
Integrative Analysis of Omics Reveals RdDM Pathway Participation in the Initiation of Rice Microspore Embryogenesis Under Cold Treatment
 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. 10 Days of Cold Treatment Is Critical for Rice MDEC Initiation
2.2. Cold-Stress-Induced Dynamic Changes in DNA Methylation Levels in Rice Microspores
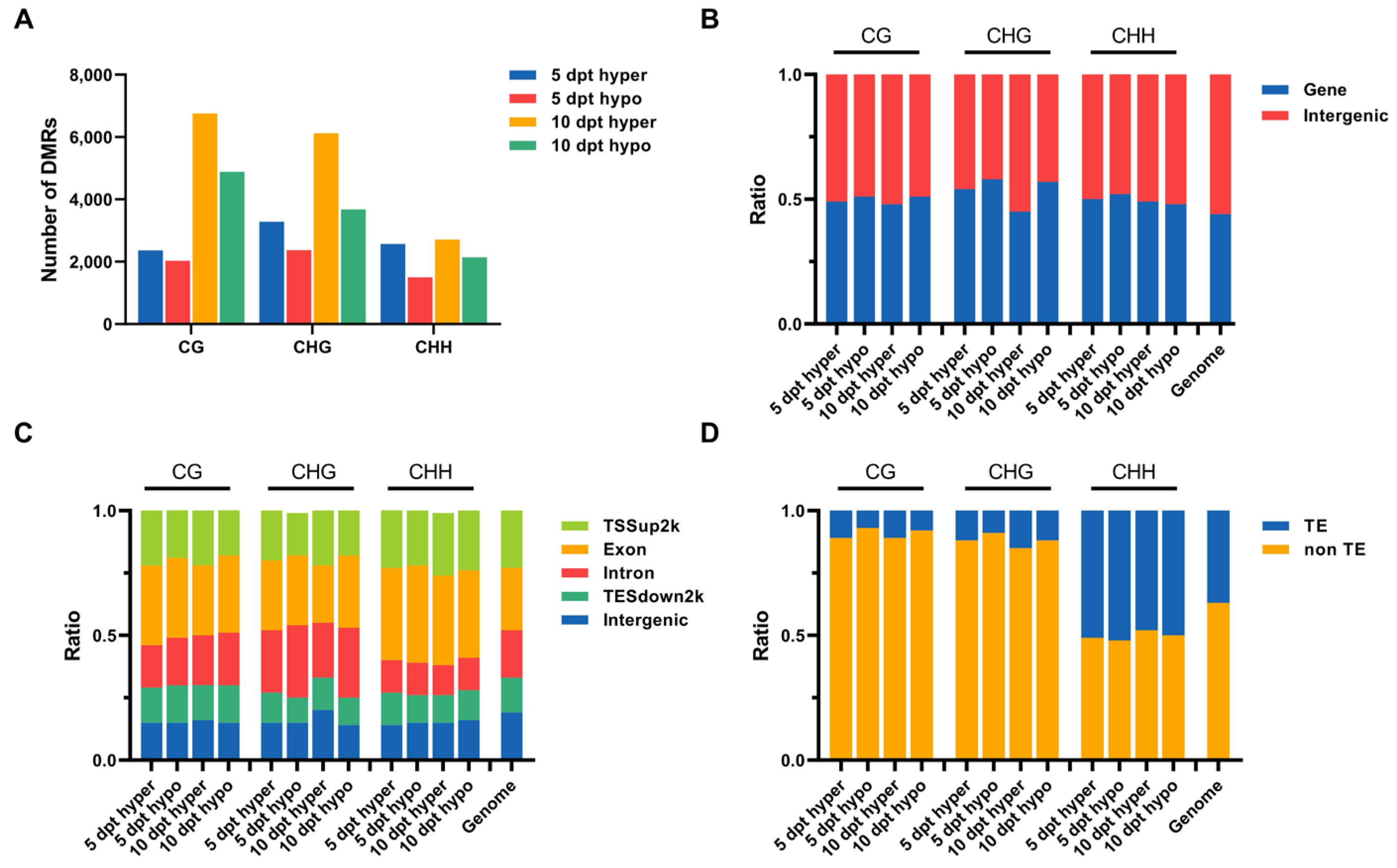
2.3. Identification of DMRs in Rice Microspores upon Cold Treatment
2.4. Association Analysis Between DMRs and sRNAs in Rice Microspores upon Cold Treatment
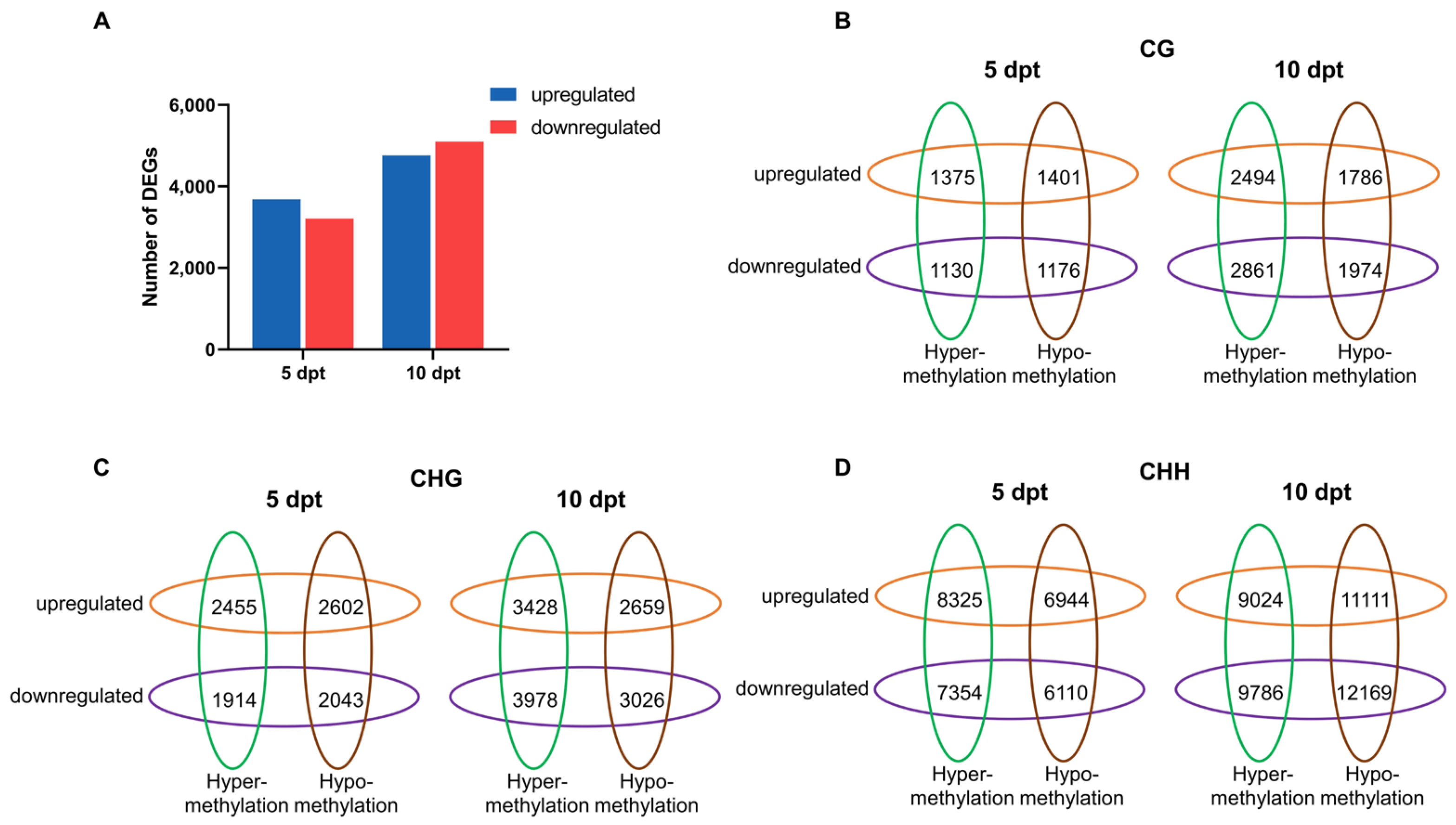
2.5. Association Analysis Between DMRs and mRNAs in Rice Microspores upon Cold Treatment
2.6. Candidate Gene Regulated by the RdDM Pathway in Rice Microspores Under Cold Stress
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Rice Microspore Collection and In Vitro Culture
5.2. Bisulphite Sequencing and Analysis
5.3. Transcriptome Sequence and Analysis
5.4. Small RNA Sequence and Analysis
5.5. Association of Methylation Sequencing with sRNA and Transcriptome Data
5.6. qRT-PCR Validation of the RNA-Seq
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| DH | doubled haploid |
| ME | microspore embryogenesis |
| MDEC | microspore-derived embryo cultures |
| RdDM | RNA-directed DNA methylation |
| sRNAs | small RNAs |
| AGO proteins | argonaute proteins |
| CXJ | Cong Xiangjing |
| BS-Seq | bisulphite sequencing |
| DMRs | differentially methylated genomic regions |
| rRNA | ribosomal RNA |
| tRNA | transfer RNA |
| snRNA | small nuclear RNA |
| snoRNA | small nucleolar RNA |
| ncRNA | non-coding RNA |
| DSRs | differentially regulated sRNAs |
| dpt | days post-treatment |
| WGBS | whole-genome bisulfite sequencing |
| RNA-seq | RNA sequencing |
| GO | gene ontology |
| DEGs | differentially expressed genes |
| CPM | counts per million mapped reads |
| FPKM | fragments per kilobase of transcript per million mapped reads |
| TSS | transcription start sites |
References
- Murovec, J.; Bohanec, B. Haploids and Doubled Haploids in Plant Breeding. In Plant Breeding; IntechOpen Limited: London, UK, 2011. [Google Scholar]
- Germana, M.A.; Lambardi, M. (Eds.) In Vitro Embryogenesis in Higher Plants; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; Volume 1359, ISBN 978-1-4939-3060-9. [Google Scholar]
- Touraev, A.; Vicente, O.; Heberle-Bors, E. Initiation of Microspore Embryogenesis by Stress. Trends Plant Sci. 1997, 2, 297–302. [Google Scholar] [CrossRef]
- Ferrie, A.M.R.; Caswell, K.L. Isolated Microspore Culture Techniques and Recent Progress for Haploid and Doubled Haploid Plant Production. Plant Cell Tissue Organ Cult. 2011, 104, 301–309. [Google Scholar] [CrossRef]
- Nic-Can, G.I.; Loyola-Vargas, V.M. The Role of the Auxins During Somatic Embryogenesis. In Somatic Embryogenesis: Fundamental Aspects and Applications; Loyola-Vargas, V.M., Ochoa-Alejo, N., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 171–182. ISBN 978-3-319-33704-3. [Google Scholar]
- Datta, S.K.; Datta, K.; Potrykus, I. Fertile Indica Rice Plants Regenerated from Protoplasts Isolated from Microspore Derived Cell Suspensions. Plant Cell Rep. 1990, 9, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Gao, M.; Cai, Q.; Cheng, X.; Shen, Y.; Liang, Z. Improved Isolated Microspore Culture Efficiency in Medium with Maltose and Optimized Growth Regulator Combination in Japonica Rice (Oryza Sativa). Plant Cell Tissue Organ Cult. 1995, 42, 245–250. [Google Scholar] [CrossRef]
- Rahman, Z.A.; Seman, Z.A.; Othman, A.N.; Ghaffar, M.B.A.; Razak, S.A.; Yusof, M.F.M.; Nasir, K.H.; Ahmad, K.; Chow, Y.L.; How, T.C.; et al. Establishment of Effective Plantlets Regeneration Protocol via Isolated Microspore Culture in Malaysian Indica Rice MR219. Plant Biotechnol. Rep. 2022, 16, 343–355. [Google Scholar] [CrossRef]
- Gao, R.; Zong, Y.; Zhang, S.; Guo, G.; Zhang, W.; Chen, Z.; Lu, R.; Liu, C.; Wang, Y.; Li, Y. Efficient Isolated Microspore Culture Protocol for Callus Induction and Plantlet Regeneration in Japonica Rice (Oryza sativa L.). Plant Methods 2024, 20, 76. [Google Scholar] [CrossRef]
- Solís, M.T.; Rodríguez-Serrano, M.; Meijón, M.; Cañal, M.J.; Cifuentes, A.; Risueño, M.C.; Testillano, P.S. DNA Methylation Dynamics and MET1a-like Gene Expression Changes during Stress-Induced Pollen Reprogramming to Embryogenesis. J. Exp. Bot. 2012, 63, 6431–6444. [Google Scholar] [CrossRef]
- Li, W.-Z.; Song, Z.-H.; Guo, B.-T.; Xu, L.-J. The effects of DNA hypomethylating drugs on androgenesis in barley (Hordeum vulgare L.). In Vitro Cell. Dev. Biol. Plant 2001, 37, 605–608. [Google Scholar] [CrossRef]
- Solís, M.T.; El-Tantawy, A.A.; Cano, V.; Risueño, M.C.; Testillano, P.S. 5-Azacytidine Promotes Microspore Embryogenesis Initiation by Decreasing Global DNA Methylation, but Prevents Subsequent Embryo Development in Rapeseed and Barley. Front. Plant Sci. 2015, 6, 472. [Google Scholar] [CrossRef]
- Kong, C.; Su, H.; Deng, S.; Ji, J.; Wang, Y.; Zhang, Y.; Yang, L.; Fang, Z.; Lv, H. Global DNA Methylation and mRNA-miRNA Variations Activated by Heat Shock Boost Early Microspore Embryogenesis in Cabbage (Brassica oleracea). Int. J. Mol. Sci. 2022, 23, 5147. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, J.-K. Epigenetic Gene Regulation in Plants and Its Potential Applications in Crop Improvement. Nat. Rev. Mol. Cell Biol. 2025, 26, 51–67. [Google Scholar] [CrossRef]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef]
- Stroud, H.; Do, T.; Du, J.; Zhong, X.; Feng, S.; Johnson, L.; Patel, D.J.; Jacobsen, S.E. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 2014, 21, 64–72. [Google Scholar] [CrossRef]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.-L.; Chen, H.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; et al. Genome-Wide High-Resolution Mapping and Functional Analysis of DNA Methylation in Arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Crisp, P.A.; Marand, A.P.; Noshay, J.M.; Zhou, P.; Lu, Z.; Schmitz, R.J.; Springer, N.M. Stable Unmethylated DNA Demarcates Expressed Genes and Their Cis-Regulatory Space in Plant Genomes. Proc. Natl. Acad. Sci. USA 2020, 117, 23991–24000. [Google Scholar] [CrossRef] [PubMed]
- Matzke, M.A.; Mosher, R.A. RNA-Directed DNA Methylation: An Epigenetic Pathway of Increasing Complexity. Nat. Rev. Genet. 2014, 15, 394–408. [Google Scholar] [CrossRef]
- D’Ario, M.; Griffiths-Jones, S.; Kim, M. Small RNAs: Big Impact on Plant Development. Trends Plant Sci. 2017, 22, 1056–1068. [Google Scholar] [CrossRef]
- Vu, T.M.; Nakamura, M.; Calarco, J.P.; Susaki, D.; Lim, P.Q.; Kinoshita, T.; Higashiyama, T.; Martienssen, R.A.; Berger, F. RNA-Directed DNA Methylation Regulates Parental Genomic Imprinting at Several Loci in Arabidopsis. Development 2013, 140, 2953–2960. [Google Scholar] [CrossRef]
- Wang, L.; Zheng, K.; Zeng, L.; Xu, D.; Zhu, T.; Yin, Y.; Zhan, H.; Wu, Y.; Yang, D.-L. Reinforcement of CHH Methylation through RNA-Directed DNA Methylation Ensures Sexual Reproduction in Rice. Plant Physiol. 2022, 188, 1189–1209. [Google Scholar] [CrossRef]
- Xu, L.; Yuan, K.; Yuan, M.; Meng, X.; Chen, M.; Wu, J.; Li, J.; Qi, Y. Regulation of Rice Tillering by RNA-Directed DNA Methylation at Miniature Inverted-Repeat Transposable Elements. Mol. Plant 2020, 13, 851–863. [Google Scholar] [CrossRef]
- Zheng, K.; Wang, L.; Zeng, L.; Xu, D.; Guo, Z.; Gao, X.; Yang, D.-L. The Effect of RNA Polymerase V on 24-Nt siRNA Accumulation Depends on DNA Methylation Contexts and Histone Modifications in Rice. Proc. Natl. Acad. Sci. USA 2021, 118, e2100709118. [Google Scholar] [CrossRef]
- Testillano, P.S. Microspore embryogenesis: Targeting the determinant factors of stress-induced cell reprogramming for crop improvement. J. Exp. Bot. 2019, 70, 2965–2978. [Google Scholar] [CrossRef]
- Li, Y.; Zong, Y.; Li, W.; Guo, G.; Zhou, L.; Xu, H.; Gao, R.; Liu, C. Transcriptomics Integrated with Metabolomics Reveals the Effect of Cold Stress on Rice Microspores. BMC Plant Biol. 2023, 23, 521. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Wang, Y.; Sun, Y.; Shan, L.; Chen, P.; Huang, J. Improvement of Isolated Microspore Culture of Barley (Hordeum Vulgare L.): The Effect of Floret Co-Culture. Plant Cell Tissue Organ Cult. 2008, 93, 21–27. [Google Scholar] [CrossRef]
- Shim, S.; Lee, H.G.; Park, O.-S.; Shin, H.; Lee, K.; Lee, H.; Huh, J.H.; Seo, P.J. Dynamic Changes in DNA Methylation Occur in TE Regions and Affect Cell Proliferation during Leaf-to-Callus Transition in Arabidopsis. Epigenetics 2022, 17, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, M.; Li, Y.; Zhang, Q.; Lindsey, K.; Daniell, H.; Jin, S.; Zhang, X. Multi-omics analyses reveal epigenomics basis for cotton somatic embryogenesis through successive regeneration acclimation process. Plant Biotechnol. J. 2019, 17, 435–450. [Google Scholar] [CrossRef]
- Gallego-Bartolomé, J. DNA methylation in plants: Mechanisms and tools for targeted manipulation. New Phytol. 2020, 227, 38–44. [Google Scholar] [CrossRef]
- Lisch, D. Epigenetic Regulation of Transposable Elements in Plants. Annu. Rev. Plant Biol. 2009, 60, 43–66. [Google Scholar] [CrossRef]
- Zemach, A.; Kim, M.Y.; Silva, P.; Rodrigues, J.A.; Dotson, B.; Brooks, M.D.; Zilberman, D. Local DNA Hypomethylation Activates Genes in Rice Endosperm. Proc. Natl. Acad. Sci. USA 2010, 107, 18729–18734. [Google Scholar] [CrossRef]
- Hu, Y.; Li, S.; Fan, X.; Song, S.; Zhou, X.; Weng, X.; Xiao, J.; Li, X.; Xiong, L.; You, A.; et al. OsHOX1 and OsHOX28 Redundantly Shape Rice Tiller Angle by Reducing HSFA2D Expression and Auxin Content. Plant Physiol. 2020, 184, 1424–1437. [Google Scholar] [CrossRef]
- Karami, O.; Aghavaisi, B.; Mahmoudi Pour, A. Molecular Aspects of Somatic-to-Embryogenic Transition in Plants. J. Chem. Biol. 2009, 2, 177–190. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Gao, R.; Zong, Y.; Guo, G.; Zhang, W.; Chen, Z.; Guo, J.; Liu, C. Integrative Analysis of Omics Reveals RdDM Pathway Participation in the Initiation of Rice Microspore Embryogenesis Under Cold Treatment. Plants 2025, 14, 2267. https://doi.org/10.3390/plants14152267
Li Y, Gao R, Zong Y, Guo G, Zhang W, Chen Z, Guo J, Liu C. Integrative Analysis of Omics Reveals RdDM Pathway Participation in the Initiation of Rice Microspore Embryogenesis Under Cold Treatment. Plants. 2025; 14(15):2267. https://doi.org/10.3390/plants14152267
Chicago/Turabian StyleLi, Yingbo, Runhong Gao, Yingjie Zong, Guimei Guo, Wenqi Zhang, Zhiwei Chen, Jiao Guo, and Chenghong Liu. 2025. "Integrative Analysis of Omics Reveals RdDM Pathway Participation in the Initiation of Rice Microspore Embryogenesis Under Cold Treatment" Plants 14, no. 15: 2267. https://doi.org/10.3390/plants14152267
APA StyleLi, Y., Gao, R., Zong, Y., Guo, G., Zhang, W., Chen, Z., Guo, J., & Liu, C. (2025). Integrative Analysis of Omics Reveals RdDM Pathway Participation in the Initiation of Rice Microspore Embryogenesis Under Cold Treatment. Plants, 14(15), 2267. https://doi.org/10.3390/plants14152267