
Impaired Function of Solute Carrier Family 19 Leads to Low Folate Levels and Lipid Droplet Accumulation in Hepatocytes
 ,
,  , , ,
, , ,  , and
, and
Abstract

1. Introduction
2. Experimental Procedures
2.1. Human Subjects
2.2. Selection of Genes and SNPs
2.3. Genotyping and Association Study
2.4. Gene Knockdown in THLE2-Cells
2.5. Folate Concentration in THLE2-Knockdown Cells
2.6. Phospholipid and Neutral Lipid Droplet Staining
2.7. Pathway-Focused Gene Expression Analysis. PCR Arrays in Knockdown THLE2-Cells
2.8. Culture of THLE2-Cells in Folic Acid Depleted Medium
2.8.1. Metabolite Extraction for Ultra-High Pressure Liquid Chromatography Coupled to Mass Spectrometry (UHPLC-MS) Analysis
2.8.2. Metabolomics Data Processing, Normalization, and Data Analysis
3. Results
3.1. SNPs Located within SLC19A1 Are Associated with NAFLD Susceptibility
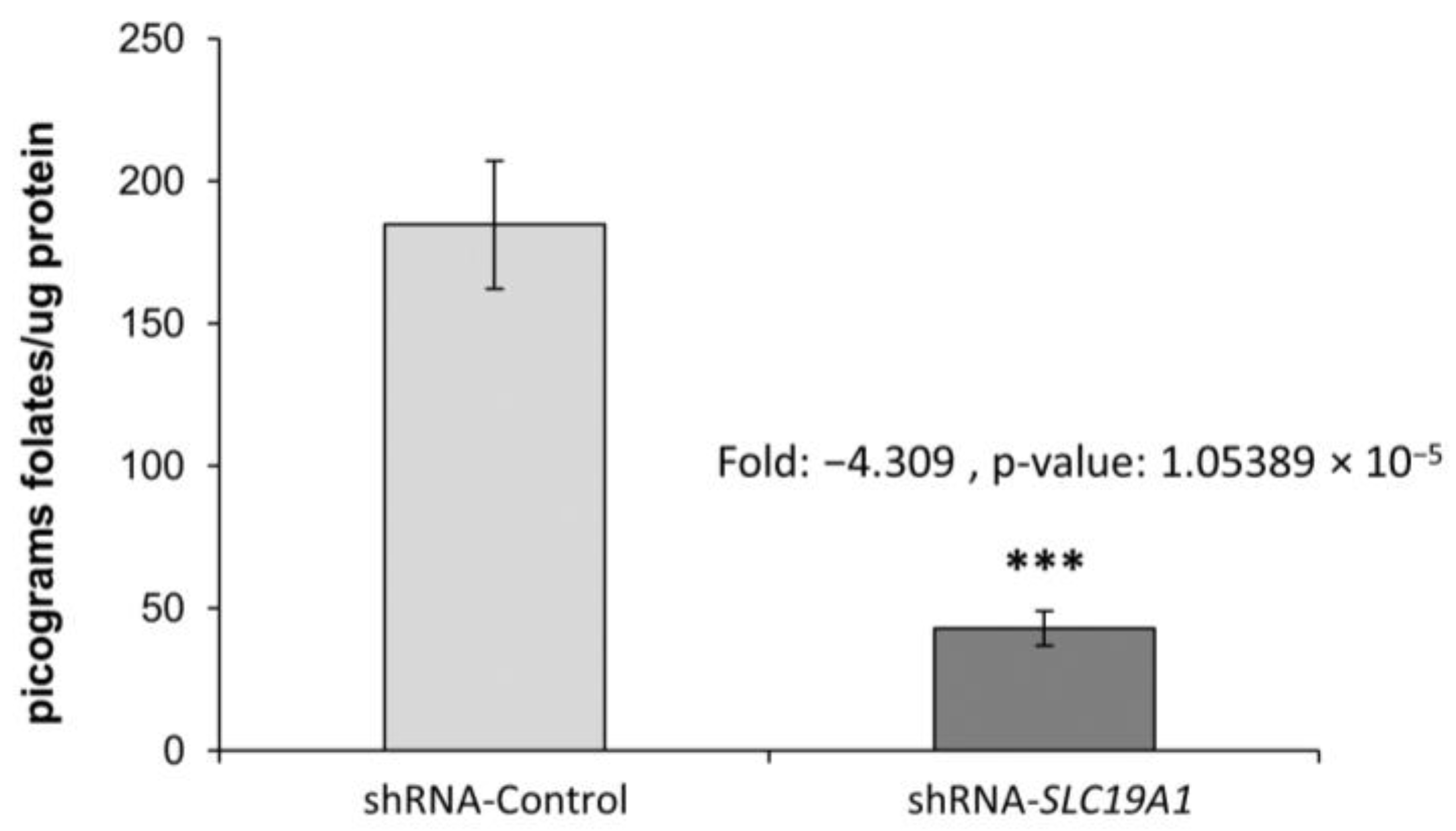
3.2. SLC19A1-Knockdown THLE2-Cells Significantly Exhibit Reduced Intracellular Folate Concentrations
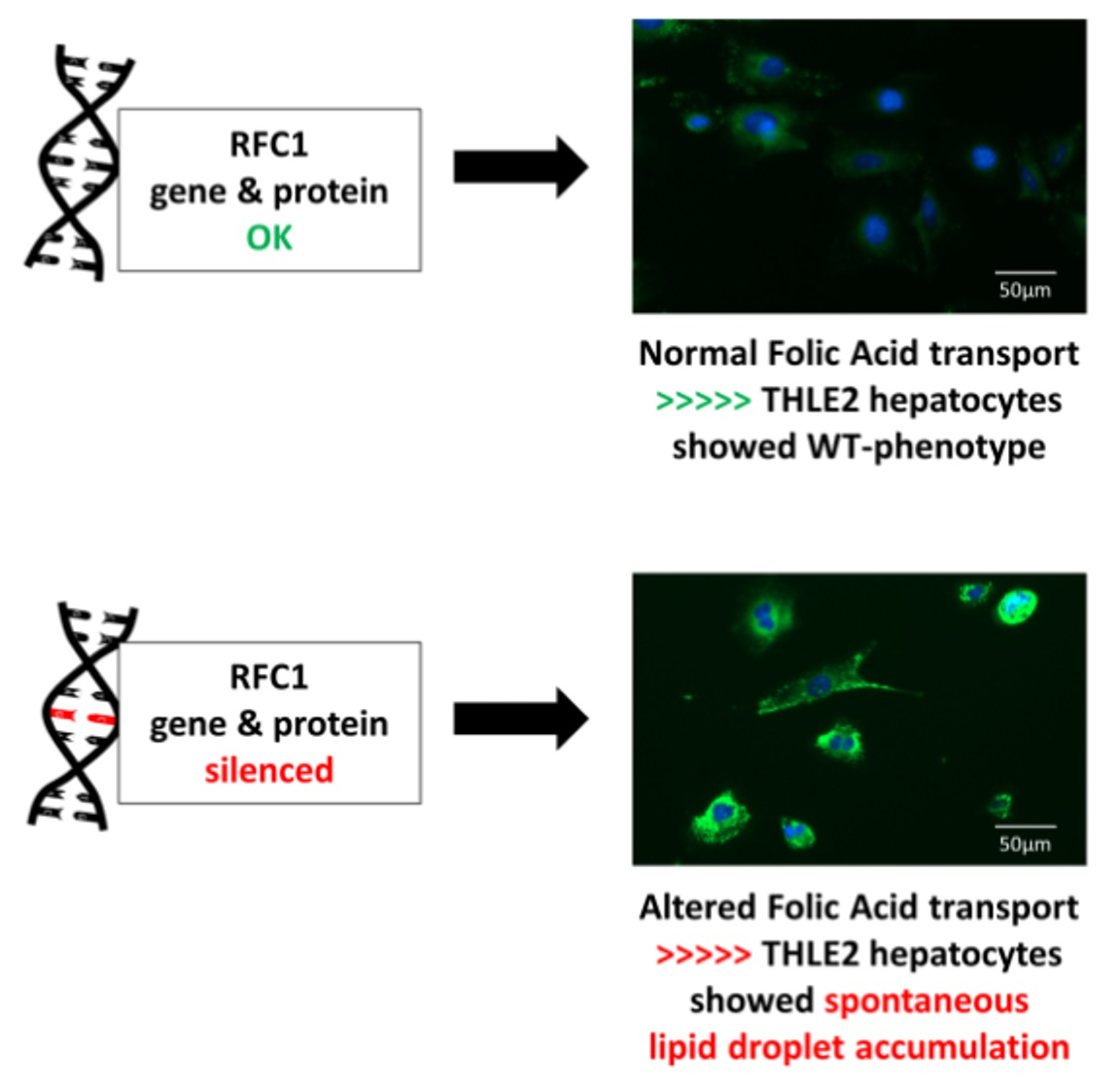
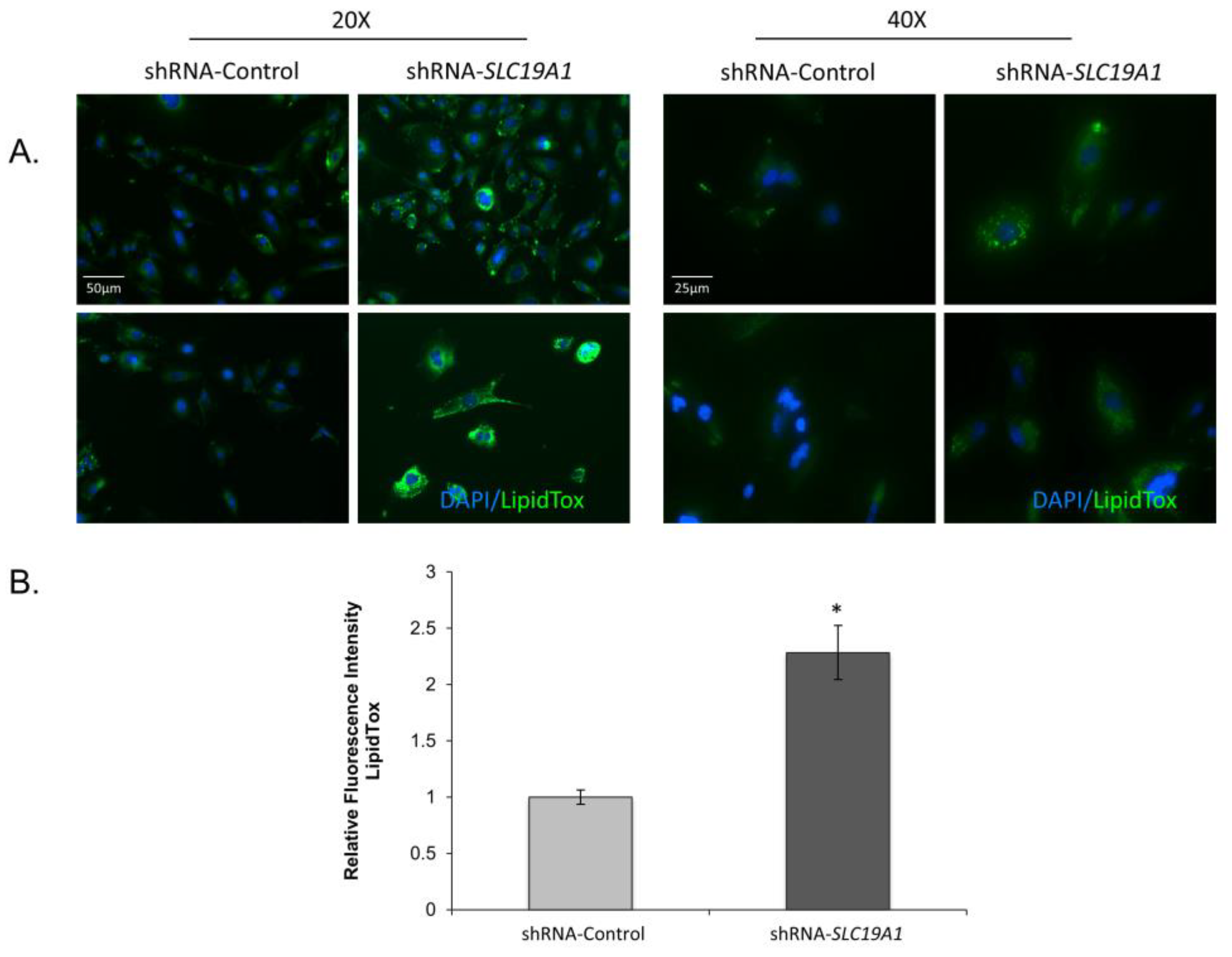
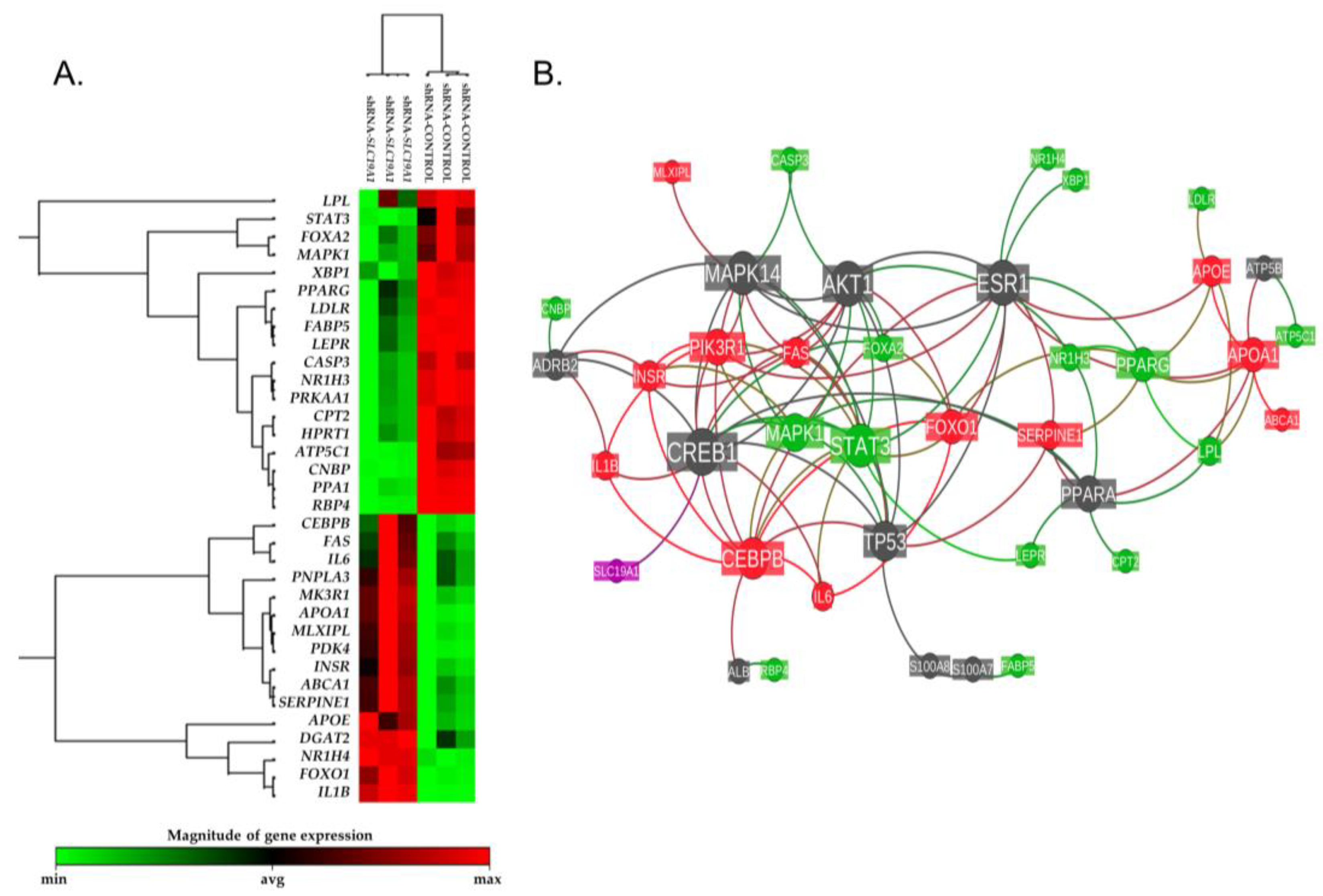
3.2.1. Pronounced, Spontaneous Lipid Droplet Accumulation in SLC19A1-Knockdown THLE2-Cells
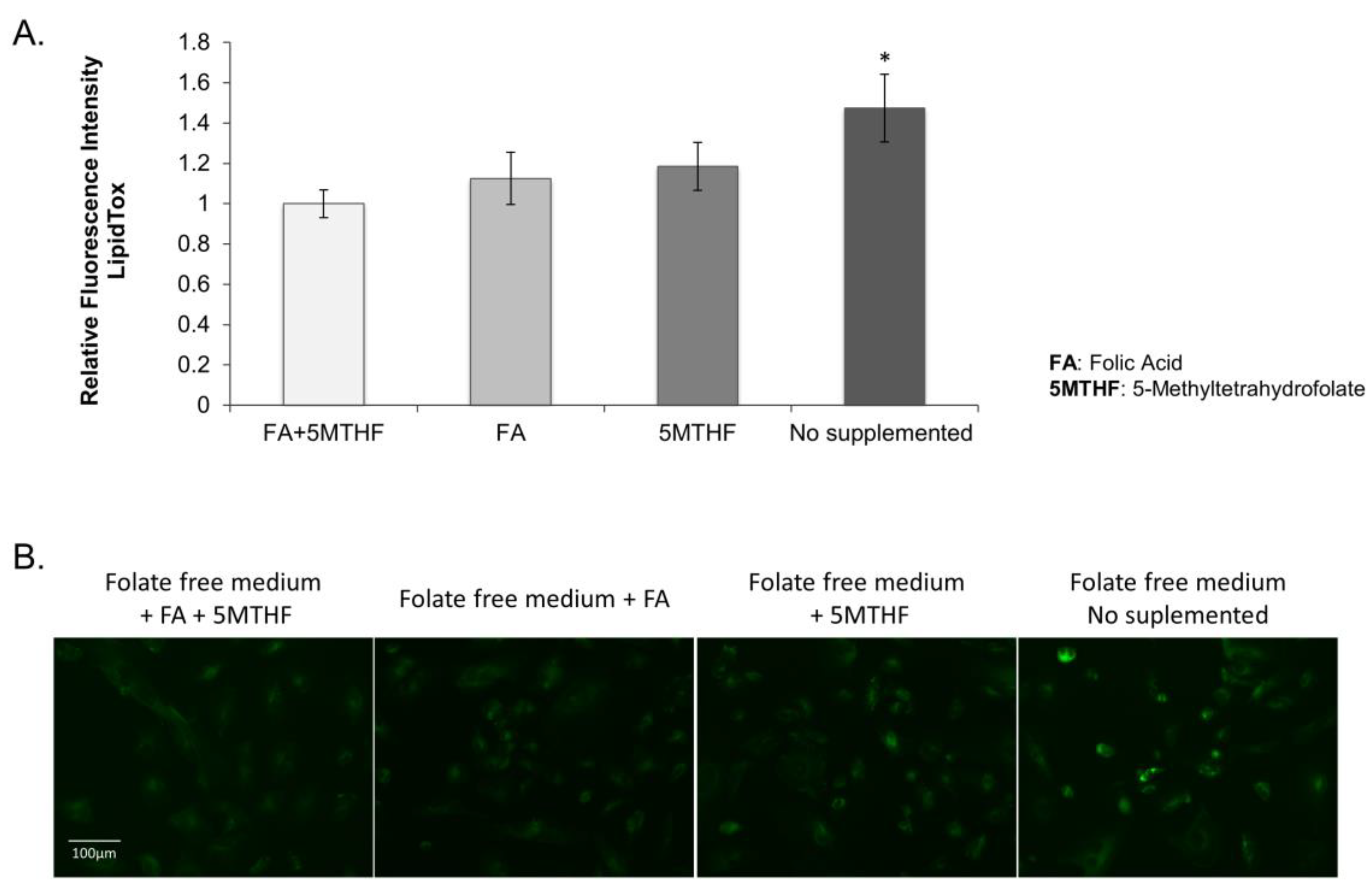
3.2.2. Lipid Droplet Accumulation in the SLC19A1-Knockdown Cells Can Be Replicated by Culturing Cells in Low Folate Medium
3.2.3. Effect of SLC19A1-Knockdown in THLE2-Cells Gene Expression
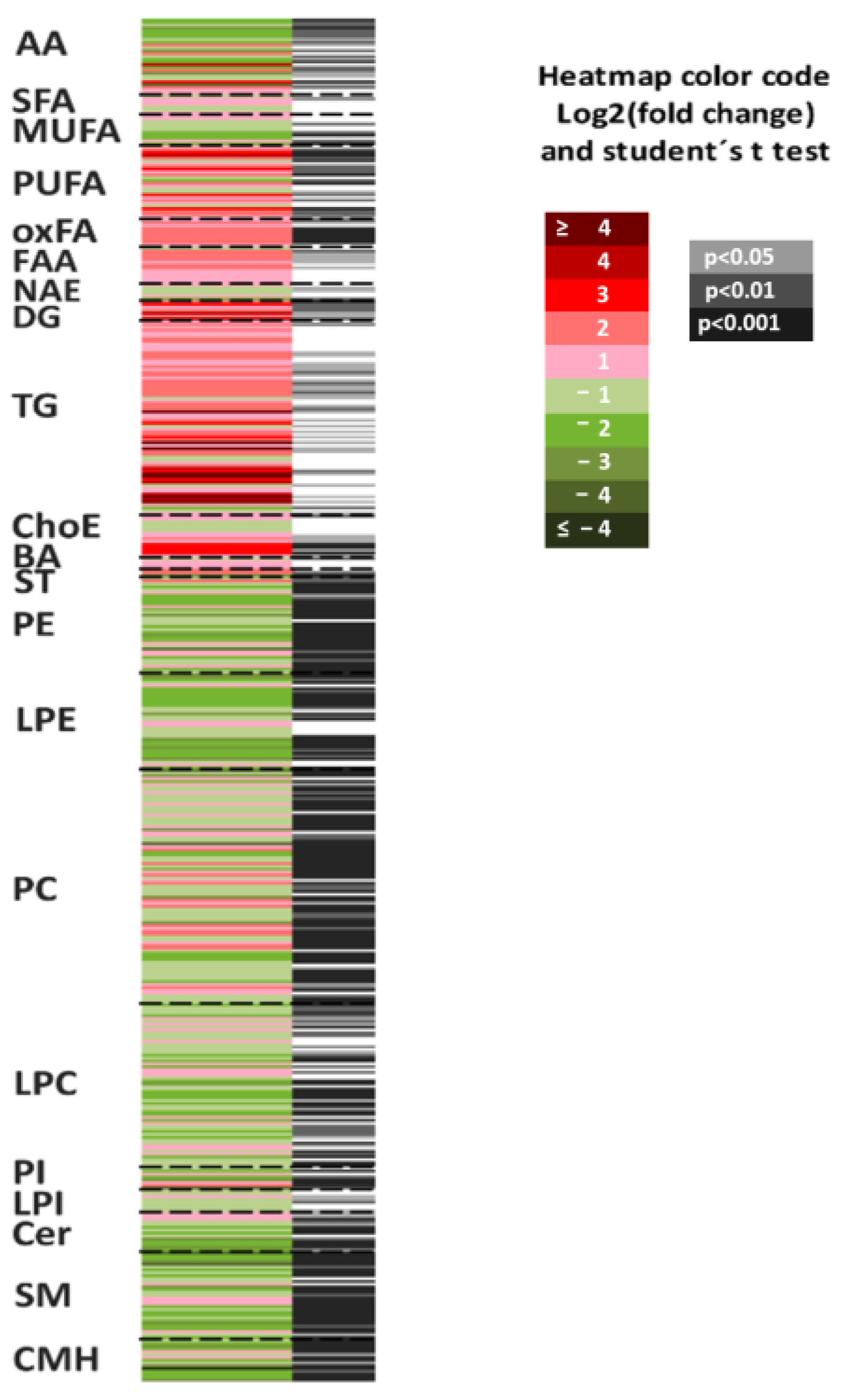
3.2.4. The Lack of SLC19A1 Results in Global Alteration of the Lipid and Amino Acid Cellular Profile
4. Discussion
4.1. Polymorphisms in Folate Transporter Genes
4.2. Phenotyping of MAFLD Patients
4.3. Low Folate Levels Contribute to Lipid Droplet Accumulation
4.4. Omics Analyses Unravelled a Potential Role of the Gene SLC19A1 in NAFLD
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALT | Alanine transaminase |
| AST | Aspartate transaminase |
| BMI | Body mass index |
| ChoE | Cholesterol esters |
| CMH | Monohexosylceramides |
| DG | Diacylglycerols, diglycerides |
| gGT | gamma glutamyl transferase |
| HCC | Hepatocellular carcinoma |
| HWE | Hardy-Weinberg equilibrium |
| MAF | Minor allele frequency |
| NAFLD | Nonalcoholic fatty liver disease |
| NASH | Nonalcoholic steatohepatitis |
| NEFA | Non-esterified fatty acids |
| MAFLD | Metabolic associated fatty liver disease |
| PC | Phosphatidylcholines |
| PE | Phosphatidylethanolamines |
| PUFA | Polyunsaturated fatty acids |
| RFC1 | Reduced folate carrier |
| SLC19A1 | solute carrier family 19 (folate transporter) member 1 (RFC1) |
| SNP | Single nucleotide polymorphism |
| TG | Triacylglycerols, triglycerides |
References
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Speliotes, E.K.; Yerges-Armstrong, L.M.; Wu, J.; Hernaez, R.; Kim, L.J.; Palmer, C.D.; Gudnason, V.; Eiriksdottir, G.; Garcia, M.E.; Launer, L.J.; et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011, 7, e1001324. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Chantada, M.; Gonzalez-Lahera, A.; Martinez-Arranz, I.; Garcia-Monzon, C.; Regueiro, M.M.; Garcia-Rodriguez, J.L.; Schlangen, K.A.; Mendibil, I.; Rodriguez-Ezpeleta, N.; Lozano, J.J.; et al. Solute carrier family 2 member 1 is involved in the development of nonalcoholic fatty liver disease. Hepatology 2013, 57, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Dorairaj, V.; Sulaiman, S.A.; Abu, N.; Abdul Murad, N.A. Nonalcoholic Fatty Liver Disease (NAFLD): Pathogenesis and Noninvasive Diagnosis. Biomedicines 2022, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Lazo, M.; Clark, J.M. The epidemiology of nonalcoholic fatty liver disease: A global perspective. Semin. Liver Dis. 2008, 28, 339–350. [Google Scholar] [CrossRef]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. Diabetologia 2016, 59, 1121–1140. [Google Scholar] [CrossRef]
- Malhi, H.; Gores, G.J. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin. Liver Dis. 2008, 28, 360–369. [Google Scholar] [CrossRef]
- Mato, J.M.; Lu, S.C. Role of S-adenosyl-L-methionine in liver health and injury. Hepatology 2007, 45, 1306–1312. [Google Scholar] [CrossRef]
- Brouwer, I.A.; van Dusseldorp, M.; Thomas, C.M.; Duran, M.; Hautvast, J.G.; Eskes, T.K.; Steegers-Theunissen, R.P. Low-dose folic acid supplementation decreases plasma homocysteine concentrations: A randomized trial. Am. J. Clin. Nutr. 1999, 69, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.F.; Bian, H.; Zhu, X.P.; Yan, H.M.; Chang, X.X.; Zhang, L.S.; Lin, H.D.; Hu, X.Q.; Gao, X. Serum folic acid levels are associated with the presence and severity of liver steatosis in Chinese adults. Clin. Nutr. 2018, 37, 1752–1758. [Google Scholar] [CrossRef] [PubMed]
- Mato, J.M.; Martinez-Chantar, M.L.; Lu, S.C. Methionine metabolism and liver disease. Annu. Rev. Nutr. 2008, 28, 273–293. [Google Scholar] [CrossRef] [PubMed]
- Mayo, R.; Crespo, J.; Martinez-Arranz, I.; Banales, J.M.; Arias, M.; Minchole, I.; Aller de la Fuente, R.; Jimenez-Aguero, R.; Alonso, C.; de Luis, D.A.; et al. Metabolomic-based noninvasive serum test to diagnose nonalcoholic steatohepatitis: Results from discovery and validation cohorts. Hepatol. Commun. 2018, 2, 807–820. [Google Scholar] [CrossRef] [PubMed]
- Schwahn, B.C.; Chen, Z.; Laryea, M.D.; Wendel, U.; Lussier-Cacan, S.; Genest, J., Jr.; Mar, M.H.; Zeisel, S.H.; Castro, C.; Garrow, T.; et al. Homocysteine-betaine interactions in a murine model of 5,10-methylenetetrahydrofolate reductase deficiency. FASEB J. 2003, 17, 512–514. [Google Scholar] [CrossRef] [PubMed]
- Welzel, T.M.; Katki, H.A.; Sakoda, L.C.; Evans, A.A.; London, W.T.; Chen, G.; O’Broin, S.; Shen, F.M.; Lin, W.Y.; McGlynn, K.A. Blood folate levels and risk of liver damage and hepatocellular carcinoma in a prospective high-risk cohort. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1279–1282. [Google Scholar] [CrossRef]
- Gulsen, M.; Yesilova, Z.; Bagci, S.; Uygun, A.; Ozcan, A.; Ercin, C.N.; Erdil, A.; Sanisoglu, S.Y.; Cakir, E.; Ates, Y.; et al. Elevated plasma homocysteine concentrations as a predictor of steatohepatitis in patients with non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2005, 20, 1448–1455. [Google Scholar] [CrossRef]
- Hirsch, S.; Poniachick, J.; Avendano, M.; Csendes, A.; Burdiles, P.; Smok, G.; Diaz, J.C.; de la Maza, M.P. Serum folate and homocysteine levels in obese females with non-alcoholic fatty liver. Nutrition 2005, 21, 137–141. [Google Scholar] [CrossRef]
- Zhao, R.; Goldman, I.D. Folate and thiamine transporters mediated by facilitative carriers (SLC19A1-3 and SLC46A1) and folate receptors. Mol. Asp. Med. 2013, 34, 373–385. [Google Scholar] [CrossRef]
- Matherly, L.H.; Hou, Z. Structure and function of the reduced folate carrier a paradigm of a major facilitator superfamily mammalian nutrient transporter. Vitam. Horm. 2008, 79, 145–184. [Google Scholar] [CrossRef]
- Alam, C.; Hoque, M.T.; Finnell, R.H.; Goldman, I.D.; Bendayan, R. Regulation of Reduced Folate Carrier (RFC) by Vitamin D Receptor at the Blood-Brain Barrier. Mol. Pharm. 2017, 14, 3848–3858. [Google Scholar] [CrossRef]
- Shaw, G.M.; Lammer, E.J.; Zhu, H.; Baker, M.W.; Neri, E.; Finnell, R.H. Maternal periconceptional vitamin use, genetic variation of infant reduced folate carrier (A80G), and risk of spina bifida. Am. J. Med. Genet. 2002, 108, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Yi, K.; Ma, Y.H.; Wang, W.; Zhang, X.; Gao, J.; He, S.E.; Xu, X.M.; Ji, M.; Guo, W.F.; You, T. The Roles of Reduced Folate Carrier-1 (RFC1) A80G (rs1051266) Polymorphism in Congenital Heart Disease: A Meta-Analysis. Med. Sci. Monit. 2021, 27, e929911. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Janney, C.G.; Di Bisceglie, A.M.; Neuschwander-Tetri, B.A.; Bacon, B.R. Nonalcoholic steatohepatitis: A proposal for grading and staging the histological lesions. Am. J. Gastroenterol. 1999, 94, 2467–2474. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Christensen, K.E.; Wu, Q.; Wang, X.; Deng, L.; Caudill, M.A.; Rozen, R. Steatosis in mice is associated with gender, folate intake, and expression of genes of one-carbon metabolism. J. Nutr. 2010, 140, 1736–1741. [Google Scholar] [CrossRef]
- Tryndyak, V.; de Conti, A.; Kobets, T.; Kutanzi, K.; Koturbash, I.; Han, T.; Fuscoe, J.C.; Latendresse, J.R.; Melnyk, S.; Shymonyak, S.; et al. Interstrain differences in the severity of liver injury induced by a choline- and folate-deficient diet in mice are associated with dysregulation of genes involved in lipid metabolism. FASEB J. 2012, 26, 4592–4602. [Google Scholar] [CrossRef]
- Barrett, J.C. Haploview: Visualization and analysis of SNP genotype data. Cold Spring Harb. Protoc. 2009, 2009, pdb-ip71. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Pfeifer, A.M.; Cole, K.E.; Smoot, D.T.; Weston, A.; Groopman, J.D.; Shields, P.G.; Vignaud, J.M.; Juillerat, M.; Lipsky, M.M.; Trump, B.F.; et al. Simian virus 40 large tumor antigen-immortalized normal human liver epithelial cells express hepatocyte characteristics and metabolize chemical carcinogens. Proc. Natl. Acad. Sci. USA 1993, 90, 5123–5127. [Google Scholar] [CrossRef]
- Salojin, K.V.; Cabrera, R.M.; Sun, W.; Chang, W.C.; Lin, C.; Duncan, L.; Platt, K.A.; Read, R.; Vogel, P.; Liu, Q.; et al. A mouse model of hereditary folate malabsorption: Deletion of the PCFT gene leads to systemic folate deficiency. Blood 2011, 117, 4895–4904. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nature protocols 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.; Caballeria, J.; Martinez-Arranz, I.; Dominguez-Diez, A.; Alonso, C.; Muntane, J.; Perez-Cormenzana, M.; Garcia-Monzon, C.; Mayo, R.; Martin-Duce, A.; et al. Obesity-dependent metabolic signatures associated with nonalcoholic fatty liver disease progression. J. Proteome Res. 2012, 11, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.M.; Valero, J.G.; Perez-Cormenzana, M.; Cano, A.; Alonso, C.; Goni, F.M. Lipidomic profile of GM95 cell death induced by Clostridium perfringens alpha-toxin. Chem. Phys. Lipids 2017, 203, 54–70. [Google Scholar] [CrossRef]
- Martinez-Arranz, I.; Mayo, R.; Perez-Cormenzana, M.; Minchole, I.; Salazar, L.; Alonso, C.; Mato, J.M. Enhancing metabolomics research through data mining. J. Proteom. 2015, 127, 275–288. [Google Scholar] [CrossRef]
- Medici, V.; Halsted, C.H. Folate, alcohol, and liver disease. Mol. Nutr. Food Res. 2013, 57, 596–606. [Google Scholar] [CrossRef] [PubMed]
- De Marco, P.; Calevo, M.G.; Moroni, A.; Arata, L.; Merello, E.; Finnell, R.H.; Zhu, H.; Andreussi, L.; Cama, A.; Capra, V. Study of MTHFR and MS polymorphisms as risk factors for NTD in the Italian population. J. Hum. Genet. 2002, 47, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Shaw, G.M.; Quach, T.; Nelson, V.; Carmichael, S.L.; Schaffer, D.M.; Selvin, S.; Yang, W. Neural tube defects associated with maternal periconceptional dietary intake of simple sugars and glycemic index. Am. J. Clin. Nutr. 2003, 78, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Zaganjor, I.; Sekkarie, A.; Tsang, B.L.; Williams, J.; Razzaghi, H.; Mulinare, J.; Sniezek, J.E.; Cannon, M.J.; Rosenthal, J. Describing the Prevalence of Neural Tube Defects Worldwide: A Systematic Literature Review. PLoS ONE 2016, 11, e0151586. [Google Scholar] [CrossRef]
- Pei, L.; Zhu, H.; Zhu, J.; Ren, A.; Finnell, R.H.; Li, Z. Genetic variation of infant reduced folate carrier (A80G) and risk of orofacial defects and congenital heart defects in China. Ann. Epidemiol. 2006, 16, 352–356. [Google Scholar] [CrossRef]
- Matherly, L.H.; Hou, Z. Folate transporter offers clues for anticancer drugs. Nature 2022, 612, 39–41. [Google Scholar] [CrossRef] [PubMed]
- Sorbi, D.; Boynton, J.; Lindor, K.D. The ratio of aspartate aminotransferase to alanine aminotransferase: Potential value in differentiating nonalcoholic steatohepatitis from alcoholic liver disease. Am. J. Gastroenterol. 1999, 94, 1018–1022. [Google Scholar] [CrossRef]
- Alnabbat, K.I.; Fardous, A.M.; Cabelof, D.C.; Heydari, A.R. Excessive Folic Acid Mimics Folate Deficiency in Human Lymphocytes. Curr. Issues Mol. Biol. 2022, 44, 1452–1462. [Google Scholar] [CrossRef]
- Chan, C.W.; Chan, P.H.; Lin, B.F. Folate Deficiency Increased Lipid Accumulation and Leptin Production of Adipocytes. Front. Nutr. 2022, 9, 852451. [Google Scholar] [CrossRef]
- Zhao, M.; Yuan, M.M.; Yuan, L.; Huang, L.L.; Liao, J.H.; Yu, X.L.; Su, C.; Chen, Y.H.; Yang, Y.Y.; Yu, H.; et al. Chronic folate deficiency induces glucose and lipid metabolism disorders and subsequent cognitive dysfunction in mice. PLoS ONE 2018, 13, e0202910. [Google Scholar] [CrossRef] [PubMed]
- Sid, V.; Shang, Y.; Siow, Y.L.; Hewage, S.M.; House, J.D.; Karmin, O. Folic Acid Supplementation Attenuates Chronic Hepatic Inflammation in High-Fat Diet Fed Mice. Lipids 2018, 53, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, M.; Singh, B.K.; Zhou, J.; Tikno, K.; Widjaja, A.; Sandireddy, R.; Arul, K.; Abdul Ghani, S.A.B.; Bee, G.G.B.; Wong, K.A.; et al. Vitamin B(12) and folate decrease inflammation and fibrosis in NASH by preventing syntaxin 17 homocysteinylation. J. Hepatol. 2022, 77, 1246–1255. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, J.; Liu, X.; Liu, R.; Wang, Y.; Huang, X.; Li, Y.; Liu, R.; Yang, X. Dietary folic acid addition reduces abdominal fat deposition mediated by alterations in gut microbiota and SCFA production in broilers. Anim. Nutr. 2023, 12, 54–62. [Google Scholar] [CrossRef]
- Kelly, K.B.; Kennelly, J.P.; Ordonez, M.; Nelson, R.; Leonard, K.; Stabler, S.; Gomez-Munoz, A.; Field, C.J.; Jacobs, R.L. Excess Folic Acid Increases Lipid Storage, Weight Gain, and Adipose Tissue Inflammation in High Fat Diet-Fed Rats. Nutrients 2016, 8, 594. [Google Scholar] [CrossRef]
- Yajnik, C.S.; Deshpande, S.S.; Jackson, A.A.; Refsum, H.; Rao, S.; Fisher, D.J.; Bhat, D.S.; Naik, S.S.; Coyaji, K.J.; Joglekar, C.V.; et al. Vitamin B12 and folate concentrations during pregnancy and insulin resistance in the offspring: The Pune Maternal Nutrition Study. Diabetologia 2008, 51, 29–38. [Google Scholar] [CrossRef]
- Akesson, B.; Fehling, C.; Jagerstad, M.; Stenram, U. Effect of experimental folate deficiency on lipid metabolism in liver and brain. Br. J. Nutr. 1982, 47, 505–520. [Google Scholar] [CrossRef] [PubMed]
- Mahamid, M.; Mahroum, N.; Bragazzi, N.L.; Shalaata, K.; Yavne, Y.; Adawi, M.; Amital, H.; Watad, A. Folate and B12 Levels Correlate with Histological Severity in NASH Patients. Nutrients 2018, 10, 440. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.; Jo, Y.; Choi, S.J.; Kim, S.M.; Kim, K.K.; Oh, B.C.; Ryu, D.; Paik, M.J.; Lee, D.H. Plasma Metabolomics and Machine Learning-Driven Novel Diagnostic Signature for Non-Alcoholic Steatohepatitis. Biomedicines 2022, 10, 1669. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Mayneris-Perxachs, J.; Boque, N.; Del Bas, J.M.; Arola, L.; Yuan, M.; Turkez, H.; Uhlen, M.; Boren, J.; Zhang, C.; et al. Combined Metabolic Activators Decrease Liver Steatosis by Activating Mitochondrial Metabolism in Hamsters Fed with a High-Fat Diet. Biomedicines 2021, 9, 1440. [Google Scholar] [CrossRef]
- Wang, D.Q.; Portincasa, P.; Neuschwander-Tetri, B.A. Steatosis in the liver. Compr. Physiol. 2013, 3, 1493–1532. [Google Scholar] [CrossRef]
- Basseri, S.; Austin, R.C. Endoplasmic reticulum stress and lipid metabolism: Mechanisms and therapeutic potential. Biochem. Res. Int. 2012, 2012, 841362. [Google Scholar] [CrossRef]
- Ruhanen, H.; Perttila, J.; Holtta-Vuori, M.; Zhou, Y.; Yki-Jarvinen, H.; Ikonen, E.; Kakela, R.; Olkkonen, V.M. PNPLA3 mediates hepatocyte triacylglycerol remodeling. J. Lipid Res. 2014, 55, 739–746. [Google Scholar] [CrossRef]
- Luo, W.J.; Cheng, T.Y.; Wong, K.I.; Fang, W.H.; Liao, K.M.; Hsieh, Y.T.; Su, K.Y. Novel therapeutic drug identification and gene correlation for fatty liver disease using high-content screening: Proof of concept. Eur. J. Pharm. Sci. 2018, 121, 106–117. [Google Scholar] [CrossRef]
- Gorden, D.L.; Ivanova, P.T.; Myers, D.S.; McIntyre, J.O.; VanSaun, M.N.; Wright, J.K.; Matrisian, L.M.; Brown, H.A. Increased diacylglycerols characterize hepatic lipid changes in progression of human nonalcoholic fatty liver disease; comparison to a murine model. PLoS ONE 2011, 6, e22775. [Google Scholar] [CrossRef]
- Clifford, A.J.; Rincon, G.; Owens, J.E.; Medrano, J.F.; Moshfegh, A.J.; Baer, D.J.; Novotny, J.A. Single nucleotide polymorphisms in CETP, SLC46A1, SLC19A1, CD36, BCMO1, APOA5, and ABCA1 are significant predictors of plasma HDL in healthy adults. Lipids Health Dis. 2013, 12, 66. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of hepatic glutathione synthesis: Current concepts and controversies. FASEB J. 1999, 13, 1169–1183. [Google Scholar] [CrossRef] [PubMed]
- Wieckowska, A.; Papouchado, B.G.; Li, Z.; Lopez, R.; Zein, N.N.; Feldstein, A.E. Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2008, 103, 1372–1379. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Ahmed, M.H. Nonalcoholic fatty liver disease and COVID-19: An epidemic that begets pandemic. World J. Clin. Cases 2021, 9, 4133–4142. [Google Scholar] [CrossRef] [PubMed]
- Chew, T.W.; Jiang, X.; Yan, J.; Wang, W.; Lusa, A.L.; Carrier, B.J.; West, A.A.; Malysheva, O.V.; Brenna, J.T.; Gregory, J.F., 3rd; et al. Folate intake, MTHFR genotype, and sex modulate choline metabolism in mice. J. Nutr. 2011, 141, 1475–1481. [Google Scholar] [CrossRef]
- da Costa, K.A.; Sanders, L.M.; Fischer, L.M.; Zeisel, S.H. Docosahexaenoic acid in plasma phosphatidylcholine may be a potential marker for in vivo phosphatidylethanolamine N-methyltransferase activity in humans. Am. J. Clin. Nutr. 2011, 93, 968–974. [Google Scholar] [CrossRef]
- Martinez-Una, M.; Varela-Rey, M.; Cano, A.; Fernandez-Ares, L.; Beraza, N.; Aurrekoetxea, I.; Martinez-Arranz, I.; Garcia-Rodriguez, J.L.; Buque, X.; Mestre, D.; et al. Excess S-adenosylmethionine reroutes phosphatidylethanolamine towards phosphatidylcholine and triglyceride synthesis. Hepatology 2013, 58, 1296–1305. [Google Scholar] [CrossRef]
- Christensen, K.E.; Mikael, L.G.; Leung, K.Y.; Levesque, N.; Deng, L.; Wu, Q.; Malysheva, O.V.; Best, A.; Caudill, M.A.; Greene, N.D.; et al. High folic acid consumption leads to pseudo-MTHFR deficiency, altered lipid metabolism, and liver injury in mice. Am. J. Clin. Nutr. 2015, 101, 646–658. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Traits | rs1051266 Genotype | Median | rs1051266 p-Value | rs3788200 Genotype | Median | rs3788200 p-Value |
|---|---|---|---|---|---|---|
| BMI (kg/m2) | AA | 29.37 | 0.0021 | GG | 28.1 | 0.0009 |
| AG | 34.65 | GA | 35.75 | |||
| GG | 40.35 | AA | 40.35 | |||
| Triglycerides (mmol/L) | AA | 1.89 | 0.0128 | GG | 1.9 | 0.0164 |
| AG | 1.89 | GA | 1.9 | |||
| GG | 1.69 | AA | 1.69 | |||
| AST/GOT (IU/L) | AA | 22 | 0.4921 | GG | 22 | 0.2502 |
| AG | 21 | GA | 21 | |||
| GG | 20.5 | AA | 20 | |||
| ALT/GPT (IU/L) | AA | 19 | 0.0142 | GG | 19 | 0.0498 |
| AG | 21 | GA | 21 | |||
| GG | 22 | AA | 22 | |||
| AST/ALT ratio | AA | 1.14 | 1.04 × 10−5 | GG | 1.12 | 9.97 × 10−6 |
| AG | 0.97 | GA | 0.97 | |||
| GG | 0.91 | AA | 0.91 | |||
| gGT (IU/L) | AA | 19.5 | 0.0258 | GG | 20 | 0.0298 |
| AG | 21 | GA | 21 | |||
| GG | 24 | AA | 24 | |||
| Glucose (mmol/L) | AA | 5.72 | 0.4609 | GG | 5.72 | 0.5239 |
| AG | 5.5 | GA | 5.5 | |||
| GG | 5.56 | AA | 5.55 | |||
| Insulin (mIU/L) | AA | 13.3 | 0.6585 | GG | 13.95 | 0.7243 |
| AG | 13.15 | GA | 13.5 | |||
| GG | 13.55 | AA | 13.5 | |||
| Total Cholesterol (mmol/L) | AA | 4.71 | 0.3726 | GG | 4.73 | 0.7426 |
| AG | 4.84 | GA | 4.83 | |||
| GG | 4.72 | AA | 4.76 | |||
| Bilirubin (µmol/L) | AA | 8 | 0.567 | GG | 8.21 | 0.3116 |
| AG | 7.01 | GA | 7.01 | |||
| GG | 7.52 | AA | 7.35 | |||
| Age (years) | AA | 36 | 0.5703 | GG | 36.5 | 0.7286 |
| AG | 36 | GA | 36 | |||
| GG | 37 | AA | 37 |
| Chemical Group | Class | Individual Notation | Student’s t-Test (p) | Log2 (Fold Change) | Fold Change |
|---|---|---|---|---|---|
| AA | Amino acids | Amino acids | 1.10 × 10−4 | −1.61 | 0.33 |
| NEFA | Non-esterified fatty acids | Non-esterified fatty acids | 2.64 × 10−3 | 0.85 | 1.80 |
| SFA | Non-esterified fatty acids | Saturated fatty acids | 8.82 × 10−1 | 0.08 | 1.06 |
| MUFA | Non-esterified fatty acids | Monounsaturated fatty acids | 1.55 × 10−1 | −0.16 | 0.89 |
| PUFA | Non-esterified fatty acids | Polyunsaturated fatty acids | 1.06 × 10−2 | 1.32 | 2.49 |
| oxFA | Oxidized fatty acids | Oxidized fatty acids | 1.87 × 10−4 | 1.45 | 2.73 |
| FAA | Fatty amides | Primary fatty amides | 5.28 × 10−2 | 0.99 | 1.98 |
| NAE | Fatty amides | N-Acyl ethanolamines | 3.25 × 10−2 | −0.39 | 0.76 |
| DG | Glycerolipids | Diglycerides | 3.35 × 10−3 | 1.35 | 2.56 |
| TG | Glycerolipids | Triglycerides | 3.49 × 10−2 | 0.73 | 1.65 |
| ChoE | Sterols | Cholesterol Esters | 1.30 × 10−2 | 1.43 | 2.69 |
| BA | Bile Acids | Bile Acids | 4.75 × 10−1 | −0.18 | 0.88 |
| ST | Sterols | Steroids | 2.12 × 10−3 | −0.84 | 0.56 |
| PE | Glycerophospholipids | Phosphatidylethanolamines | 1.93 × 10−5 | −0.24 | 0.85 |
| LPE | Glycerophospholipids | Lysophosphatidylethanolamines | 5.14 × 10−5 | −1.19 | 0.44 |
| PC | Glycerophospholipids | Phosphatidylcholines | 5.03 × 10−3 | −0.05 | 0.97 |
| LPC | Glycerophospholipids | Lysophosphatidylcholines | 8.39 × 10−3 | −0.94 | 0.52 |
| PI | Glycerophospholipids | Phosphatidylinositol | 1.33 × 10−2 | 0.34 | 1.27 |
| LPI | Glycerophospholipids | Lysophosphatidylinositol | 6.80 × 10−2 | −0.28 | 0.83 |
| LPG | Glycerophospholipids | Lysophosphatidylglycerol | 1.10 × 10−2 | −0.96 | 0.52 |
| Cer | Sphingolipids | Ceramides | 3.35 × 10−5 | −1.06 | 0.48 |
| SM | Sphingolipids | Sphingomyelins | 4.81 × 10−7 | −0.67 | 0.63 |
| CMH | Sphingolipids | Monohexosylceramides | 2.04 × 10−6 | −1.63 | 0.32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cano, A.; Vazquez-Chantada, M.; Conde-Vancells, J.; Gonzalez-Lahera, A.; Mosen-Ansorena, D.; Blanco, F.J.; Clément, K.; Aron-Wisnewsky, J.; Tran, A.; Gual, P.; et al. Impaired Function of Solute Carrier Family 19 Leads to Low Folate Levels and Lipid Droplet Accumulation in Hepatocytes. Biomedicines 2023, 11, 337. https://doi.org/10.3390/biomedicines11020337
Cano A, Vazquez-Chantada M, Conde-Vancells J, Gonzalez-Lahera A, Mosen-Ansorena D, Blanco FJ, Clément K, Aron-Wisnewsky J, Tran A, Gual P, et al. Impaired Function of Solute Carrier Family 19 Leads to Low Folate Levels and Lipid Droplet Accumulation in Hepatocytes. Biomedicines. 2023; 11(2):337. https://doi.org/10.3390/biomedicines11020337
Chicago/Turabian StyleCano, Ainara, Mercedes Vazquez-Chantada, Javier Conde-Vancells, Aintzane Gonzalez-Lahera, David Mosen-Ansorena, Francisco J. Blanco, Karine Clément, Judith Aron-Wisnewsky, Albert Tran, Philippe Gual, and et al. 2023. "Impaired Function of Solute Carrier Family 19 Leads to Low Folate Levels and Lipid Droplet Accumulation in Hepatocytes" Biomedicines 11, no. 2: 337. https://doi.org/10.3390/biomedicines11020337
APA StyleCano, A., Vazquez-Chantada, M., Conde-Vancells, J., Gonzalez-Lahera, A., Mosen-Ansorena, D., Blanco, F. J., Clément, K., Aron-Wisnewsky, J., Tran, A., Gual, P., García-Monzón, C., Caballería, J., Castro, A., Martínez-Chantar, M. L., Mato, J. M., Zhu, H., Finnell, R. H., & Aransay, A. M. (2023). Impaired Function of Solute Carrier Family 19 Leads to Low Folate Levels and Lipid Droplet Accumulation in Hepatocytes. Biomedicines, 11(2), 337. https://doi.org/10.3390/biomedicines11020337