyloidosis. J. Cardiovasc. Electrophysiol. 2016, 27, 1167–1173. [Google Scholar] [CrossRef]
- Quinn, J.H.; Sviggum, E.B.; La Nou, A.T.; Burkhamer, K.J. Point-of-Care Ultrasound Leads to a Rare Incidental Diagnosis in a Patient with Acute Kidney Injury. Jaapa 2022, 35, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Fatima, K.; Uddin, Q.S.; Tharwani, Z.H.; Kashif, M.A.B.; Javaid, S.S.; Kumar, P.; Zia, M.T.; Javed, M.; Butt, M.S.; Asim, Z. Concomitant Transthyretin Cardiac Amyloidosis in Patients Undergoing TAVR for Aortic Stenosis: A Systemic Review and Meta-Analysis. Int. J. Cardiol. 2024, 402, 131854. [Google Scholar] [CrossRef]
- Aimo, A.; Tomasoni, D.; Porcari, A.; Vergaro, G.; Castiglione, V.; Passino, C.; Adamo, M.; Bellicini, M.G.; Lombardi, C.M.; Nardi, M.; et al. Left Ventricular Wall Thickness and Severity of Cardiac Disease in Women and Men with Transthyretin Amyloidosis. Eur. J. Heart Fail. 2023, 25, 510–514. [Google Scholar] [CrossRef]
- Melero Polo, J.; Roteta Unceta-Barrenechea, A.; Revilla Martí, P.; Pérez-Palacios, R.; Gracia Gutiérrez, A.; Bueno Juana, E.; Andrés Gracia, A.; Atienza Ayala, S.; Aibar Arregui, M.Á. Echocardiographic Markers of Cardiac Amyloidosis in Patients with Heart Failure and Left Ventricular Hypertrophy. Cardiol. J. 2023, 30, 266–275. [Google Scholar] [CrossRef]
- Aimo, A.; Merlo, M.; Porcari, A.; Georgiopoulos, G.; Pagura, L.; Vergaro, G.; Sinagra, G.; Emdin, M.; Rapezzi, C. Redefining the Epidemiology of Cardiac Amyloidosis. A Systematic Review and Meta-Analysis of Screening Studies. Eur. J. Heart Fail. 2022, 24, 2342–2351. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Ha, S.; Kim, Y. Cardiac Amyloidosis Imaging with Amyloid Positron Emission Tomography: A Systematic Review and Meta-Analysis. J. Nucl. Cardiol. 2020, 27, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Ricci, F.; Ceriello, L.; Khanji, M.Y.; Dangas, G.; Bucciarelli-Ducci, C.; Di Mauro, M.; Fedorowski, A.; Zimarino, M.; Gallina, S. Prognostic Significance of Cardiac Amyloidosis in Patients with Aortic Stenosis. JACC Cardiovasc. Imaging 2021, 14, 293–295. [Google Scholar] [CrossRef]
- Cai, S.; Haghbayan, H.; Chan, K.K.W.; Deva, D.P.; Jimenez-Juan, L.; Connelly, K.A.; Ng, M.-Y.; Yan, R.T.; Yan, A.T. Tissue Mapping by Cardiac Magnetic Resonance Imaging for the Prognostication of Cardiac Amyloidosis: A Systematic Review and Meta-Analysis. Int. J. Cardiol. 2024, 403, 131892. [Google Scholar] [CrossRef]
- See, A.S.Y.; Ho, J.S.-Y.; Chan, M.Y.; Lim, Y.C.; Yeo, T.-C.; Chai, P.; Wong, R.C.C.; Lin, W.; Sia, C.-H. Prevalence and Risk Factors of Cardiac Amyloidosis in Heart Failure: A Systematic Review and Meta-Analysis. Heart Lung Circ. 2022, 31, 1450–1462. [Google Scholar] [CrossRef]
- Ho, J.S.-Y.; Kor, Q.; Kong, W.K.; Lim, Y.C.; Chan, M.Y.-Y.; Syn, N.L.; Ngiam, J.N.; Chew, N.W.; Yeo, T.-C.; Chai, P.; et al. Prevalence and Outcomes of Concomitant Cardiac Amyloidosis and Aortic Stenosis: A Systematic Review and Meta-Analysis. Hell. J. Cardiol. 2022, 64, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, N.; Roshankar, G.; Draycott, L.; Jimenez-Zepeda, V.; Fine, N.; Chan, D.; Han, D.; Miller, R.J.H. Diagnostic Accuracy of Bone Scintigraphy Imaging for Transthyretin Cardiac Amyloidosis: Systematic Review and Meta-Analysis. J. Nucl. Cardiol. 2023, 30, 2464–2476. [Google Scholar] [CrossRef]
- Kang, Y.; Qu, N.; Zhang, Z.; Zhang, Q.; Chen, X.; Fu, M. Tolerability and Effectiveness of Beta-Blockers in Patients with Cardiac Amyloidosis: A Systematic Review and Meta-Analysis. Int. J. Cardiol. 2024, 402, 131813. [Google Scholar] [CrossRef] [PubMed]
- de Campos, D.; Saleiro, C.; Botelho, A.; Costa, M.; Gonçalves, L.; Teixeira, R. Aortic Valve Intervention for Aortic Stenosis and Cardiac Amyloidosis: A Systematic Review and Meta-Analysis. Future Cardiol. 2022, 18, 477–486. [Google Scholar] [CrossRef]
- Kwok, C.S.; Choy, C.H.; Pinney, J.; Townend, J.N.; Whelan, C.; Fontana, M.; Gillmore, J.D.; Steeds, R.P.; Moody, W.E. Effect of Beta-Blockade on Mortality in Patients with Cardiac Amyloidosis: A Systematic Review and Meta-Analysis. ESC Heart Fail. 2024, 11, 3901–3910. [Google Scholar] [CrossRef]
- Khan, L.A.; Shaikh, F.H.; Khan, M.S.; Zafar, B.; Farooqi, M.; Bold, B.; Aslam, H.M.; Essam, N.; Noor, I.; Siddique, A.; et al. Artificial Intelligence-Enhanced Electrocardiogram for the Diagnosis of Cardiac Amyloidosis: A Systemic Review and Meta-Analysis. Curr. Probl. Cardiol. 2024, 49, 102860. [Google Scholar] [CrossRef]
- Cannata, F.; Chiarito, M.; Pinto, G.; Villaschi, A.; Sanz-Sánchez, J.; Fazzari, F.; Regazzoli, D.; Mangieri, A.; Bragato, R.M.; Colombo, A.; et al. Transcatheter Aortic Valve Replacement in Aortic Stenosis and Cardiac Amyloidosis: A Systematic Review and Meta-Analysis. ESC Heart Fail. 2022, 9, 3188–3197. [Google Scholar] [CrossRef]
- Sun, H.; Shi, Z.; Liu, W. The Value of the Electrocardiogram in the Recognition of Cardiac Amyloidosis: A Systematic Meta-Analysis. BMC Cardiovasc. Disord. 2024, 24, 488. [Google Scholar] [CrossRef]
- Halawa, A.; Woldu, H.G.; Kacey, K.G.; Alpert, M.A. Effect of ICD Implantation on Cardiovascular Outcomes in Patients with Cardiac Amyloidosis: A Systematic Review and Meta-Anaylsis. J. Cardiovasc. Electrophysiol. 2020, 31, 1749–1758. [Google Scholar] [CrossRef]
- Kato, S.; Azuma, M.; Horita, N.; Utsunomiya, D. Monitoring the Efficacy of Tafamidis in ATTR Cardiac Amyloidosis by MRI-ECV: A Systematic Review and Meta-Analysis. Tomography 2024, 10, 1303–1311. [Google Scholar] [CrossRef]
- Wang, J.; Chen, D.; Dong, F.; Chi, H. Diagnostic Sensitivity of Abdominal Fat Aspiration Biopsy for Cardiac Amyloidosis: A Systematic Review and Meta-Analysis. Int. J. Surg. Pathol. 2024, 32, 286–293. [Google Scholar] [CrossRef]
- Albulushi, A.; Buraiki, J.A.; Aly, G.; Al-Wahshi, Y.; Jahangirifard, A. Role of Biomarkers in Early Diagnosis and Prognosis of Cardiac Amyloidosis: A Systematic Review and Meta-Analysis. Curr. Probl. Cardiol. 2025, 50, 102883. [Google Scholar] [CrossRef] [PubMed]
- Treglia, G.; Glaudemans, A.W.J.M.; Bertagna, F.; Hazenberg, B.P.C.; Erba, P.A.; Giubbini, R.; Ceriani, L.; Prior, J.O.; Giovanella, L.; Slart, R.H.J.A. Diagnostic Accuracy of Bone Scintigraphy in the Assessment of Cardiac Transthyretin-Related Amyloidosis: A Bivariate Meta-Analysis. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1945–1955. [Google Scholar] [CrossRef]
- Wu, Z.; Yu, C. Diagnostic Performance of CMR, SPECT, and PET Imaging for the Detection of Cardiac Amyloidosis: A Meta-Analysis. BMC Cardiovasc. Disord. 2021, 21, 482. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Leng, Z. An Analysis Regarding the Article “Artificial Intelligence-Enhanced Electrocardiogram for the Diagnosis of Cardiac Amyloidosis: A Systemic Review and Meta-Analysis”. Curr. Probl. Cardiol. 2024, 49, 102866. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Hu, H.; Cui, W. Performance of Bone Tracer for Diagnosis and Differentiation of Transthyretin Cardiac Amyloidosis: A Systematic Review and Meta-Analysis. Diagn. Interv. Radiol. 2021, 27, 802–810. [Google Scholar] [CrossRef]
- Riley, J.M.; Junarta, J.; Ullah, W.; Siddiqui, M.U.; Anzelmi, A.; Ruge, M.; Vishnevsky, A.; Alvarez, R.J.; Ruggiero, N.J.; Rajapreyar, I.N.; et al. Transcatheter Aortic Valve Implantation in Cardiac Amyloidosis and Aortic Stenosis. Am. J. Cardiol. 2023, 198, 101–107. [Google Scholar] [CrossRef]
- Cho, S.-G.; Han, S. Prognostic Value of Bone Scintigraphy in Cardiac Amyloidosis: A Systematic Review and Meta-Analysis. Clin. Nucl. Med. 2025, 50, e34–e40. [Google Scholar] [CrossRef]
- Zhang, Y.; Chaolu, H. Diagnostic Role of NT-proBNP in Patients with Cardiac Amyloidosis Involvement: A Meta-Analysis. Arq. Bras. Cardiologia 2022, 119, 212–222. [Google Scholar] [CrossRef]
- Zhao, L.; Tian, Z.; Fang, Q. Diagnostic Accuracy of Cardiovascular Magnetic Resonance for Patients with Suspected Cardiac Amyloidosis: A Systematic Review and Meta-Analysis. BMC Cardiovasc. Disord. 2016, 16, 129. [Google Scholar] [CrossRef]
- Jaiswal, V.; Joshi, A.; Ishak, A.; Nataraj, M.; Ang, S.P.; Khan, N.; Daneshvar, F.; Aguilera-Alvarez, V.H.; Verma, D.; Shrestha, A.B.; et al. Meta-Analysis of Post-Transcatheter Aortic Valve Replacement Outcomes in Patients with Cardiac Amyloidosis and Aortic Stenosis. Int. J. Surg. 2023, 109, 2872–2874. [Google Scholar] [CrossRef]
- Lee, C.-Y.; Nabeshima, Y.; Kitano, T.; Yang, L.-T.; Takeuchi, M. Diagnostic Accuracy and Prognostic Value of Relative Apical Sparing in Cardiac Amyloidosis—Systematic Review and Meta-Analysis. Circ. J. 2024, 89, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.K.M.; Brizneda, M.V.; Kwon, D.H.; Popovic, Z.B.; Flamm, S.D.; Hanna, M.; Griffin, B.P.; Xu, B. Reference Ranges, Diagnostic and Prognostic Utility of Native T1 Mapping and Extracellular Volume for Cardiac Amyloidosis: A Meta-Analysis. J. Magn. Reson. Imaging 2021, 53, 1458–1468. [Google Scholar] [CrossRef] [PubMed]
- American College of Cardiology. A Study to Evaluate Patisiran in Participants with Transthyretin Amyloidosis with Cardiomyopathy. 2023. Available online: https://www.acc.org/Latest-in-Cardiology/Clinical-Trials/2023/10/30/15/55/apollo-b (accessed on 8 May 2025).
- European Society of Cardiology: Vutrisiran Offers a New Lifeline to Patients with Progressive Heart Condition. Available online: https://www.escardio.org/The-ESC/Press-Office/Press-releases/Vutrisiran-offers-a-new-lifeline-to-patients-with-progressive-heart-condition (accessed on 8 May 2025).
- Roberts, J.R.; Lan, M.L.; Mank, V.M.F.; Talavera, F.; Brent, L.H.; Besa, E.C.; Wolf, R.E.; Pumerantz, A.; Middleman, C.F.; Sprowl, G.M. Strategies for Shortening Time to Diagnosis of Transthyretin Amyloidosis. Medscape. Available online: https://emedicine.medscape.com/article/335301-treatment?form=fpf (accessed on 16 May 2025).
- Attruby: Uses, Dosage, Side Effects, Warnings. Available online: https://www.drugs.com/attruby.html (accessed on 8 May 2025).
- Gillmore, J.D.; Taubel, J.; Gane, E. First-in-Human in vivo CRISPR/Cas9 Editing of the TTR Gene by NTLA-2001 in Patients with Transthyretin (ATTR) Amyloidosis with Cardiomyopathy. In Proceedings of the American Heart Association Scientific Sessions, Chicago, IL, USA, 5–7 November 2022; Available online: https://www.acc.org/-/media/Clinical/PDF-Files/Approved-PDFs/2022/11/01/AHA22/Nov05/Nov05-5pm-First-in-Human-in-vivo-Crispr-Cas9-Editing-aha-2022.pdf (accessed on 11 April 2025).
- Kittleson, M.M.; Ruberg, F.L.; Ambardekar, A.V.; Brannagan, T.H.; Cheng, R.K.; Clarke, J.O.; Dember, L.M.; Frantz, J.G.; Hershberger, R.E.; Maurer, M.S.; et al. 2023 ACC Expert Consensus Decision Pathway on Comprehensive Multidisciplinary Care for the Patient with Cardiac Amyloidosis. JACC 2023, 81, 1076–1126. [Google Scholar] [CrossRef]
- Kumar, S.K.; Callander, N.S.; Adekola, K.; Anderson, L.D.; Baljevic, M.; Campagnaro, E.; Castillo, J.J.; Costello, C.; D’Angelo, C.; Devarakonda, S.; et al. Systemic Light Chain Amyloidosis, Version 2.2023, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2023, 21, 67–81. [Google Scholar]
- Palladini, G.; Milani, P.; Merlini, G. Management of AL Amyloidosis in 2020. Blood 2020, 136, 2620–2627. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure: Developed by the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure of the European Society of Cardiology (ESC) with the Special Contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.-A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the Diagnosis and Management of Atrial Fibrillation Developed in Collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the Diagnosis and Management of Atrial Fibrillation of the European Society of Cardiology (ESC) Developed with the Special Contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2021, 42, 373–498. [Google Scholar] [CrossRef]
- Mallus, M.T.; Rizzello, V. Treatment of Amyloidosis: Present and Future. Eur. Heart J. Suppl. 2023, 25, B99–B103. [Google Scholar] [CrossRef]
- Otto, C.M.; Nishimura, R.A.; Bonow, R.O.; Carabello, B.A.; Erwin, J.P.; Gentile, F.; Jneid, H.; Krieger, E.V.; Mack, M.; McLeod, C.; et al. 2020 ACC/AHA Guideline for the Management of Patients with Valvular Heart Disease: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2021, 143, e72–e227. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).