
Genome Survey and Chromosome-Level Draft Genome Assembly of Glycine max var. Dongfudou 3: Insights into Genome Characteristics and Protein Deficiencies


Abstract
:1. Introduction
2. Results
2.1. Sequencing and Data Cleaning
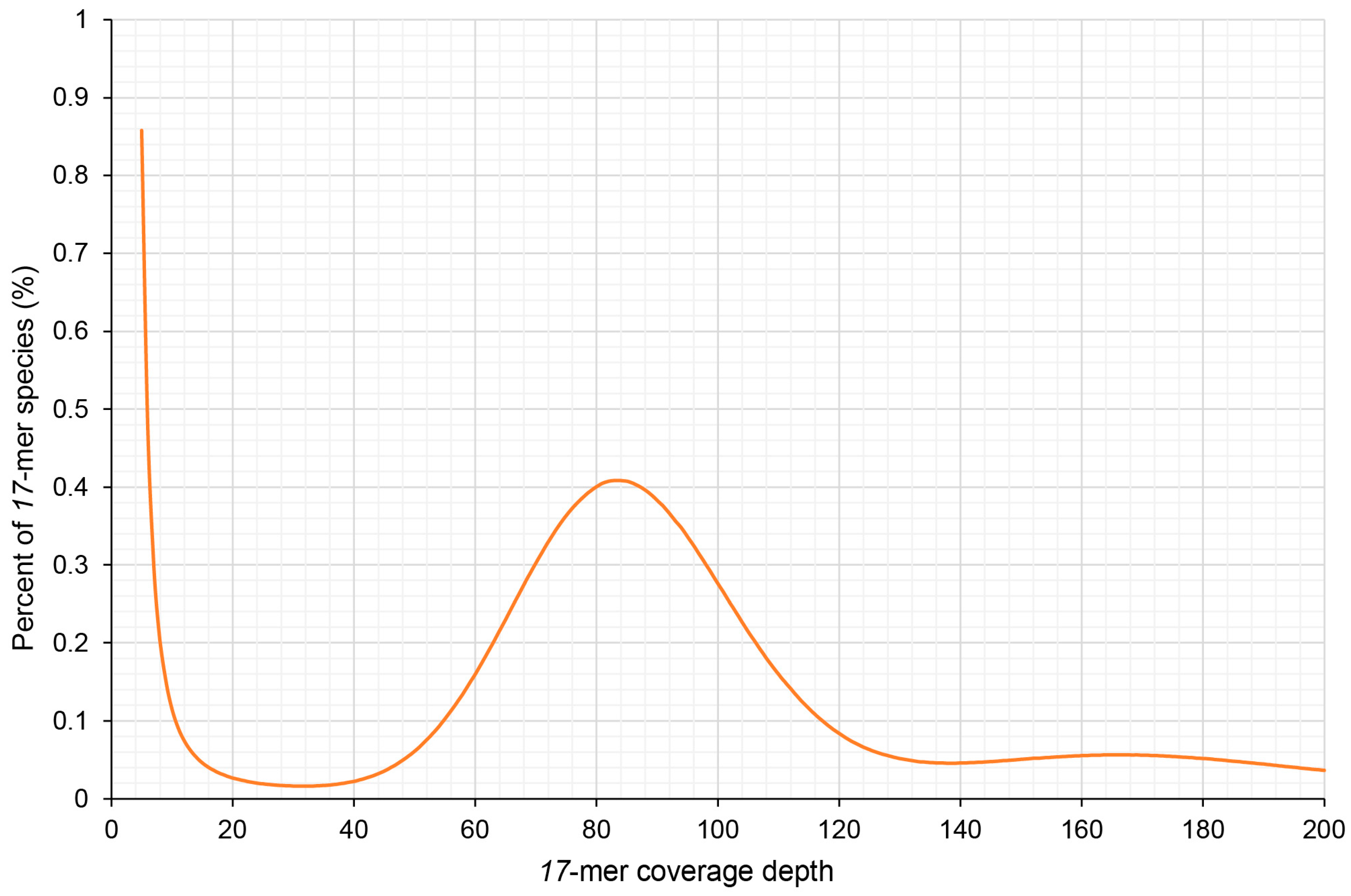
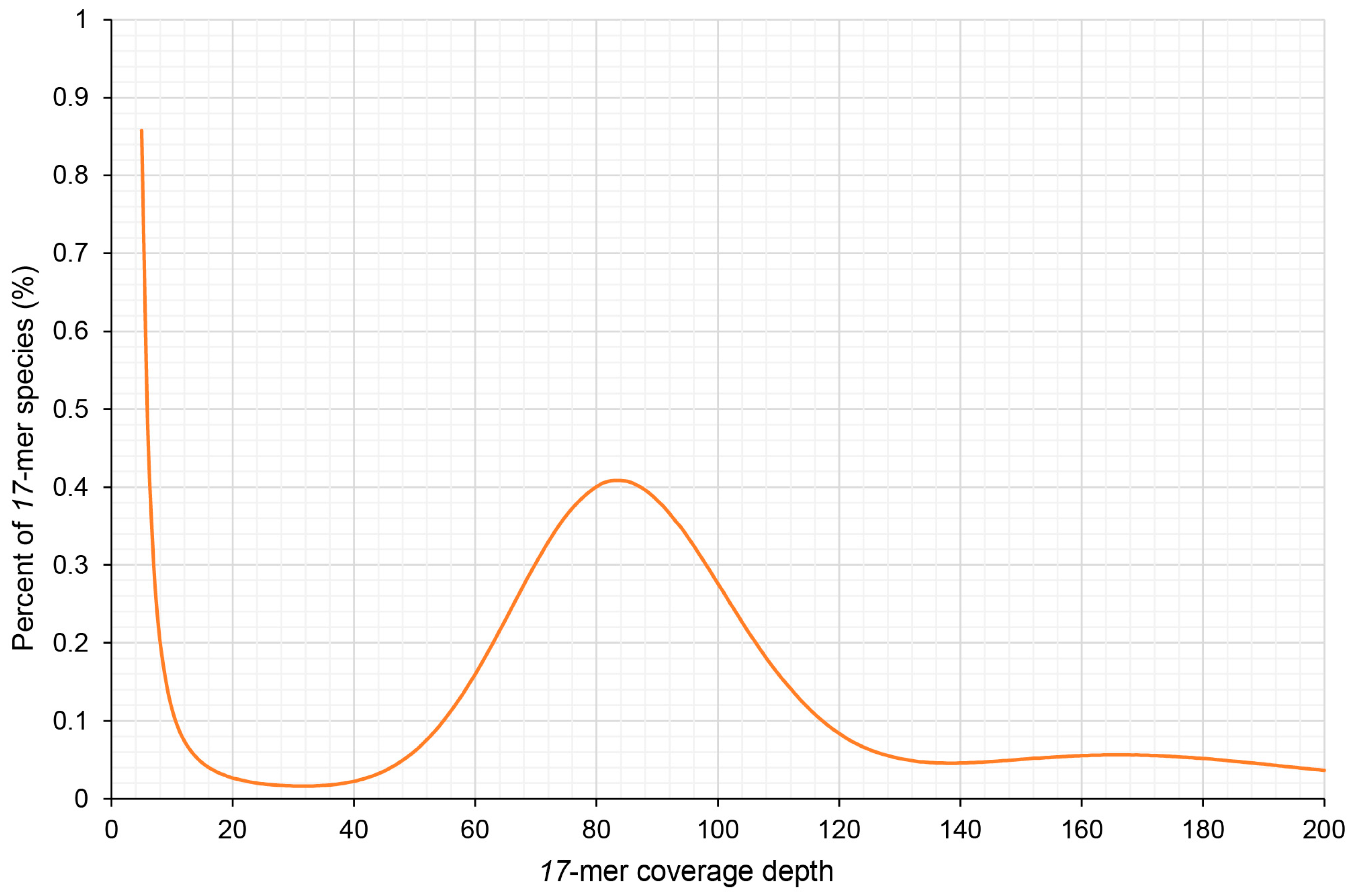
2.2. Genome Size Estimation by k-mer Analysis
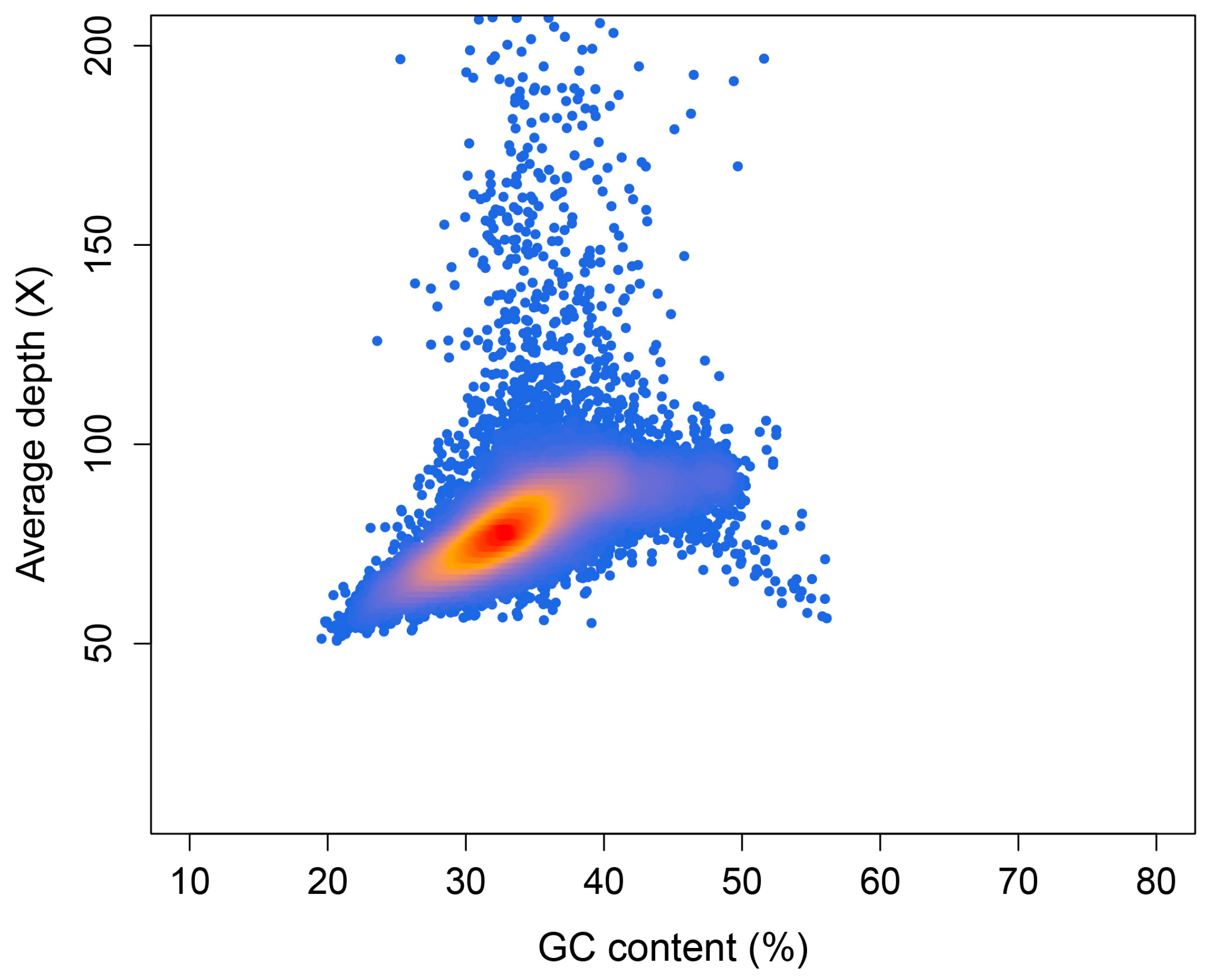
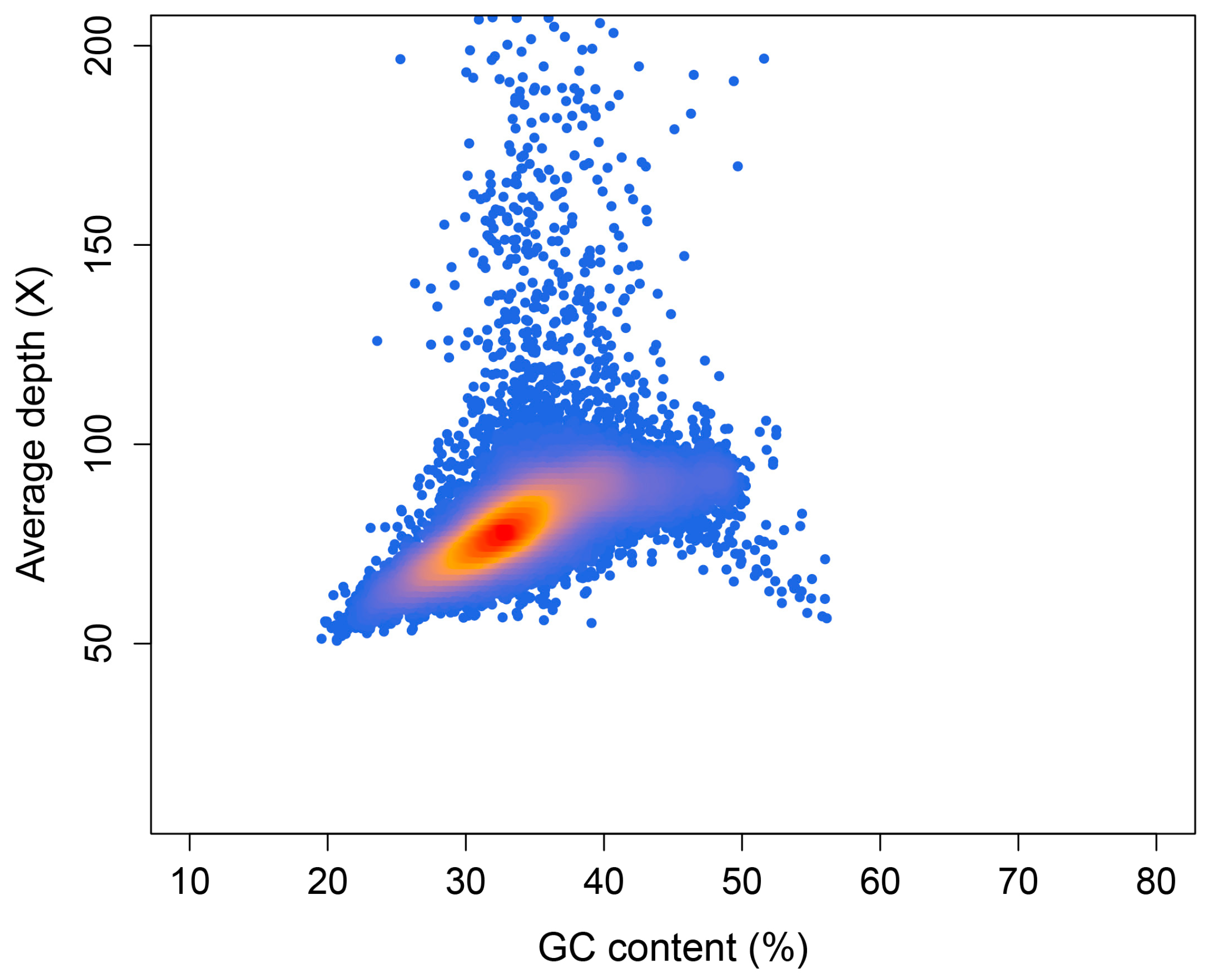
2.3. Preliminary Genome Assembly and GC Content Assessment
2.4. Reference-Guided Chromosome Anchoring and Gene Annotation
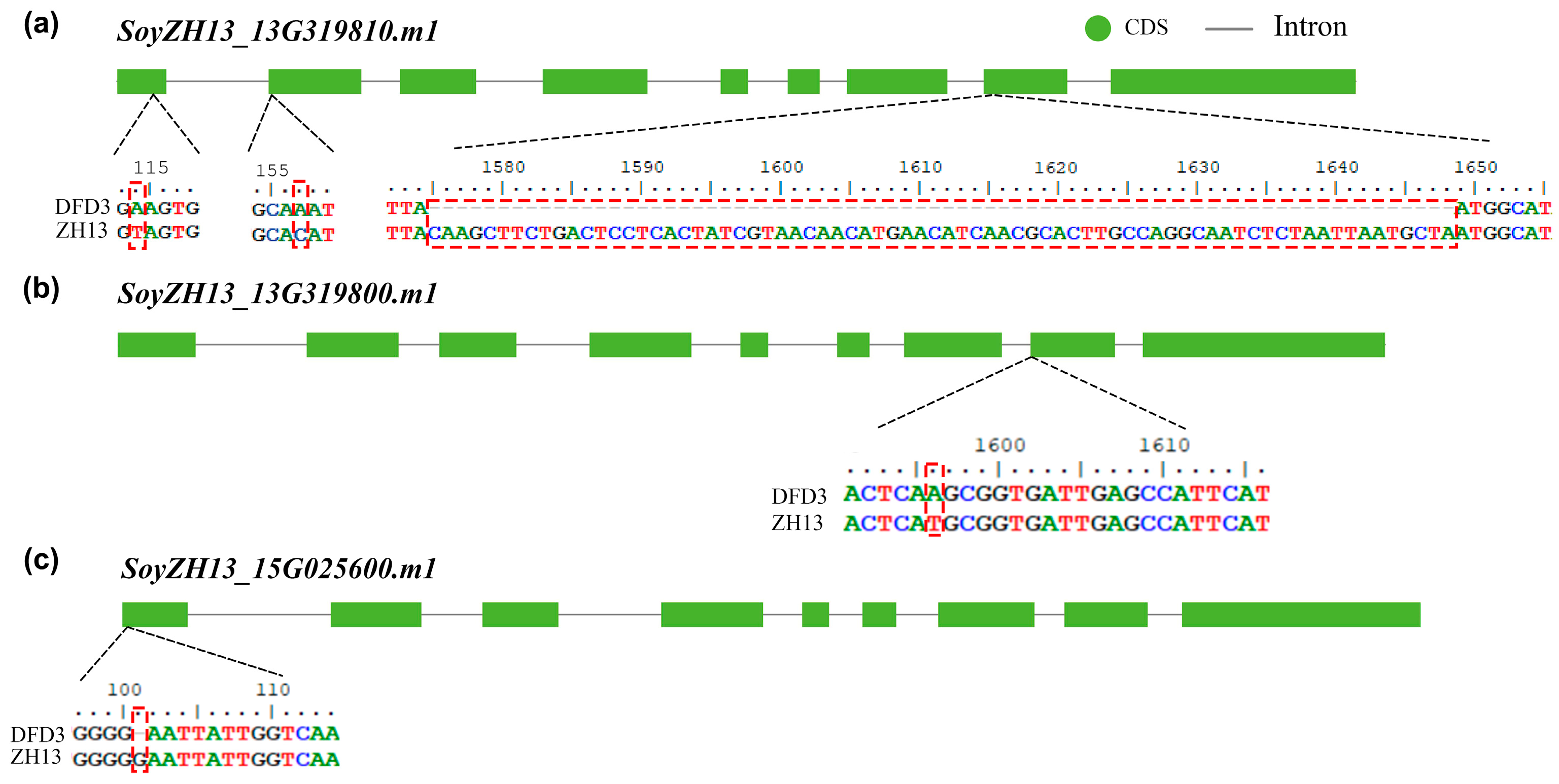
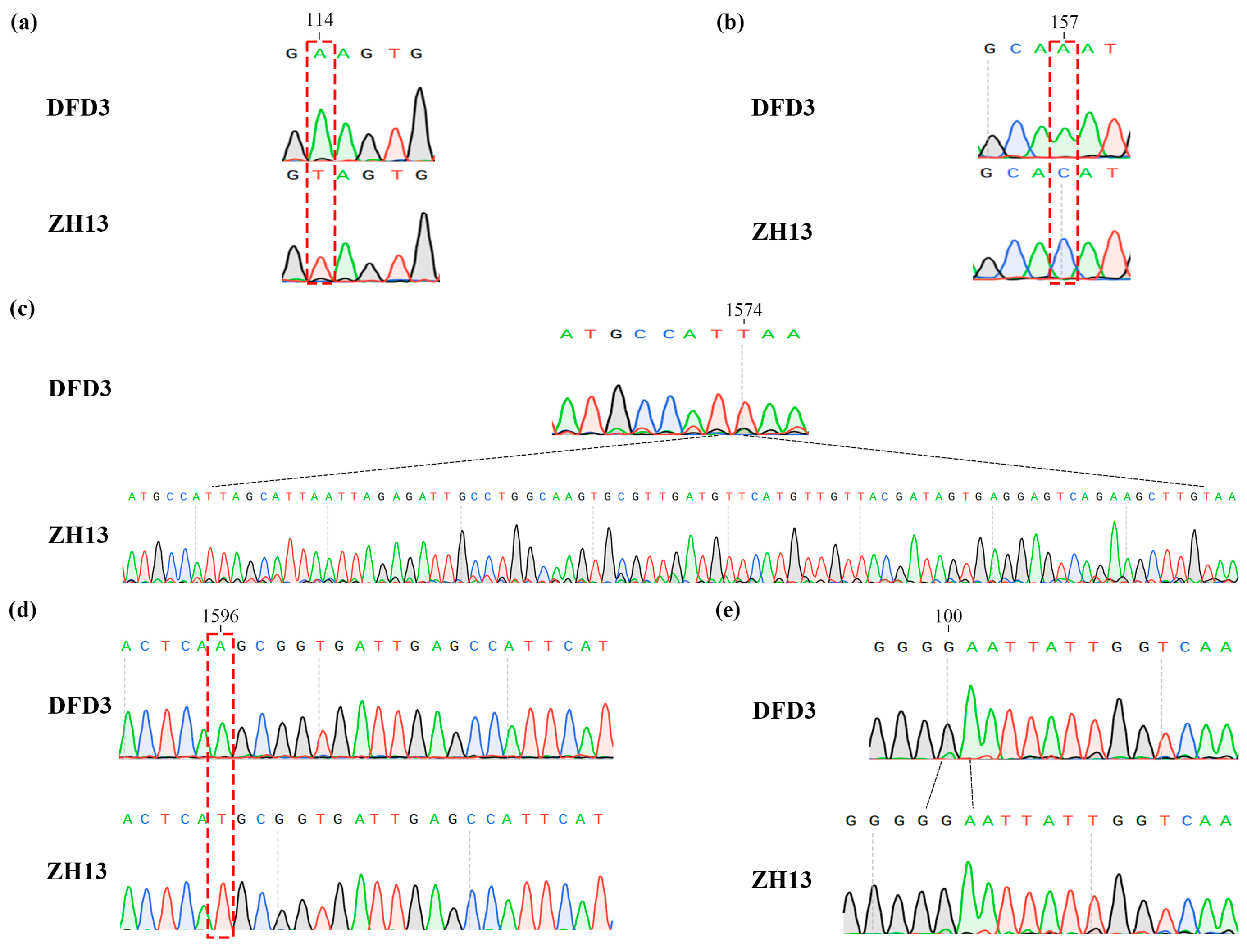
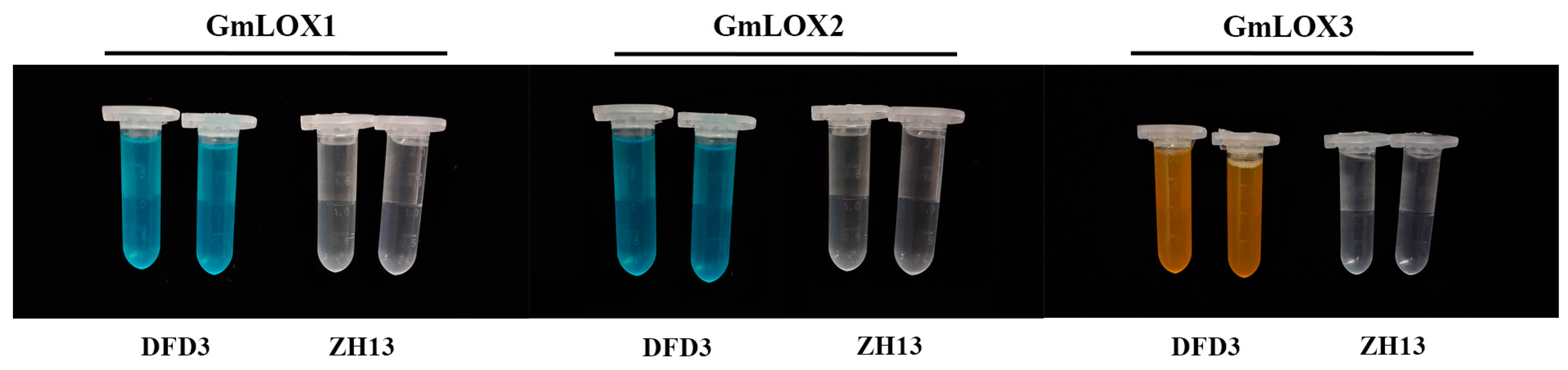
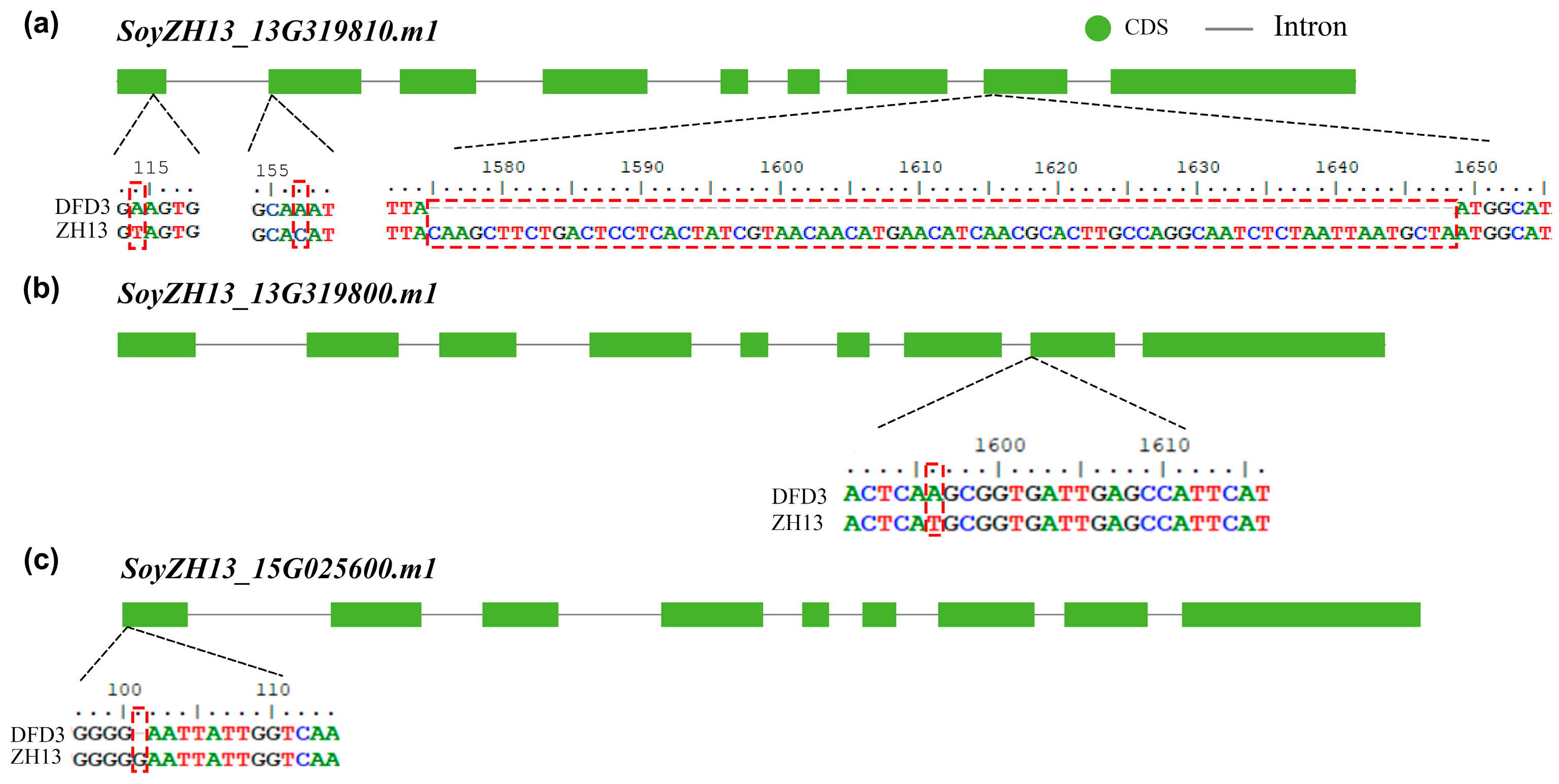
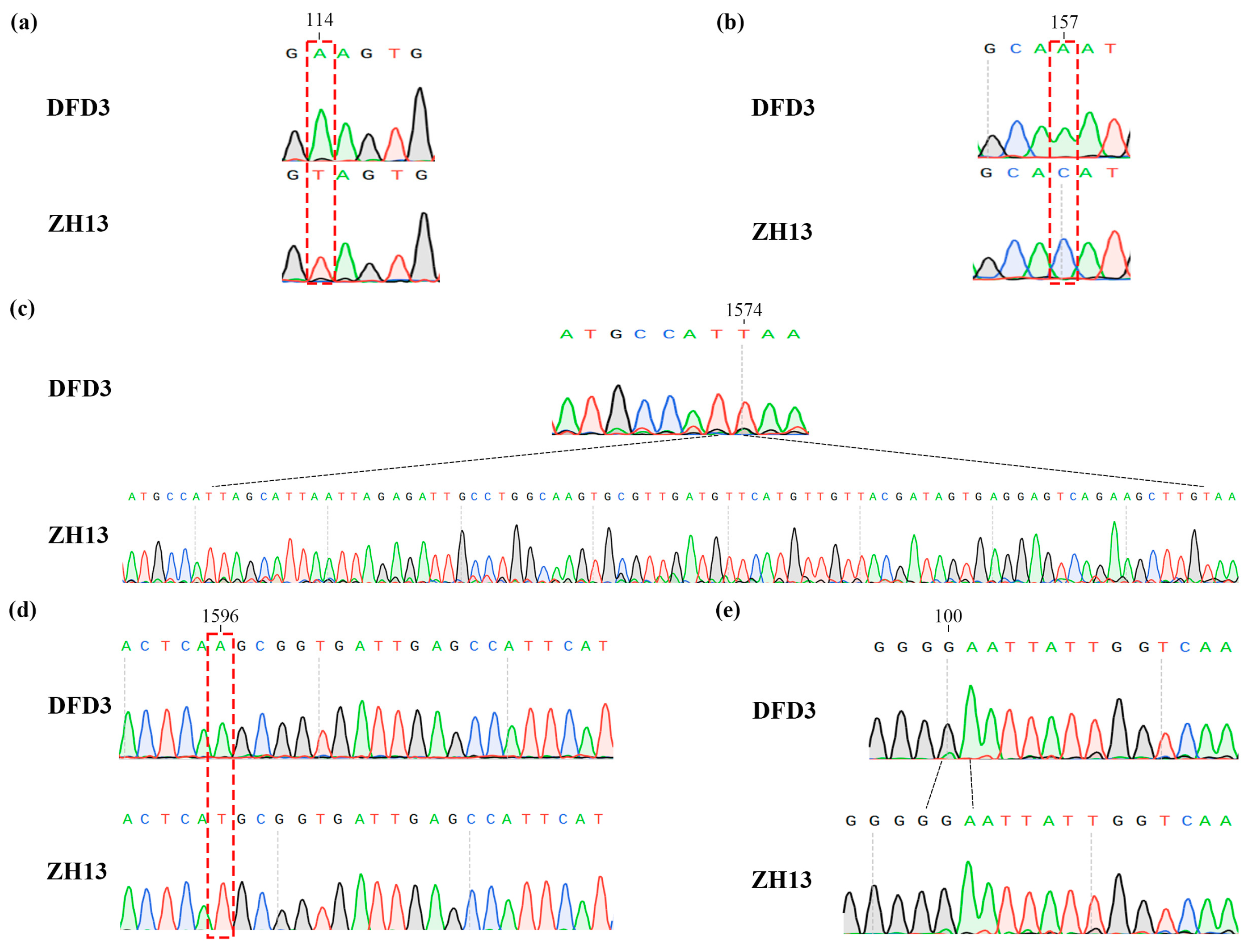
2.5. Mutation Analysis of Lipoxygenase Deficiency
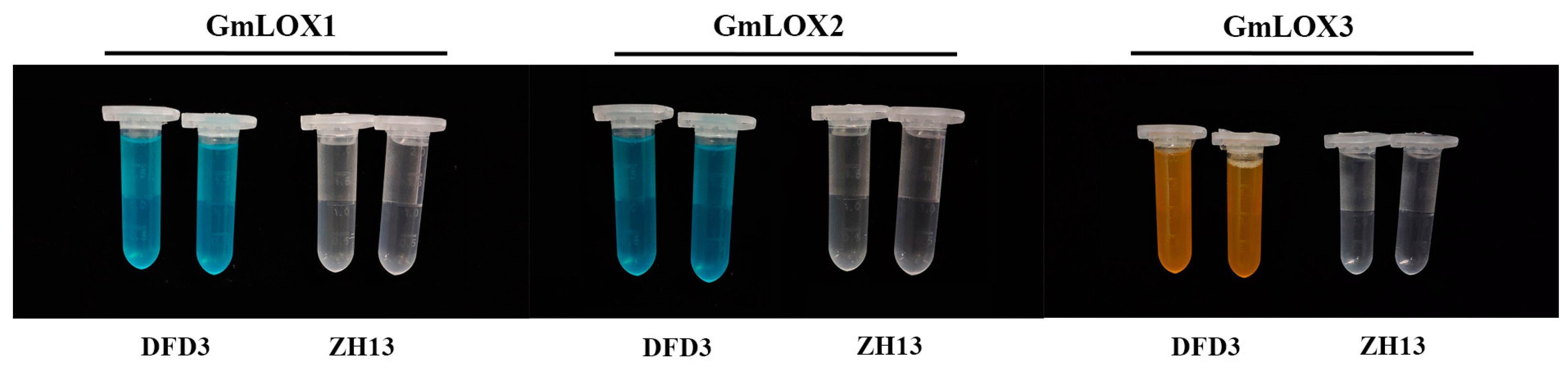
2.6. Impact of GmLox Genes Variations on Transcription and Translation Processes
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Sequencing
4.2. Quality Control and Data Cleaning
4.3. Genome Size and Repeat Rate Estimation by k-mer Analysis
4.4. Preliminary Genome Assembly and GC Content Analysis
4.5. Fast Reference-Guided Chromosome Anchoring and Gene Annotation
4.6. Sequence Variations of Seed Lipoxygenases and Their Detection at the Transcriptional and Translational Levels
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bilyeu, K.; Ratnaparkhe, M.B.; Kole, C. Genetics, Genomics, and Breeding of Soybean, 1st ed.; CRC Press: Boca Raton, FL, USA, 2010; pp. 1–2. [Google Scholar] [CrossRef]
- Roy, S.; Liu, W.; Nandety, R.S.; Crook, A.; Mysore, K.S.; Pislariu, C.I.; Frugoli, J.; Dickstein, R.; Udvardi, M.K. Celebrating 20 years of genetic discoveries in legume nodulation and symbiotic nitrogen fixation. Plant Cell 2020, 32, 15–41. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Ma, Q.; Chen, Y.; Tian, B.; Xu, L.; Bai, Y.; Chen, W.-F.; Li, X. Variation in rhizosphere microbial communities and its asso-ciation with the symbiotic efficiency of rhizobia in soybean. ISME J. 2020, 14, 1915–1928. [Google Scholar] [CrossRef] [PubMed]
- Novikova, L.; Seferova, I.; Matvienko, I.; Shchedrina, Z.; Vishnyakova, M. Photoperiod and temperature sensitivity in early soybean accessions from the vir collection in leningrad province of the Russian Federation. Turk. J. Agric. For. 2022, 46, 947–954. [Google Scholar] [CrossRef]
- Lin, X.-Y.; Liu, B.-H.; Weller, J.L.; Abe, J.; Kong, F.-J. Molecular mechanisms for the photoperiodic regulation of flowering in soybean. J. Integr. Plant Biol. 2021, 63, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-H.; Qin, C.; Wang, L.; Jiao, C.-Z.; Hong, H.-L.; Tian, Y.; Li, Y.-F.; Xing, G.-N.; Wang, J.; Gu, Y.-Z.; et al. Genome-wide signatures of the geographic expansion and breeding of soybean. Sci. China Life Sci. 2023, 66, 350–365. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.-X.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.-J.; Thelen, J.J.; Cheng, J.-L.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.-T.; Liu, J.; Geng, H.-Y.; Zhang, J.-X.; Liu, Y.-C.; Zhang, H.-K.; Xing, S.-L.; Du, J.-C.; Ma, S.-S.; Tian, Z.-X. De novo assembly of a Chinese soybean genome. Sci. China Life Sci. 2018, 61, 871–884. [Google Scholar] [CrossRef]
- Shen, Y.-T.; Du, H.-L.; Liu, Y.-C.; Ni, L.-B.; Wang, Z.; Liang, C.-Z.; Tian, Z.-X. Update soybean Zhonghuang 13 genome to a golden reference. Sci. China Life Sci. 2019, 62, 1257–1260. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Du, H.-L.; Li, P.-C.; Shen, Y.-T.; Peng, H.; Liu, S.-L.; Zhou, G.-A.; Zhang, H.-K.; Liu, Z.; Shi, M.; et al. Pan-genome of wild and cultivated soybeans. Cell 2020, 182, 162–176. [Google Scholar] [CrossRef]
- Lang, D.-D.; Zhang, S.-L.; Ren, P.-P.; Liang, F.; Sun, Z.-Y.; Meng, G.-L.; Tan, Y.-T.; Li, X.-K.; Lai, Q.-H.; Han, L.-L.; et al. Comparison of the two up-to-date sequencing technologies for genome assembly: HiFi reads of Pacific Biosciences Sequel II system and ultralong reads of Oxford Nanopore. Gigascience 2020, 9, 1–7. [Google Scholar] [CrossRef]
- Davies, C.S.; Nielsen, S.S.; Nielsen, N.C. Flavor improvement of soybean preparations by genetic removal of lipoxygenase-2. J. Am. Oil Chem. Soc. 1987, 64, 1428–1433. [Google Scholar] [CrossRef]
- Wang, J.; Kuang, H.-Q.; Zhang, Z.-H.; Yang, Y.-Q.; Yan, L.; Zhang, M.-C.; Song, S.-K.; Guan, Y.-F. Generation of seed lipoxygenase-free soybean using CRISPR-Cas9. Crop J. 2020, 8, 432–439. [Google Scholar] [CrossRef]
- Doležel, J.; Bartoš, J. Plant DNA flow cytometry and estimation of nuclear genome size. Ann. Bot. 2005, 95, 99–110. [Google Scholar] [CrossRef]
- Doležel, J.; Greilhuber, J.; Lucretti, S.; Meister, A.; Lysák, M.A.; Nardi, L.; Obermayer, R. Plant genome size estimation by flow cytometry: Inter-laboratory comparison. Ann. Bot. 1998, 82, 17–26. [Google Scholar] [CrossRef]
- Liu, B.-H.; Shi, Y.-J.; Yuan, J.-Y.; Hu, X.-S.; Zhang, H.; Li, N.; Li, Z.-Y.; Chen, Y.-X.; Mu, D.-S.; Fan, W. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv 2013, arXiv:1308.2012. [Google Scholar] [CrossRef]
- Lutz, K.A.; Wang, W.-Q.; Zdepski, A.; Michael, T.P. Isolation and analysis of high quality nuclear DNA with reduced organellar DNA for plant genome sequencing and resequencing. BMC Biotechnol. 2011, 11, 54. [Google Scholar] [CrossRef]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 1–29. [Google Scholar] [CrossRef]
- Chevigny, N.; Schatz-Daas, D.; Lotfi, F.; Gualberto, J.M. DNA repair and the stability of the plant mitochondrial genome. Int. J. Mol. Sci. 2020, 21, 328. [Google Scholar] [CrossRef]
- Martin, W. Gene transfer from organelles to the nucleus: Frequent and in big chunks. Proc. Natl. Acad. Sci. USA 2003, 100, 8612–8614. [Google Scholar] [CrossRef]
- Shih, P.M.; Matzke, N.J. Primary endosymbiosis events date to the later Proterozoic with cross-calibrated phylogenetic dating of duplicated ATPase proteins. Proc. Natl. Acad. Sci. USA 2013, 110, 12355–12360. [Google Scholar] [CrossRef]
- Hildebrand, F.; Meyer, A.; Eyre-Walker, A. Evidence of selection upon genomic GC-Content in bacteria. PLoS Genet. 2010, 6, e1001107. [Google Scholar] [CrossRef]
- Singh, R.; Ming, R.; Yu, Q. Comparative Analysis of GC content variations in plant genomes. Trop. Plant Biol. 2016, 9, 136–149. [Google Scholar] [CrossRef]
- Wang, S.; Chen, S.; Liu, C.-X.; Liu, Y.; Zhao, X.-Y.; Yang, C.-P.; Qu, G.-Z. Genome survey sequencing of Betula platyphylla. Forests 2019, 10, 826. [Google Scholar] [CrossRef]
- Almeida Lopes, K.B.; Carpentieri Pipolo, V.; Oro, T.H.; Stefani Pagliosa, E.; Degrassi, G. Culturable endophytic bacterial communities associated with field-grown soybean. J. Appl. Microbiol. 2016, 120, 740–755. [Google Scholar] [CrossRef]
- Hung, P.Q.; Kumar, S.M.; Govindsamy, V.; Annapurna, K. Isolation and characterization of endophytic bacteria from wild and cultivated soybean varieties. Biol. Fertil. Soils 2007, 44, 155–162. [Google Scholar] [CrossRef]
- Liao, S.-M.; Du, Q.-S.; Meng, J.-Z.; Pang, Z.-W.; Huang, R.-B. The multiple roles of histidine in protein interactions. Chem. Cent. J. 2013, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Boyington, J.C.; Gaffney, B.J.; Amzel, L.M. The three-dimensional structure of an arachidonic acid 15-lipoxygenase. Science 1993, 260, 1482–1486. [Google Scholar] [CrossRef]
- Steczko, J.; Donoho, G.P.; Clemens, J.C.; Dixon, J.E.; Axelrod, B. Conserved histidine residues in soybean lipoxygenase: Functional consequences of their replacement. Biochemistry 1992, 31, 4053. [Google Scholar] [CrossRef]
- Steczko, J.; Axelrod, B. Identification of the iron-binding histidine residues in soybean lipoxygenase L-1. Biochem. Biophys. Res. Commun. 1992, 186, 686–689. [Google Scholar] [CrossRef]
- Wang, W.H.; Takano, T.; Shibata, D.; Kitamura, K.; Takeda, G. Molecular basis of a null mutation in soybean lipoxygenase 2: Substitution of glutamine for an iron-ligand histidine. Proc. Natl. Acad. Sci. USA 1994, 91, 5828–5832. [Google Scholar] [CrossRef] [PubMed]
- Chikhi, R.; Medvedev, P. Informed and automated k-mer size selection for genome assembly. Bioinformatics 2014, 30, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Alonge, M.; Lebeigle, L.; Kirsche, M.; Jenike, K.; Ou, S.; Aganezov, S.; Wang, X.-G.; Lippman, Z.B.; Schatz, M.C.; Soyk, S. Automated assembly scaffolding using RagTag elevates a new tomato system for high-throughput genome editing. Genome Biol. 2022, 23, 258. [Google Scholar] [CrossRef] [PubMed]
- Shumate, A.; Salzberg, S.L. Liftoff: Accurate mapping of gene annotations. Bioinformatics 2021, 37, 1639–1643. [Google Scholar] [CrossRef] [PubMed]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 2020, 36, 2251–2252. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Zhang, J.; Ng, C.; Jiang, Y.; Wang, X.-X.; Wang, S.-D.; Wang, S. Genome-wide identification and analysis of LOX genes in soybean cultivar “Zhonghuang 13”. Front. Genet. 2022, 13, 1020554. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Narvel, J.M.; Fehr, W.R.; Weldon, L.C. Analysis of soybean seed lipoxygenases. Crop Sci. 2000, 40, 838–840. [Google Scholar] [CrossRef]
- Suda, I.; Hajika, M.; Nishiba, Y.; Furuta, S.; Igita, K. Simple and rapid method for the selective detection of individual lipoxygenase isoenzymes in soybean seeds. J. Agric. Food. Chem. 1995, 43, 742–747. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
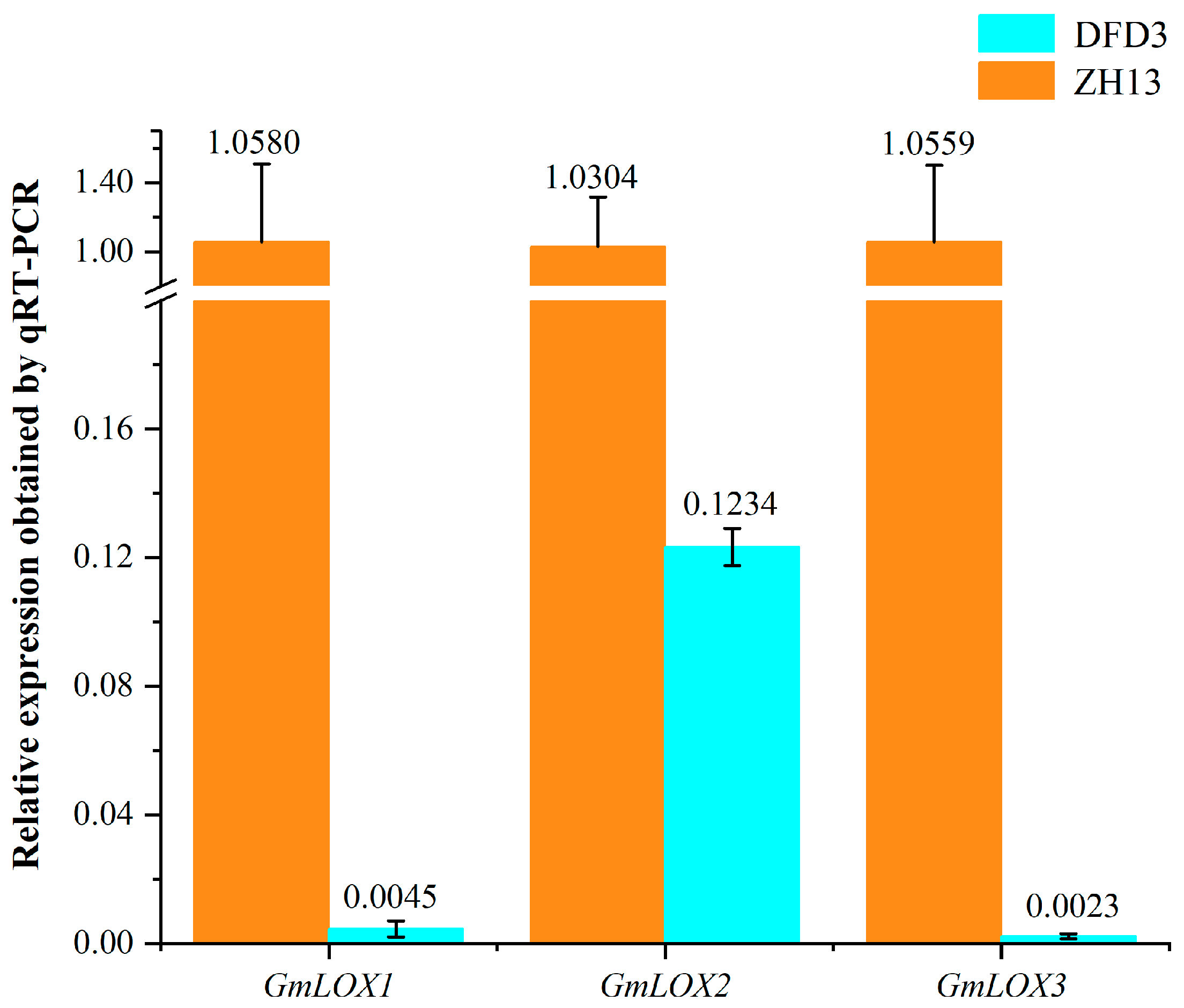



{kind=link}
{kind=link}
{kind=link}
{kind=link}
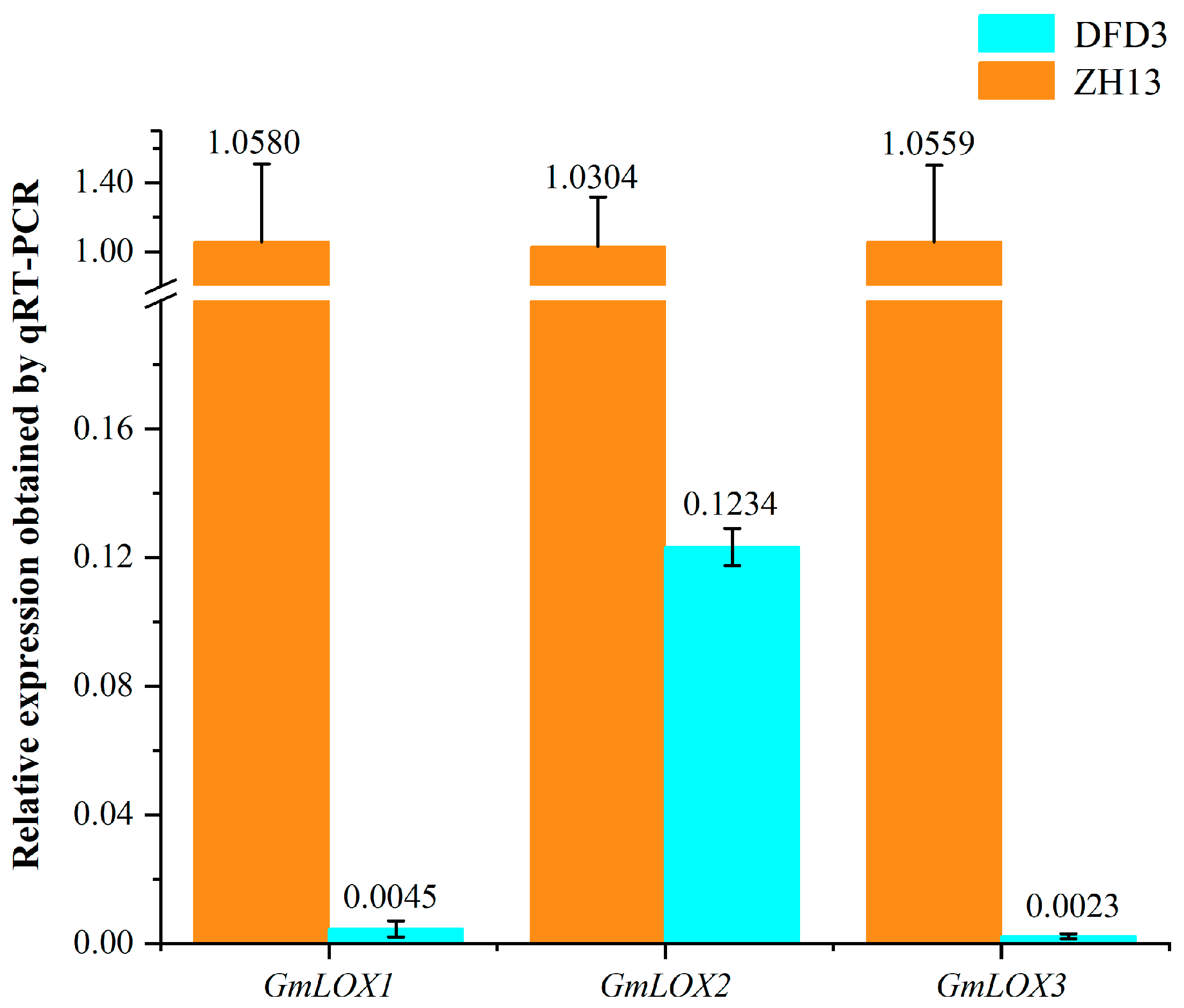{kind=link}
{kind=link}
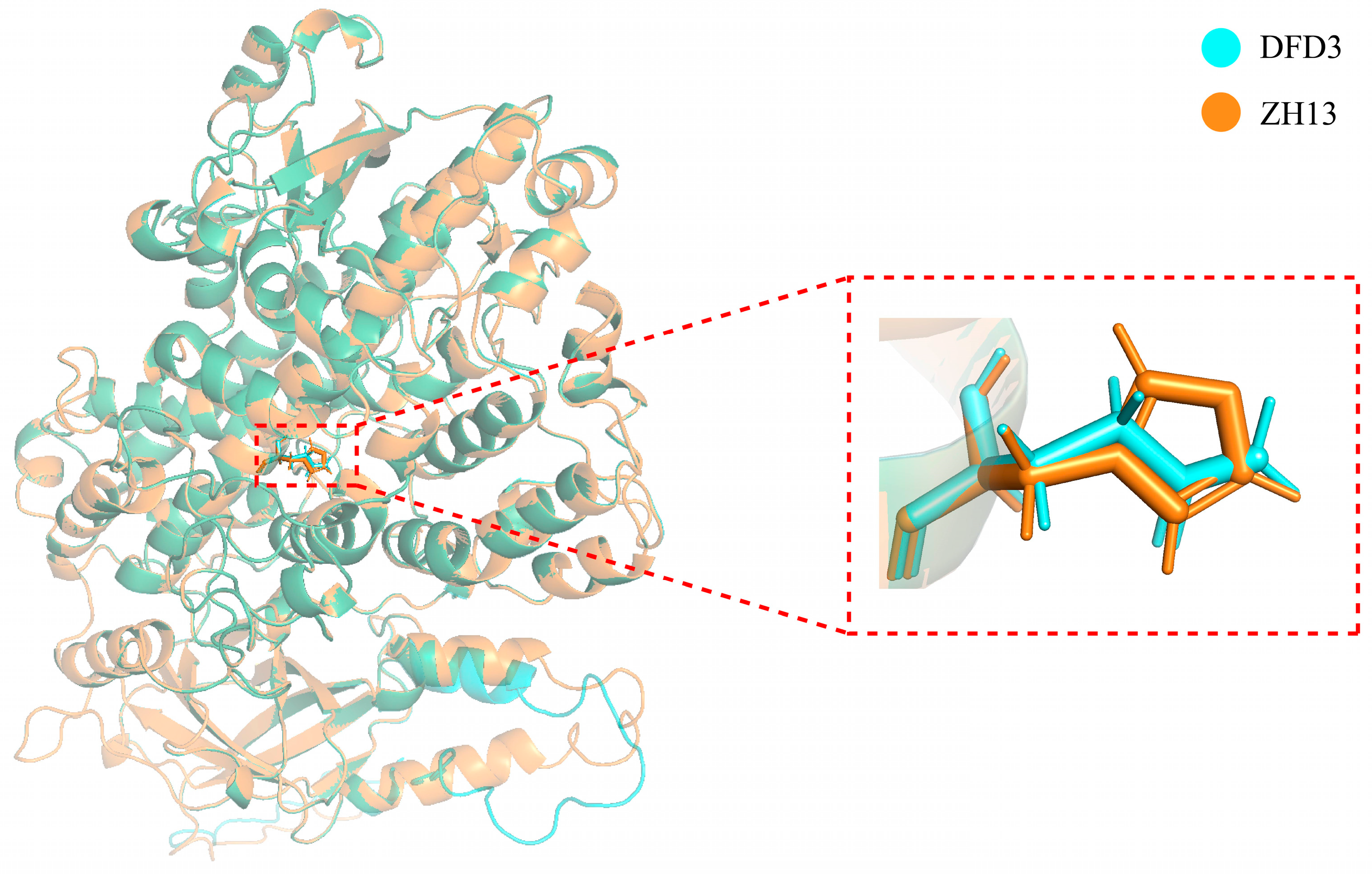{kind=link}
{kind=link}
| Item | Value |
|---|---|
| Assembled genome size (Mb) | 916.00 |
| Number of contigs | 60,936 |
| Number of Scaffolds | 20 |
| N50 of contigs (bp) | 39,370 |
| N90 of contigs (bp) | 9078 |
| Longest contig (bp) | 298,193 |
| GC content (%) | 34.3 |
| BUSCO complete of the genome (%) | 99.5 |
| BUSCO complete of the genes (%) | 99.1 |
| Number of protein-coding genes | 46,446 |
| Number of genes annotated to GO terms | 30,797 |
| Number of genes annotated to KEGG terms | 19,737 |
| Primer Name 1 | Primer |
|---|---|
| GmLox1-snp-F | AGCTTTGGTTGATTTTCTCACAGGT |
| GmLox1-snp-R | CGGGGATTCCCATGCTTCCG |
| GmLox1-del-F | CGAGGTAAACATGCGAAGCG |
| GmLox1-del-R | GCAGCCCATATCTCCAGTCC |
| GmLox2-snp-F | TGCCACATCCTGCTGGGGA |
| GmLox2-snp-R | CCGCTGAAGACATCTCAACGGAA |
| GmLox3-del-F | TACCACCAGGGGCTGTGCTT |
| GmLox3-del-R | GAAGACACACAAGGAGGACACGC |
| GmLox1-qPCR-F | ATTGGTTAAATACTCATGCGGC |
| GmLox1-qPCR-R | CCGAAGACATCTCCACAGAATA |
| GmLox2-qPCR-F | TCCTGAACAGAGGAGGAGGG |
| GmLox2-qPCR-R | GTGCCTATGAGTCCCCCAAC |
| GmLox3-qPCR-F | GCTTGGGGGTCTTCTCCATAG |
| GmLox3-qPCR-R | GCTGGAGAGACACGGATCG |
| GmCYP2-qPCR-F | CAAAAACCCTGTCACGCAGT |
| GmCYP2-qPCR-R | CACTTTCTCTCAAGGGCACCA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, Y.; Li, Y.; Zhang, J.; Song, Y.; Jiang, Y.; Tong, X.; Bi, Y.; Wang, S.; Wang, S. Genome Survey and Chromosome-Level Draft Genome Assembly of Glycine max var. Dongfudou 3: Insights into Genome Characteristics and Protein Deficiencies. Plants 2023, 12, 2994. https://doi.org/10.3390/plants12162994
Duan Y, Li Y, Zhang J, Song Y, Jiang Y, Tong X, Bi Y, Wang S, Wang S. Genome Survey and Chromosome-Level Draft Genome Assembly of Glycine max var. Dongfudou 3: Insights into Genome Characteristics and Protein Deficiencies. Plants. 2023; 12(16):2994. https://doi.org/10.3390/plants12162994
Chicago/Turabian StyleDuan, Yajuan, Yue Li, Jing Zhang, Yongze Song, Yan Jiang, Xiaohong Tong, Yingdong Bi, Shaodong Wang, and Sui Wang. 2023. "Genome Survey and Chromosome-Level Draft Genome Assembly of Glycine max var. Dongfudou 3: Insights into Genome Characteristics and Protein Deficiencies" Plants 12, no. 16: 2994. https://doi.org/10.3390/plants12162994
APA StyleDuan, Y., Li, Y., Zhang, J., Song, Y., Jiang, Y., Tong, X., Bi, Y., Wang, S., & Wang, S. (2023). Genome Survey and Chromosome-Level Draft Genome Assembly of Glycine max var. Dongfudou 3: Insights into Genome Characteristics and Protein Deficiencies. Plants, 12(16), 2994. https://doi.org/10.3390/plants12162994