
Surveying the Proteome-Wide Landscape of Mitoxantrone and Examining Drug Sensitivity in BRCA1-Deficient Ovarian Cancer Using Quantitative Proteomics

 , , , , ,
, , , , ,  and
and
Abstract
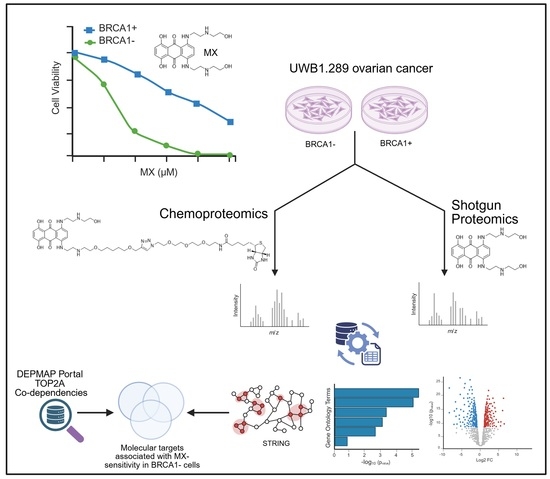
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Whole Cell Lysate Proteomics
2.3. Affinity Purification of MXP Binding Proteins
2.4. Chemoproteomics
2.5. Proteomics Analysis
2.6. DEPMAP
2.7. ProteinSimple (Bio-Techne) Peggy Sue Simple Western (Supplemental)
3. Results and Discussion
3.1. Comprehensive Molecular Characterization of MX Treatment in BRCA1− UWB1.289 Cells Using Quantitative Shotgun Proteomics
3.2. Exploring the Target Landscape of MX in BRCA1− and BRCA1+ UWB1.289 Ovarian Cancer Cells Using MXP
3.3. Identifying Essential Gene Relationships That Influence Cellular Response and May Incite Synthetic Lethal Interactions upon MX Treatment
4. Limitations and Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Damiani, R.M.; Moura, D.J.; Viau, C.M.; Caceres, R.A.; Henriques, J.A.P.; Saffi, J. Pathways of cardiac toxicity: Comparison between chemotherapeutic drugs doxorubicin and mitoxantrone. Arch. Toxicol. 2016, 90, 2063–2076. [Google Scholar] [CrossRef]
- Evison, B.J.; Sleebs, B.E.; Watson, K.G.; Phillips, D.R.; Cutts, S.M. Mitoxantrone, More than Just Another Topoisomerase II Poison. Med. Res. Rev. 2016, 36, 248–299. [Google Scholar] [CrossRef] [PubMed]
- Fortune, J.M.; Osheroff, N. Topoisomerase II as a target for anticancer drugs: When enzymes stop being nice. Prog. Nucleic Acid. Res. Mol. Biol. 2000, 64, 221–253. [Google Scholar] [CrossRef] [PubMed]
- Wyhs, N.; Walker, D.; Giovinazzo, H.; Yegnasubramanian, S.; Nelson, W.G. Time-Resolved Fluorescence Resonance Energy Transfer Assay for Discovery of Small-Molecule Inhibitors of Methyl-CpG Binding Domain Protein 2. J. Biomol. Screen. 2014, 19, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Zhang, W.; Li, L.; Xie, Y.; Li, W.; Huang, N. A new target for an old drug: Identifying mitoxantrone as a nanomolar inhibitor of PIM1 kinase via kinome-wide selectivity modeling. J. Med. Chem. 2013, 56, 2619–2629. [Google Scholar] [CrossRef]
- Minko, I.G.; Moellmer, S.A.; Luzadder, M.M.; Tomar, R.; Stone, M.P.; McCullough, A.K.; Lloyd, R.S. Interaction of mitoxantrone with abasic sites—DNA strand cleavage and inhibition of apurinic/apyrimidinic endonuclease 1, APE1. DNA Repair 2024, 133, 103606. [Google Scholar] [CrossRef]
- Al-Mugotir, M.; Lovelace, J.J.; George, J.; Bessho, M.; Pal, D.; Struble, L.; Kolar, C.; Rana, S.; Natarajan, A.; Bessho, T.; et al. Selective killing of homologous recombination-deficient cancer cell lines by inhibitors of the RPA:RAD52 protein-protein interaction. PLoS ONE 2021, 16, e0248941. [Google Scholar] [CrossRef]
- Wallin, S.; Singh, S.; Borgstahl, G.E.O.; Natarajan, A. Design, synthesis, and evaluation of a mitoxantrone probe (MXP) for biological studies. Bioorg. Med. Chem. Lett. 2023, 94, 129465. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef]
- Toh, M.; Ngeow, J. Homologous Recombination Deficiency: Cancer Predispositions and Treatment Implications. Oncologist 2021, 26, e1526–e1537. [Google Scholar] [CrossRef]
- Shi, Z.; Chen, B.; Han, X.; Gu, W.; Liang, S.; Wu, L. Genomic and molecular landscape of homologous recombination deficiency across multiple cancer types. Sci. Rep. 2023, 13, 8899. [Google Scholar] [CrossRef]
- Murai, J.; Pommier, Y. BRCAness, Homologous Recombination Deficiencies, and Synthetic Lethality. Cancer Res. 2023, 83, 1173–1174. [Google Scholar] [CrossRef]
- Peng, G.; Chun-Jen Lin, C.; Mo, W.; Dai, H.; Park, Y.Y.; Kim, S.M.; Peng, Y.; Mo, Q.; Siwko, S.; Hu, R.; et al. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat. Commun. 2014, 5, 3361. [Google Scholar] [CrossRef]
- Li, X.; Zou, L. BRCAness, DNA gaps, and gain and loss of PARP inhibitor-induced synthetic lethality. J. Clin. Investig. 2024, 134, e181062. [Google Scholar] [CrossRef]
- DelloRusso, C.; Welcsh, P.L.; Wang, W.; Garcia, R.L.; King, M.C.; Swisher, E.M. Functional characterization of a novel BRCA1-null ovarian cancer cell line in response to ionizing radiation. Mol. Cancer Res. 2007, 5, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.; D’Costa, Z.; Ray, S.; Gorski, J.; Harkin, D.P.; Mullan, P.; Panov, K.I. The identification of a novel role for BRCA1 in regulating RNA polymerase I transcription. Oncotarget 2016, 7, 68097–68110. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.I.; Gorski, J.J.; Barros, E.M.; Irwin, G.W.; Manti, L.; Powell, A.J.; Pellagatti, A.; Lukashchuk, N.; McCance, D.J.; McCluggage, W.G.; et al. Identification of a BRCA1-mRNA splicing complex required for efficient DNA repair and maintenance of genomic stability. Mol. Cell 2014, 54, 445–459. [Google Scholar] [CrossRef]
- Scully, R.; Anderson, S.F.; Chao, D.M.; Wei, W.; Ye, L.; Young, R.A.; Livingston, D.M.; Parvin, J.D. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc. Natl. Acad. Sci. USA 1997, 94, 5605–5610. [Google Scholar] [CrossRef]
- Tarsounas, M.; Sung, P. The antitumorigenic roles of BRCA1-BARD1 in DNA repair and replication. Nat. Rev. Mol. Cell Biol. 2020, 21, 284–299. [Google Scholar] [CrossRef]
- Wu, D.; Huang, H.; Chen, T.; Gai, X.; Li, Q.; Wang, C.; Yao, J.; Liu, Y.; Cai, S.; Yu, X. The BRCA1/BARD1 complex recognizes pre-ribosomal RNA to facilitate homologous recombination. Cell Discov. 2023, 9, 99. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Nesvizhskii, A.I.; Kolker, E.; Aebersold, R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002, 74, 5383–5392. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef] [PubMed]
- Nesvizhskii, A.I. Computational and informatics strategies for identification of specific protein interaction partners in affinity purification mass spectrometry experiments. Proteomics 2012, 12, 1639–1655. [Google Scholar] [CrossRef]
- Kuenzi, B.M.; Borne, A.L.; Li, J.; Haura, E.B.; Eschrich, S.A.; Koomen, J.M.; Rix, U.; Stewart, P.A. APOSTL: An Interactive Galaxy Pipeline for Reproducible Analysis of Affinity Proteomics Data. J. Proteome Res. 2016, 15, 4747–4754. [Google Scholar] [CrossRef]
- Stewart, P.A.; Kuenzi, B.M.; Mehta, S.; Kumar, P.; Johnson, J.E.; Jagtap, P.; Griffin, T.J.; Haura, E.B. The Galaxy Platform for Reproducible Affinity Proteomic Mass Spectrometry Data Analysis. Methods Mol. Biol. 2019, 1977, 249–261. [Google Scholar] [CrossRef]
- Choi, H.; Larsen, B.; Lin, Z.Y.; Breitkreutz, A.; Mellacheruvu, D.; Fermin, D.; Qin, Z.S.; Tyers, M.; Gingras, A.C.; Nesvizhskii, A.I. SAINT: Probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods 2011, 8, 70–73. [Google Scholar] [CrossRef]
- Knight, J.D.; Liu, G.; Zhang, J.P.; Pasculescu, A.; Choi, H.; Gingras, A.C. A web-tool for visualizing quantitative protein-protein interaction data. Proteomics 2015, 15, 1432–1436. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.D.R.; Choi, H.; Gupta, G.D.; Pelletier, L.; Raught, B.; Nesvizhskii, A.I.; Gingras, A.C. ProHits-viz: A suite of web tools for visualizing interaction proteomics data. Nat. Methods 2017, 14, 645–646. [Google Scholar] [CrossRef] [PubMed]
- Mellacheruvu, D.; Wright, Z.; Couzens, A.L.; Lambert, J.P.; St-Denis, N.A.; Li, T.; Miteva, Y.V.; Hauri, S.; Sardiu, M.E.; Low, T.Y.; et al. The CRAPome: A contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 2013, 10, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Arafeh, R.; Shibue, T.; Dempster, J.M.; Hahn, W.C.; Vazquez, F. The present and future of the Cancer Dependency Map. Nat. Rev. Cancer 2025, 25, 59–73. [Google Scholar] [CrossRef]
- McFarland, J.M.; Ho, Z.V.; Kugener, G.; Dempster, J.M.; Montgomery, P.G.; Bryan, J.G.; Krill-Burger, J.M.; Green, T.M.; Vazquez, F.; Boehm, J.S.; et al. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun. 2018, 9, 4610. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Dempster, J.M.; Boyle, I.; Vazquez, F.; Root, D.E.; Boehm, J.S.; Hahn, W.C.; Tsherniak, A.; McFarland, J.M. Chronos: A cell population dynamics model of CRISPR experiments that improves inference of gene fitness effects. Genome Biol. 2021, 22, 343. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691. [Google Scholar] [CrossRef]
- Elhamamsy, A.R.; Metge, B.J.; Alsheikh, H.A.; Shevde, L.A.; Samant, R.S. Ribosome Biogenesis: A Central Player in Cancer Metastasis and Therapeutic Resistance. Cancer Res. 2022, 82, 2344–2353. [Google Scholar] [CrossRef]
- Zisi, A.; Bartek, J.; Lindstrom, M.S. Targeting Ribosome Biogenesis in Cancer: Lessons Learned and Way Forward. Cancers 2022, 14, 2126. [Google Scholar] [CrossRef]
- Khan, S.N.; Danishuddin, M.; Khan, A.U. Inhibition of transcription factor assembly and structural stability on mitoxantrone binding with DNA. Biosci. Rep. 2010, 30, 331–340. [Google Scholar] [CrossRef]
- Khan, S.N.; Yennamalli, R.; Subbarao, N.; Khan, A.U. Mitoxantrone induced impediment of histone acetylation and structural flexibility of the protein. Cell Biochem. Biophys. 2011, 60, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Panova, T.; Miller, G.; Volkov, A.; Porter, A.C.; Russell, J.; Panov, K.I.; Zomerdijk, J.C. Topoisomerase IIalpha promotes activation of RNA polymerase I transcription by facilitating pre-initiation complex formation. Nat. Commun. 2013, 4, 1598. [Google Scholar] [CrossRef] [PubMed]
- Ruff, S.E.; Logan, S.K.; Garabedian, M.J.; Huang, T.T. Roles for MDC1 in cancer development and treatment. DNA Repair 2020, 95, 102948. [Google Scholar] [CrossRef]
- Majka, J.; Burgers, P.M. The PCNA-RFC families of DNA clamps and clamp loaders. Prog. Nucleic Acid. Res. Mol. Biol. 2004, 78, 227–260. [Google Scholar] [CrossRef]
- Shiomi, Y.; Nishitani, H. Control of Genome Integrity by RFC Complexes; Conductors of PCNA Loading onto and Unloading from Chromatin during DNA Replication. Genes 2017, 8, 52. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Cheok, C.F. RIF1: A novel regulatory factor for DNA replication and DNA damage response signaling. DNA Repair 2014, 15, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, K.; Tojo, A.; Kobayashi, H.; Shimizu, K.; Kamimura, Y.; Horikoshi, Y.; Fukuto, A.; Sun, J.; Yasui, M.; Honma, M.; et al. Dose-dependent effects of histone methyltransferase NSD2 on site-specific double-strand break repair. Genes Cells 2024, 29, 951–965. [Google Scholar] [CrossRef]
- Burger, K.; Muhl, B.; Harasim, T.; Rohrmoser, M.; Malamoussi, A.; Orban, M.; Kellner, M.; Gruber-Eber, A.; Kremmer, E.; Holzel, M.; et al. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J. Biol. Chem. 2010, 285, 12416–12425. [Google Scholar] [CrossRef]
- Chegini, N.; Safa, A.R.; Tseng, M.T. Acute effects of mitoxantrone on the template activity of isolated nuclei from the T-47D human breast tumor cell line. Cancer Lett. 1984, 21, 329–336. [Google Scholar] [CrossRef]
- Bruno, P.M.; Liu, Y.; Park, G.Y.; Murai, J.; Koch, C.E.; Eisen, T.J.; Pritchard, J.R.; Pommier, Y.; Lippard, S.J.; Hemann, M.T. A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat. Med. 2017, 23, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Pietrzak, M.; Smith, S.C.; Geralds, J.T.; Hagg, T.; Gomes, C.; Hetman, M. Nucleolar disruption and apoptosis are distinct neuronal responses to etoposide-induced DNA damage. J. Neurochem. 2011, 117, 1033–1046. [Google Scholar] [CrossRef] [PubMed]
- Damiani, R.M.; Moura, D.J.; Viau, C.M.; Brito, V.; Moras, A.M.; Henriques, J.A.P.; Saffi, J. Influence of PARP-1 inhibition in the cardiotoxicity of the topoisomerase 2 inhibitors doxorubicin and mitoxantrone. Toxicol. Vitr. 2018, 52, 203–213. [Google Scholar] [CrossRef]
- Brandao, S.R.; Carvalho, F.; Amado, F.; Ferreira, R.; Costa, V.M. Insights on the molecular targets of cardiotoxicity induced by anticancer drugs: A systematic review based on proteomic findings. Metabolism 2022, 134, 155250. [Google Scholar] [CrossRef] [PubMed]
- Hajihassan, Z.; Rabbani-Chadegani, A. Interaction of mitoxantrone, as an anticancer drug, with chromatin proteins, core histones and H1, in solution. Int. J. Biol. Macromol. 2011, 48, 87–92. [Google Scholar] [CrossRef]
- Hajihassan, Z.; Rabbani-Chadegani, A. Studies on the binding affinity of anticancer drug mitoxantrone to chromatin, DNA and histone proteins. J. Biomed. Sci. 2009, 16, 31. [Google Scholar] [CrossRef]
- Li, S.; Li, R.; Ma, Y.; Zhang, C.; Huang, T.; Zhu, S. Transcriptome analysis of differentially expressed genes and pathways associated with mitoxantrone treatment prostate cancer. J. Cell. Mol. Med. 2019, 23, 1987–2000. [Google Scholar] [CrossRef]
- Wang, Z.; Hou, R.; Wang, S.; Chen, M.; Zheng, D.; Zhang, Z.; Bai, L.; Chang, C.; Zhou, S. FGFBP1 promotes triple-negative breast cancer progression through the KLK10-AKT axis. Biochem. Biophys. Res. Commun. 2025, 763, 151763. [Google Scholar] [CrossRef]
- Shao, Z.; Abdel-Maksoud, M.A.; Saleh, I.A.; Al-Hawadi, J.S.; Zomot, N.; Almutairi, S.M.; Chen, J. Comprehensive multi-omics analysis and prognostic significance of fibroblast growth factor binding protein 1 (FGFBP1) in pancreatic adenocarcinoma. Am. J. Transl. Res. 2024, 16, 4101–4119. [Google Scholar] [CrossRef]
- Zhang, Z.; Qin, Y.; Ji, S.; Xu, W.; Liu, M.; Hu, Q.; Ye, Z.; Fan, G.; Yu, X.; Liu, W.; et al. FGFBP1-mediated crosstalk between fibroblasts and pancreatic cancer cells via FGF22/FGFR2 promotes invasion and metastasis of pancreatic cancer. Acta Biochim. Biophys. Sin. 2021, 53, 997–1008. [Google Scholar] [CrossRef]
- Schmidt, M.O.; Garman, K.A.; Lee, Y.G.; Zuo, C.; Beck, P.J.; Tan, M.; Aguilar-Pimentel, J.A.; Ollert, M.; Schmidt-Weber, C.; Fuchs, H.; et al. The Role of Fibroblast Growth Factor-Binding Protein 1 in Skin Carcinogenesis and Inflammation. J. Invest. Dermatol. 2018, 138, 179–188. [Google Scholar] [CrossRef]
- Zhang, W.; Zhou, Y.; Li, C.; Xu, S.; Li, M.; Liu, W.; Ma, Y.; Wang, H. The Expression and Prognostic Value of FGF2, FGFR3, and FGFBP1 in Esophageal Squamous Cell Carcinoma. Anal. Cell. Pathol. 2020, 2020, 2872479. [Google Scholar] [CrossRef]
- Ho, C.K.; Law, S.L.; Chiang, H.; Hsu, M.L.; Wang, C.C.; Wang, S.Y. Inhibition of microtubule assembly is a possible mechanism of action of mitoxantrone. Biochem. Biophys. Res. Commun. 1991, 180, 118–123. [Google Scholar] [CrossRef]
- Seki, A.; Fang, G. CKAP2 is a spindle-associated protein degraded by APC/C-Cdh1 during mitotic exit. J. Biol. Chem. 2007, 282, 15103–15113. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.U.; Park, Y.S.; Seong, Y.S.; Kang, D.; Bae, C.D.; Park, J. Functional importance of the anaphase-promoting complex-Cdh1-mediated degradation of TMAP/CKAP2 in regulation of spindle function and cytokinesis. Mol. Cell. Biol. 2007, 27, 3667–3681. [Google Scholar] [CrossRef] [PubMed]
- Soliman, T.N.; Keifenheim, D.; Parker, P.J.; Clarke, D.J. Cell cycle responses to Topoisomerase II inhibition: Molecular mechanisms and clinical implications. J. Cell Biol. 2023, 222, e202209125. [Google Scholar] [CrossRef] [PubMed]
- Foo, T.K.; Xia, B. BRCA1-Dependent and Independent Recruitment of PALB2-BRCA2-RAD51 in the DNA Damage Response and Cancer. Cancer Res. 2022, 82, 3191–3197. [Google Scholar] [CrossRef]
- Ibanez-Cabellos, J.S.; Seco-Cervera, M.; Picher-Latorre, C.; Perez-Machado, G.; Garcia-Gimenez, J.L.; Pallardo, F.V. Acute depletion of telomerase components DKC1 and NOP10 induces oxidative stress and disrupts ribosomal biogenesis via NPM1 and activation of the P53 pathway. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118845. [Google Scholar] [CrossRef]
- Choudhury, P.; Hackert, P.; Memet, I.; Sloan, K.E.; Bohnsack, M.T. The human RNA helicase DHX37 is required for release of the U3 snoRNP from pre-ribosomal particles. RNA Biol. 2019, 16, 54–68. [Google Scholar] [CrossRef]
- Kobayashi, W.; Takizawa, Y.; Aihara, M.; Negishi, L.; Ishii, H.; Kurumizaka, H. Structural and biochemical analyses of the nuclear pore complex component ELYS identify residues responsible for nucleosome binding. Commun. Biol. 2019, 2, 163. [Google Scholar] [CrossRef] [PubMed]
- Roychowdhury, A.; Joret, C.; Bourgeois, G.; Heurgue-Hamard, V.; Lafontaine, D.L.J.; Graille, M. The DEAH-box RNA helicase Dhr1 contains a remarkable carboxyl terminal domain essential for small ribosomal subunit biogenesis. Nucleic Acids Res. 2019, 47, 7548–7563. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Reyes, Y.; Fonseca-Rodriguez, C.; Freire, R.; Smits, V.A.J. DDX37 and DDX50 Maintain Genome Stability by Preventing Transcription-dependent R-loop Formation. J. Mol. Biol. 2025, 437, 169061. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.C.; Qi, L.; Mitra, P.; Huang, Y.; Hu, Y.; Li, R. R-loop functions in Brca1-associated mammary tumorigenesis. Proc. Natl. Acad. Sci. USA 2024, 121, e2403600121. [Google Scholar] [CrossRef]
- Bader, A.S.; Hawley, B.R.; Wilczynska, A.; Bushell, M. The roles of RNA in DNA double-strand break repair. Br. J. Cancer 2020, 122, 613–623. [Google Scholar] [CrossRef]
- Veras, I.; Rosen, E.M.; Schramm, L. Inhibition of RNA polymerase III transcription by BRCA1. J. Mol. Biol. 2009, 387, 523–531. [Google Scholar] [CrossRef]
- Koh, G.C.C.; Boushaki, S.; Zhao, S.J.; Pregnall, A.M.; Sadiyah, F.; Badja, C.; Memari, Y.; Georgakopoulos-Soares, I.; Nik-Zainal, S. The chemotherapeutic drug CX-5461 is a potent mutagen in cultured human cells. Nat. Genet. 2024, 56, 23–26. [Google Scholar] [CrossRef]
- Mars, J.C.; Tremblay, M.G.; Valere, M.; Sibai, D.S.; Sabourin-Felix, M.; Lessard, F.; Moss, T. The chemotherapeutic agent CX-5461 irreversibly blocks RNA polymerase I initiation and promoter release to cause nucleolar disruption, DNA damage and cell inviability. NAR Cancer 2020, 2, zcaa032. [Google Scholar] [CrossRef]
- Sanij, E.; Hannan, K.M.; Xuan, J.; Yan, S.; Ahern, J.E.; Trigos, A.S.; Brajanovski, N.; Son, J.; Chan, K.T.; Kondrashova, O.; et al. CX-5461 activates the DNA damage response and demonstrates therapeutic efficacy in high-grade serous ovarian cancer. Nat. Commun. 2020, 11, 2641. [Google Scholar] [CrossRef]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Santos, N.D.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Hurley, L.H. A first-in-class clinical G-quadruplex-targeting drug. The bench-to-bedside translation of the fluoroquinolone QQ58 to CX-5461 (Pidnarulex). Bioorg Med. Chem. Lett. 2022, 77, 129016. [Google Scholar] [CrossRef]
- Tripathi, S.; Pradeep, T.P.; Barthwal, R. Molecular Recognition of Parallel DNA Quadruplex d(TTAGGGT)4 by Mitoxantrone: Binding with 1:2 Stoichiometry Leading to Thermal Stabilization and Telomerase Inhibition. ChemBioChem 2016, 17, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bandla, C.; Kundu, D.J.; Kamatchinathan, S.; Bai, J.; Hewapathirana, S.; John, N.S.; Prakash, A.; Walzer, M.; Wang, S.; et al. The PRIDE database at 20 years: 2025 update. Nucleic Acids Res. 2025, 53, D543–D553. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enrichment Factor (EF) | ||
|---|---|---|
| BRCA1− | BRCA1+ | |
| TOP2A | 1.92 | 1.01 |
| MDC1 | Enriched | Enriched |
| RFC1 | 5.25 | 10.60 |
| RFC4 | 3.61 | 0.80 |
| NSD2 | 11.05 | Enriched |
| RIF1 | 0.76 | 0.58 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wallin, S.; Pandithar, S.; Singh, S.; Kumar, S.; Natarajan, A.; Borgstahl, G.E.O.; Woods, N. Surveying the Proteome-Wide Landscape of Mitoxantrone and Examining Drug Sensitivity in BRCA1-Deficient Ovarian Cancer Using Quantitative Proteomics. Proteomes 2025, 13, 61. https://doi.org/10.3390/proteomes13040061
Wallin S, Pandithar S, Singh S, Kumar S, Natarajan A, Borgstahl GEO, Woods N. Surveying the Proteome-Wide Landscape of Mitoxantrone and Examining Drug Sensitivity in BRCA1-Deficient Ovarian Cancer Using Quantitative Proteomics. Proteomes. 2025; 13(4):61. https://doi.org/10.3390/proteomes13040061
Chicago/Turabian StyleWallin, Savanna, Sneha Pandithar, Sarbjit Singh, Siddhartha Kumar, Amarnath Natarajan, Gloria E. O. Borgstahl, and Nicholas Woods. 2025. "Surveying the Proteome-Wide Landscape of Mitoxantrone and Examining Drug Sensitivity in BRCA1-Deficient Ovarian Cancer Using Quantitative Proteomics" Proteomes 13, no. 4: 61. https://doi.org/10.3390/proteomes13040061
APA StyleWallin, S., Pandithar, S., Singh, S., Kumar, S., Natarajan, A., Borgstahl, G. E. O., & Woods, N. (2025). Surveying the Proteome-Wide Landscape of Mitoxantrone and Examining Drug Sensitivity in BRCA1-Deficient Ovarian Cancer Using Quantitative Proteomics. Proteomes, 13(4), 61. https://doi.org/10.3390/proteomes13040061