Millet Could Be both a Weed and Serve as a Virus Reservoir in Crop Fields



Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
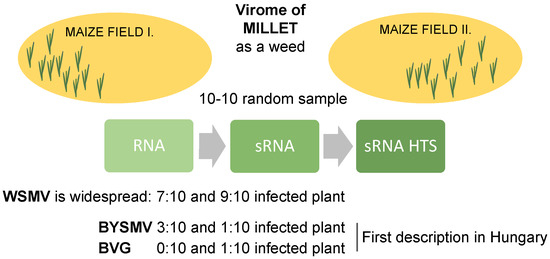
2.1. sRNA HTS of Millet Populations at Two Locations
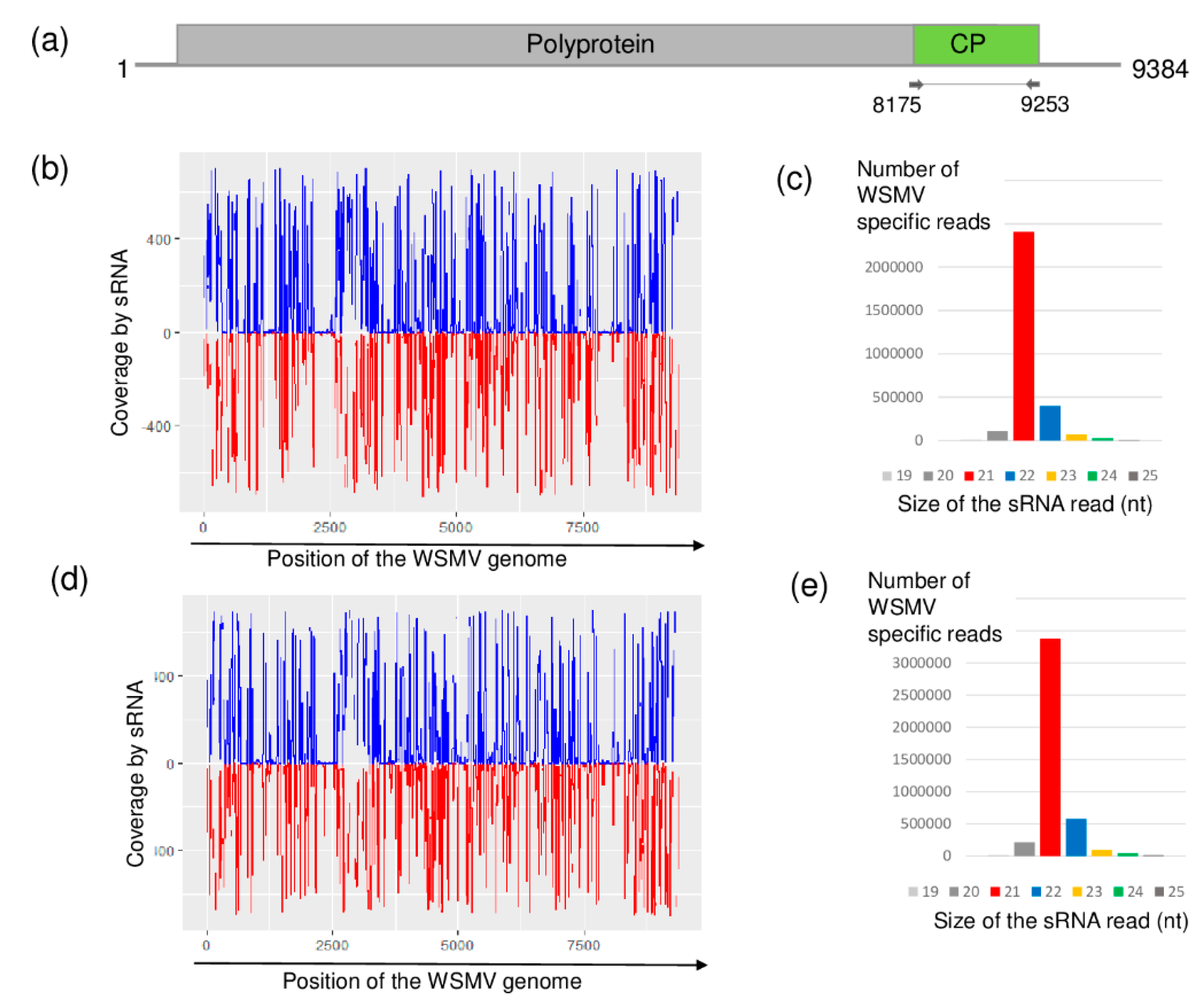
2.2. Millet Weed Is Highly Infected with WSMV
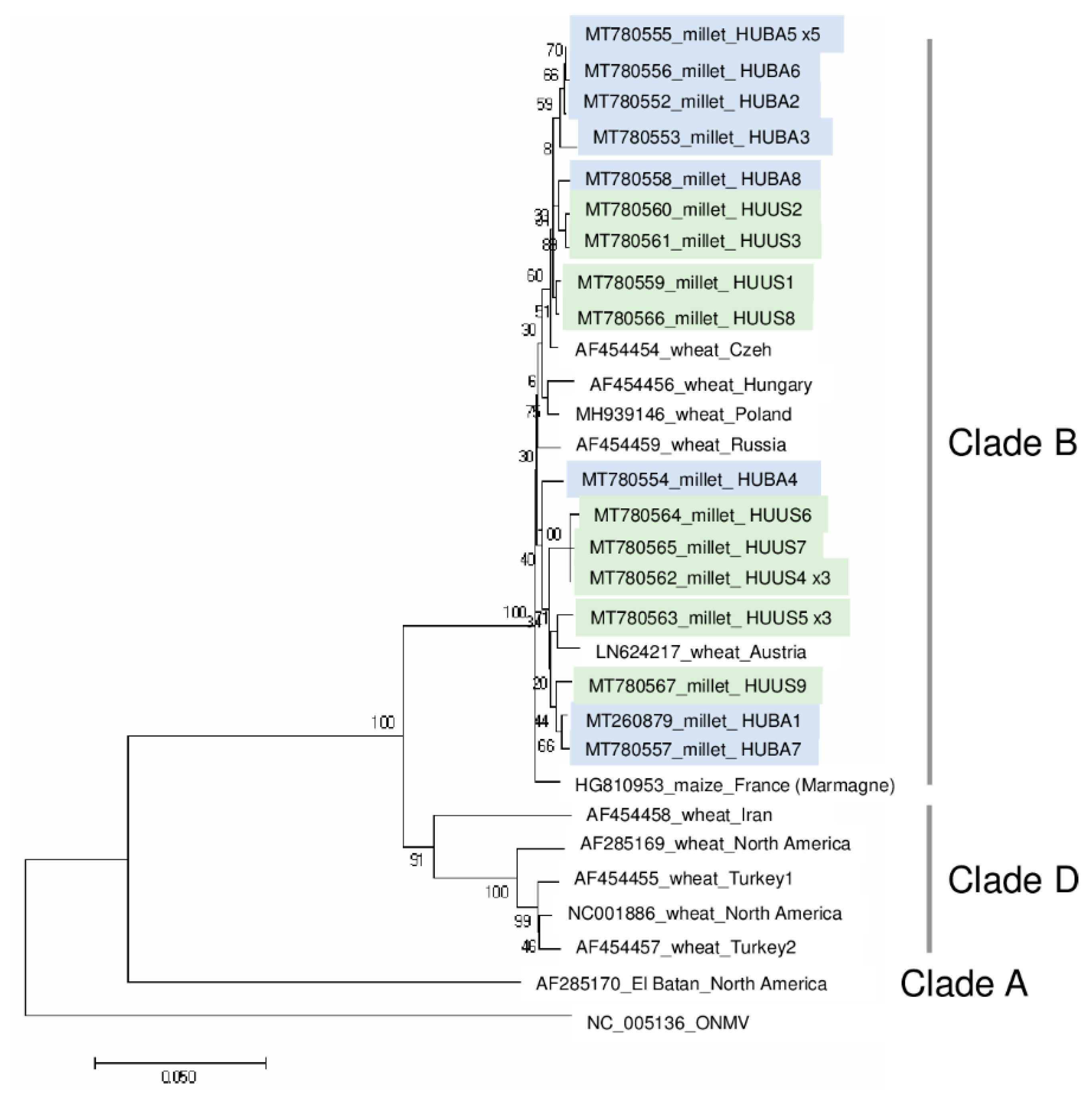
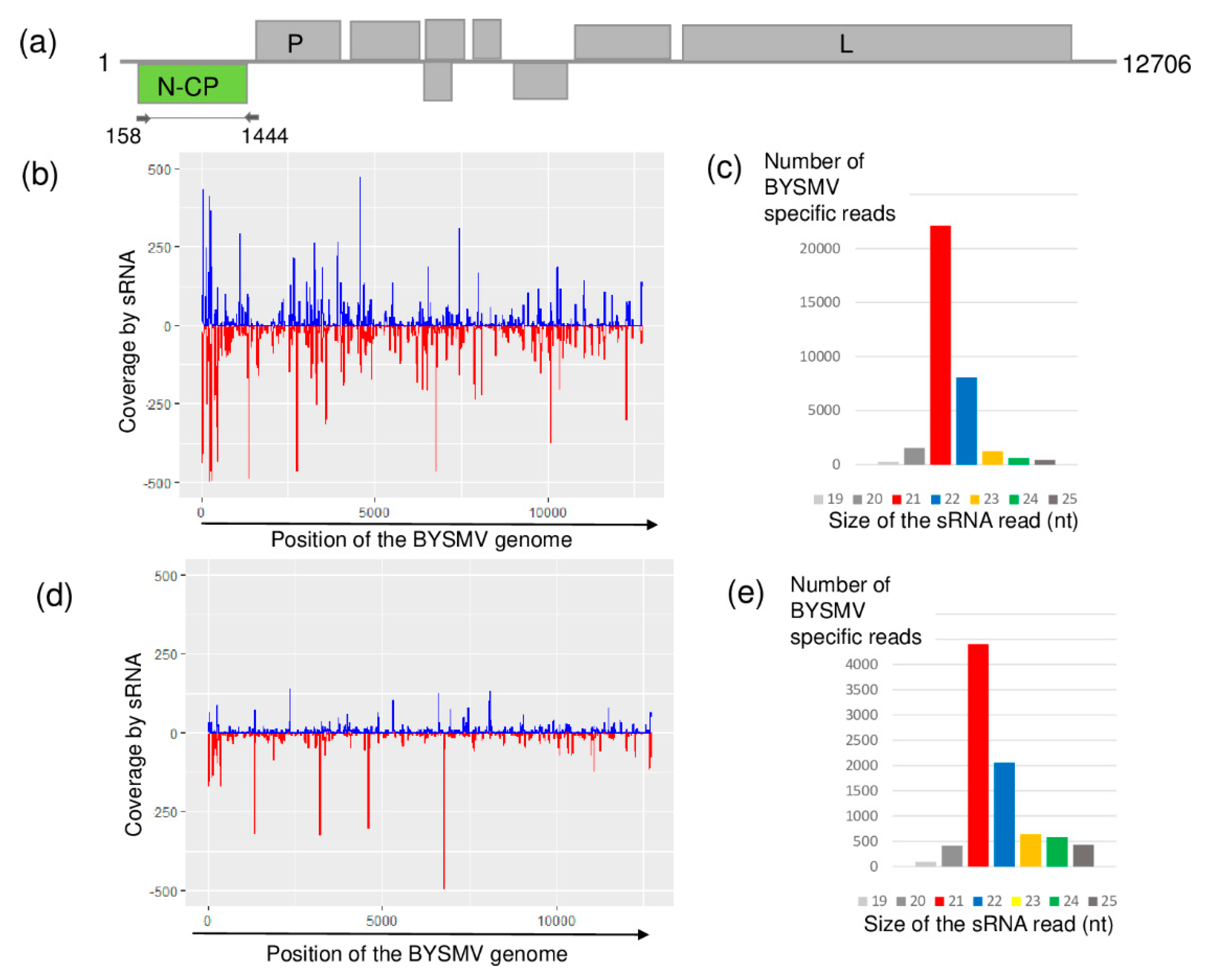
2.3. Different Strains of BYSMV Were Present at the Two Locations
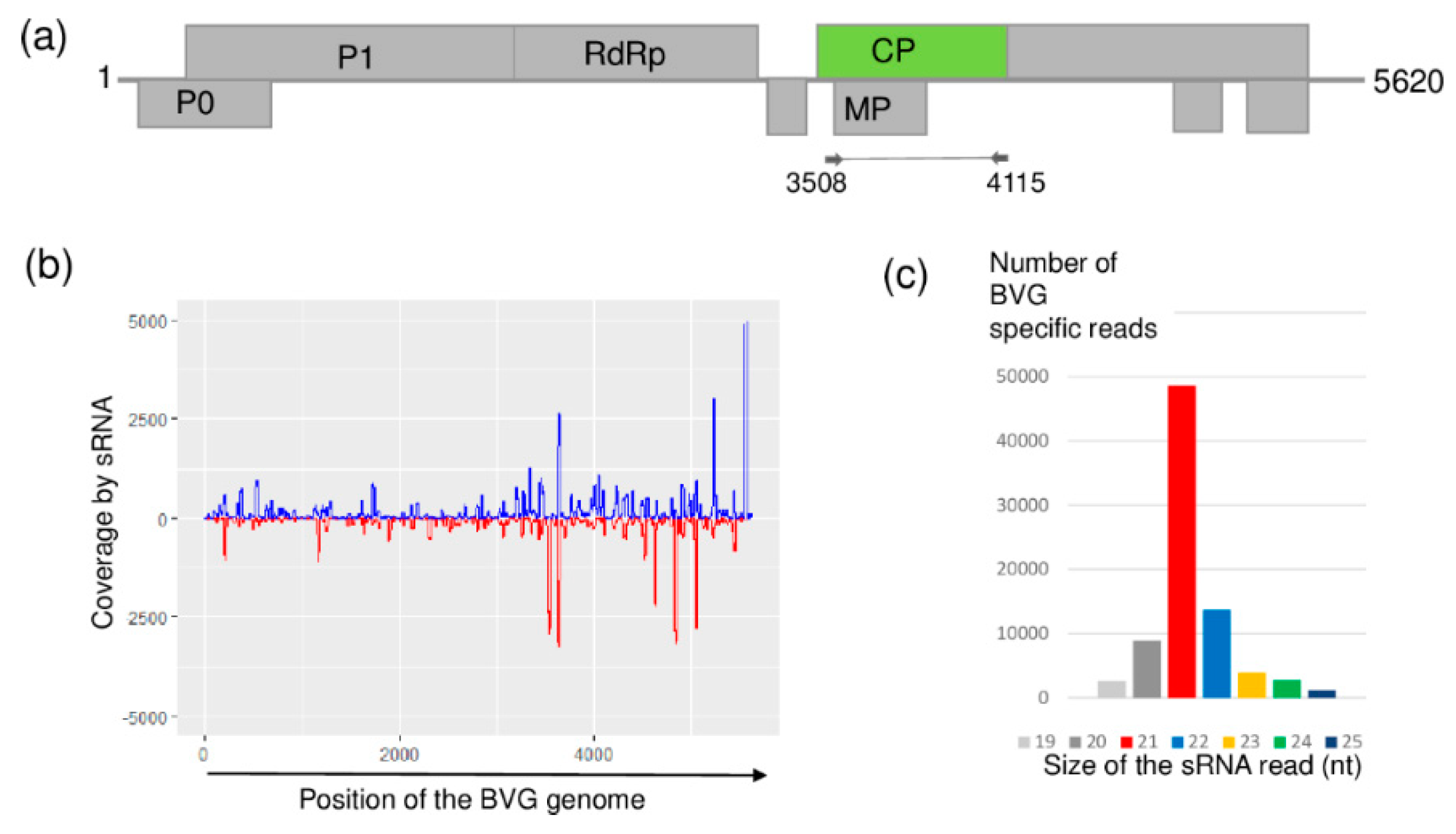
2.4. BVG, Which Had Not Previously Been Described in Hungary, Is Present at US
3. Materials and Methods
3.1. Plant Material and Sample Preparation
3.2. sRNA Library Preparation and Sequencing
3.3. Pipeline for Data Evaluation of HTS Results (Bioinformatics)
3.4. Virus Diagnostics by RT-PCR
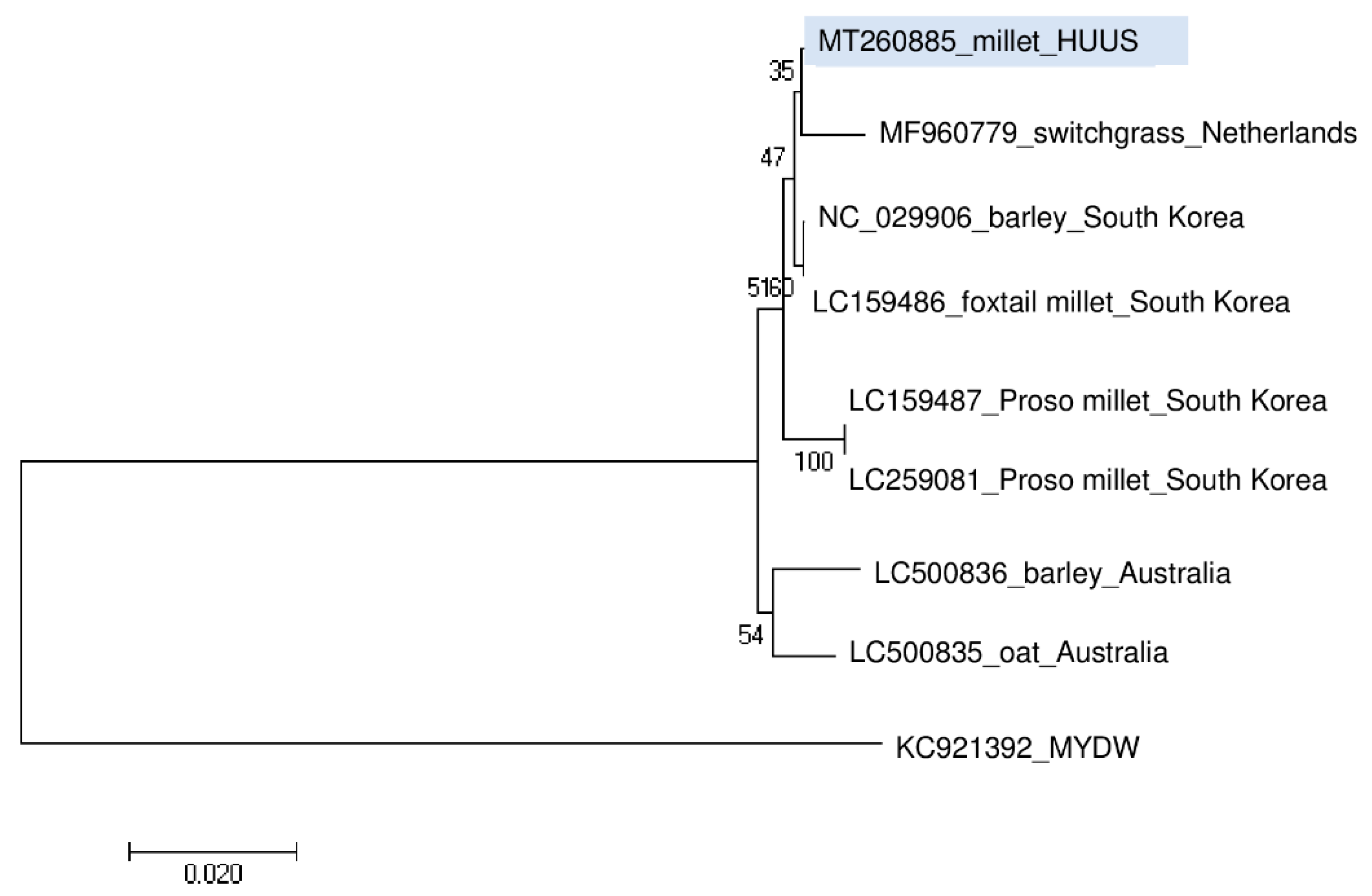
3.5. Phylogenetic Analysis of the Detected Viral Strains
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ryves, T.B.; Clement, E.J.; Foster, M.C. Alien Grasses of the British Isles; BSBI: London, UK, 1996. [Google Scholar]
- Novák, J.; Dancza, R.; Szentey, I.; Karamán, L. Magyarország szántóföldjeinek gyomnövényzete. In Ötödik Országos Szántóföldi Gyomfelvételezés; FVM: Budapest, Hungary, 2009; pp. 71–75. [Google Scholar]
- Moravcová, L.; Pysek, P.; Jarosik, V.H.V.Z.P. Reproductive characteristics of neophytes in the czech republic, traits of invasive and non-invasive species. Preslia 2010, 82, 365–390. [Google Scholar]
- Van De Wouw, M.; Jorge, M.A.; Bierwirth, J.H. Characterization of a collection of perennial panicum species. Trop. Grassl. 2008, 42, 40–53. [Google Scholar]
- Novák, J.; Dancza, R.; Szentey, I.; Karaman, L. Az ötödik országos gyomfelvételezés magyarország szántóföldjein. In Vidékfejlesztési Miniszt. Élelmiszerlánc- Felügyeleti FőosztályNövény-És Talajvédelmi; Osztály: Budapest, Hungary, 2011. [Google Scholar]
- Czimber, G.; Csala, G. Adatok a monokultúrás kukoricavetésekben gyomosodást okozó köles (Panicum miliaceum L.) terjedéséről. Növénytermelés 1974, 23, 207–217. [Google Scholar]
- Hunyadi, K. Szántóföldi Gyomnövények és Biológiájuk; Mezőgazdasági Kiadó: Budapest, Hungary, 1988. [Google Scholar]
- Czimber, F.; Hartmann, G. Köles nemzetség (panicum spp.). In Veszélyes 48, Veszélyes, Nehezen Irtható Gyomnövények és Ellenük való Védekezés; Benécsné bárdi, G., et al., Eds.; Mezôföldi Agrofórum Kft.: Szekszárd, Hungary, 2006; pp. 218–224. [Google Scholar]
- Bocz, M.; Késmárki, E.; Kováts, I.; Ruzsányi, A.; Szabó, L.S. Szántóföldi Növénytermesztés; Mezőgazda Kiadó: Budapest, Hungary, 1992; pp. 354–356. [Google Scholar]
- Wilson, R.G.; Westra, P. Wild proso millet (Panicum miliaceum) interference in corn (Zea mays). Weed Sci. 1991, 39, 217–220. [Google Scholar] [CrossRef]
- Pásztor, G.; Szabó, R.; Takács, A.; Henézi, Á.; Nádasy, E. The natural viral infections of the weedy Panicum miliaceum (L.). Columella J. Agric. Environ. Sci. 2017, 4, 35–38. [Google Scholar]
- Roossinck, M.J.; Martin, D.P.; Roumagnac, P. Plant virus metagenomics: Advances in virus discovery. Phytopathology 2015, 105, 716–727. [Google Scholar] [CrossRef]
- Pooggin, M.M. Small RNA-Omics for Plant Virus Identification, Virome Reconstruction, and Antiviral Defense Characterization. Front. Microbiol. 2018, 9, 2779. [Google Scholar] [CrossRef]
- Massart, S.; Chiumenti, M.; De Jonghe, K.; Glover, R.; Haegeman, A.; Koloniuk, I.; Komínek, P.; Kreuze, J.; Kutnjak, D.; Lotos, L.; et al. Virus detection by high-throughput sequencing of small rnas: Large-scale performance testing of sequence analysis strategies. Phytopathology 2018, 109, 488–497. [Google Scholar] [CrossRef]
- Molnár, A.; Csorba, T.; Lakatos, L.; Várallyay, É.; Lacomme, C.; Burgyán, J. Plant virus-derived small interfering rnas originate predominantly from highly structured single-stranded viral rnas. J. Virol. 2005, 79, 7812. [Google Scholar] [CrossRef]
- Singh, K.; JaroŠOvÁ, J.; Fousek, J.; Chen, H.; Kundu, J.K. Virome identification in wheat in the czech republic using small rna deep sequencing. J. Integr. Agric. 2020, 19, 1825–1833. [Google Scholar] [CrossRef]
- Golyaev, V.; Candresse, T.; Rabenstein, F.; Pooggin, M.M. Plant virome reconstruction and antiviral rnai characterization by deep sequencing of small rnas from dried leaves. Sci. Rep. 2019, 9, 19268. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, P.; Charles-Dominique, T.; Barakat, M.; Ortet, P.; Fernandez, E.; Filloux, D.; Hartnady, P.; Rebelo, T.A.; Cousins, S.R.; Mesleard, F.; et al. Geometagenomics illuminates the impact of agriculture on the distribution and prevalence of plant viruses at the ecosystem scale. ISME J. 2018, 12, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Nyitrai, Á.; Gaborjanyi, R. Wheat streak mosaic a new virus disease of wheats in hungary. Cereal Res. Commun. 1988, 16, 261–263. [Google Scholar]
- Navia, D.; de Mendonça, R.S.; Skoracka, A.; Szydło, W.; Knihinicki, D.; Hein, G.L.; da Silva Pereira, P.R.V.; Truol, G.; Lau, D. Wheat curl mite, aceria tosichella, and transmitted viruses: An expanding pest complex affecting cereal crops. Exp. Appl. Acarol. 2013, 59, 95–143. [Google Scholar] [CrossRef]
- Chalupniková, J.; Kundu, J.K.; Singh, K.; Bartaková, P.; Beoni, E. Wheat streak mosaic virus: Incidence in field crops, potential reservoir within grass species and uptake in winter wheat cultivars. J. Integr. Agric. 2017, 16, 523–531. [Google Scholar] [CrossRef]
- Singh, K.; Wegulo, S.N.; Skoracka, A.; Kundu, J.K. Wheat streak mosaic virus: A century old virus with rising importance worldwide. Mol. Plant Pathol. 2018, 19, 2193–2206. [Google Scholar] [CrossRef]
- Rabenstein, F.; Seifers, D.L.; Schubert, J.; French, R.; Stenger, D.C. Phylogenetic relationships, strain diversity and biogeography of tritimoviruses. J. Gen. Virol. 2002, 83, 895–906. [Google Scholar] [CrossRef]
- Conti, M. Investigations on a bullet-shaped virus of cereals isolated in italy from planthoppers. J. Phytopathol. 1969, 66, 275–279. [Google Scholar] [CrossRef]
- Yan, T.; Zhu, J.-R.; Di, D.; Gao, Q.; Zhang, Y.; Zhang, A.; Yan, C.; Miao, H.; Wang, X.-B. Characterization of the complete genome of barley yellow striate mosaic virus reveals a nested gene encoding a small hydrophobic protein. Virology 2015, 478, 112–122. [Google Scholar] [CrossRef]
- Makkouk, K.M.; Bertschinger, L.; Conti, M.; Bolay, N.; Dusunceli, F. Barley yellow striate mosaic rhabdovirus naturally infects cereal crops in the anatolian plateau of turkey. J. Phytopathol. 1996, 144, 413–415. [Google Scholar] [CrossRef]
- Almasi, R.; Afsharifar, A.; Niazi, A.; Pakdel, A.; Izadpanah, K. Analysis of the complete nucleotide sequence of the polymerase gene of barley yellow striate mosaic virus–iranian isolate. J. Phytopathol. 2010, 158, 351–356. [Google Scholar] [CrossRef]
- Izadpanah, K.; Ebrahim-Nesbat, F.; Afsharifar, A.R. Barley yellow striate mosaic virus as the cause of a major disease of wheat and millet in iran. J. Phytopathol. 1991, 131, 290–296. [Google Scholar] [CrossRef]
- Makkouk, K.M.; Kumari, S.G.; Ghulam, W.; Attar, N. First record of barley yellow striate mosaic virus affecting wheat summer-nurseries in syria. Plant Dis. 2004, 88, 83. [Google Scholar] [CrossRef]
- Di, D.P.; Zhang, Y.L.; Yan, C.; Yan, T.; Zhang, A.H.; Yang, F.; Cao, X.L.; Li, D.W.; Lu, Y.G.; Wang, X.B.; et al. First report of barley yellow striate mosaic virus on wheat in china. Plant Dis. 2014, 98, 1450. [Google Scholar] [CrossRef]
- Lockhart, B.E.L.; El-Maataoui, M.; Carroll, T.W.; Lennon, A.M.; Zaske, S.K. Identification of barley yellow striate mosaic virus in morocco and its field detection by enzyme immune assay. Plant Dis. 1986, 70, 1113–1117. [Google Scholar] [CrossRef]
- Dietzgen, R.G.; Higgins, C.M. Complete genome sequence of maize sterile stunt virus. Arch. Virol. 2019, 164, 1221–1223. [Google Scholar] [CrossRef]
- Zhao, F.; Lim, S.; Yoo, R.H.; Igori, D.; Kim, S.-M.; Kwak, D.Y.; Kim, S.L.; Lee, B.C.; Moon, J.S. The complete genomic sequence of a tentative new polerovirus identified in barley in south korea. Arch. Virol. 2016, 161, 2047–2050. [Google Scholar] [CrossRef]
- Park, C.Y.; Oh, J.H.; Min, H.G.; Lee, H.K.; Lee, S.H. First report of barley virus g in proso millet (Panicum miliaceum) in korea. Plant Dis. 2016, 101, 393. [Google Scholar] [CrossRef]
- Oh, J.; Park, C.Y.; Min, H.G.; Lee, H.K.; Yeom, Y.A.; Yoon, Y.; Lee, S.H. First report of barley virus g in foxtail millet (Setaria italica) in korea. Plant Dis. 2017, 101, 1061. [Google Scholar] [CrossRef]
- Nancarrow, N.; Aftab, M.; Zheng, L.; Maina, S.; Freeman, A.; Rodoni, B.; Spackman, M.; Trębicki, P. First report of barley virus g in australia. Plant Dis. 2019, 103, 1799. [Google Scholar] [CrossRef]
- Kumar, L.M.; Foster, J.A.; McFarland, C.; Malapi-Wight, M. First report of barley virus g in switchgrass (Panicum virgatum). Plant Dis. 2017, 102, 466. [Google Scholar] [CrossRef]
- Schubert, J.; Ziegler, A.; Rabenstein, F. First detection of wheat streak mosaic virus in germany: Molecular and biological characteristics. Arch. Virol. 2015, 160, 1761–1766. [Google Scholar] [CrossRef]
- Kúdela, O.; Kúdelová, M.; Nováková, S.; Glasa, M. First report of wheat streak mosaic virus in slovakia. Plant Dis. 2008, 92, 1365. [Google Scholar] [CrossRef]
- Trzmiel, K.; Szydło, W.; Zarzyńska-Nowak, A.; Jeżewska, M. First report of brome mosaic virus (bmv) and wheat streak mosaic virus (wsmv) co-infection in triticale plants in poland. Plant Dis. 2015, 99, 1290. [Google Scholar] [CrossRef]
- Coutts, B.A.; Banovic, M.; Kehoe, M.A.; Severtson, D.L.; Jones, R.A.C. Epidemiology of wheat streak mosaic virus in wheat in a mediterranean-type environment. Eur. J. Plant Pathol. 2014, 140, 797–813. [Google Scholar] [CrossRef]
- Pecman, A.; Kutnjak, D.; Gutiérrez-Aguirre, I.; Adams, I.; Fox, A.; Boonham, N.; Ravnikar, M. Next generation sequencing for detection and discovery of plant viruses and viroids: Comparison of two approaches. Front. Microbiol. 2017, 8, 1998. [Google Scholar] [CrossRef]
- White, J.L.; Kaper, J.M. A simple method for detection of viral satellite rnas in small plant tissue samples. J. Virol. Methods 1989, 23, 83–93. [Google Scholar] [CrossRef]
- Czotter, N.; Molnár, J.; Pesti, R.; Demián, E.; Baráth, D.; Varga, T.; Várallyay, É. Use of sirnas for diagnosis of viruses associated to woody plants in nurseries and stock collections. In Viral Metagenomics: Methods and Protocols; Pantaleo, V., Chiumenti, M., Eds.; Springer: New York, NY, USA, 2018; pp. 115–130. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Jukes, T.H.; Cantor, C.R. Chapter 24—Evolution of protein molecules. In Mammalian Protein Metabolism; Munro, H.N., Ed.; Academic Press: Cambridge, MA, USA, 1969; pp. 21–132. [Google Scholar]
- Kondo, H.; Fujita, M.; Hisano, H.; Hyodo, K.; Andika, I.B.; Suzuki, N. Virome analysis of aphid populations that infest the barley field: The discovery of two novel groups of nege/kita-like viruses and other novel rna viruses. Front. Microbiol. 2020, 11, 509. [Google Scholar] [CrossRef] [PubMed]
- Szabó, A.-K.; Várallyay, É.; Demian, E.; Hegyi, A.; Galbács, Z.N.; Kiss, J.; Bálint, J.; Loxdale, H.D.; Balog, A. Local aphid species infestation on invasive weeds affects virus infection of nearest crops under different management systems—A preliminary study. Front. Plant Sci. 2020, 11, 1–11. [Google Scholar]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasztor, G.; Galbacs N., Z.; Kossuth, T.; Demian, E.; Nadasy, E.; Takacs, A.P.; Varallyay, E. Millet Could Be both a Weed and Serve as a Virus Reservoir in Crop Fields. Plants 2020, 9, 954. https://doi.org/10.3390/plants9080954
Pasztor G, Galbacs N. Z, Kossuth T, Demian E, Nadasy E, Takacs AP, Varallyay E. Millet Could Be both a Weed and Serve as a Virus Reservoir in Crop Fields. Plants. 2020; 9(8):954. https://doi.org/10.3390/plants9080954
Chicago/Turabian StylePasztor, György, Zsuzsanna Galbacs N., Tamas Kossuth, Emese Demian, Erzsebet Nadasy, Andras P. Takacs, and Eva Varallyay. 2020. "Millet Could Be both a Weed and Serve as a Virus Reservoir in Crop Fields" Plants 9, no. 8: 954. https://doi.org/10.3390/plants9080954
APA StylePasztor, G., Galbacs N., Z., Kossuth, T., Demian, E., Nadasy, E., Takacs, A. P., & Varallyay, E. (2020). Millet Could Be both a Weed and Serve as a Virus Reservoir in Crop Fields. Plants, 9(8), 954. https://doi.org/10.3390/plants9080954