
Soybean Protein and Oil Variants Identified through a Forward Genetic Screen for Seed Composition


Abstract
1. Introduction
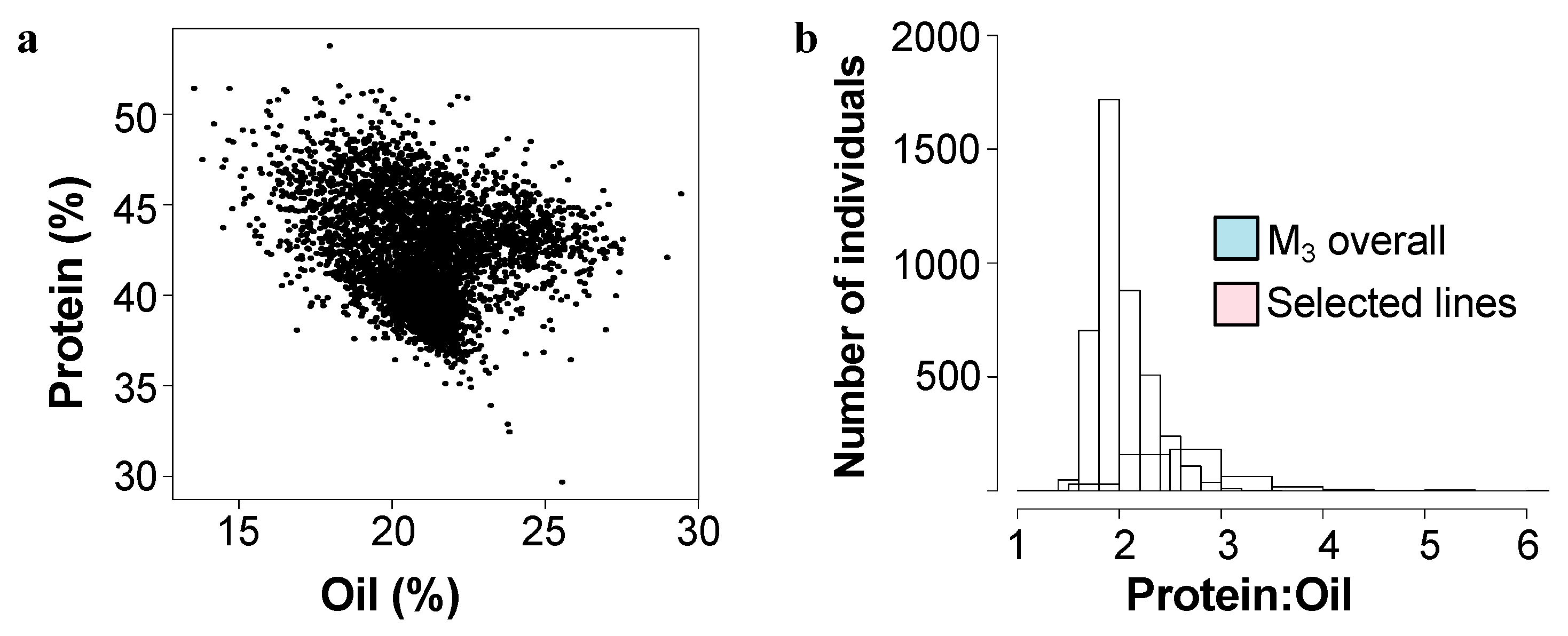
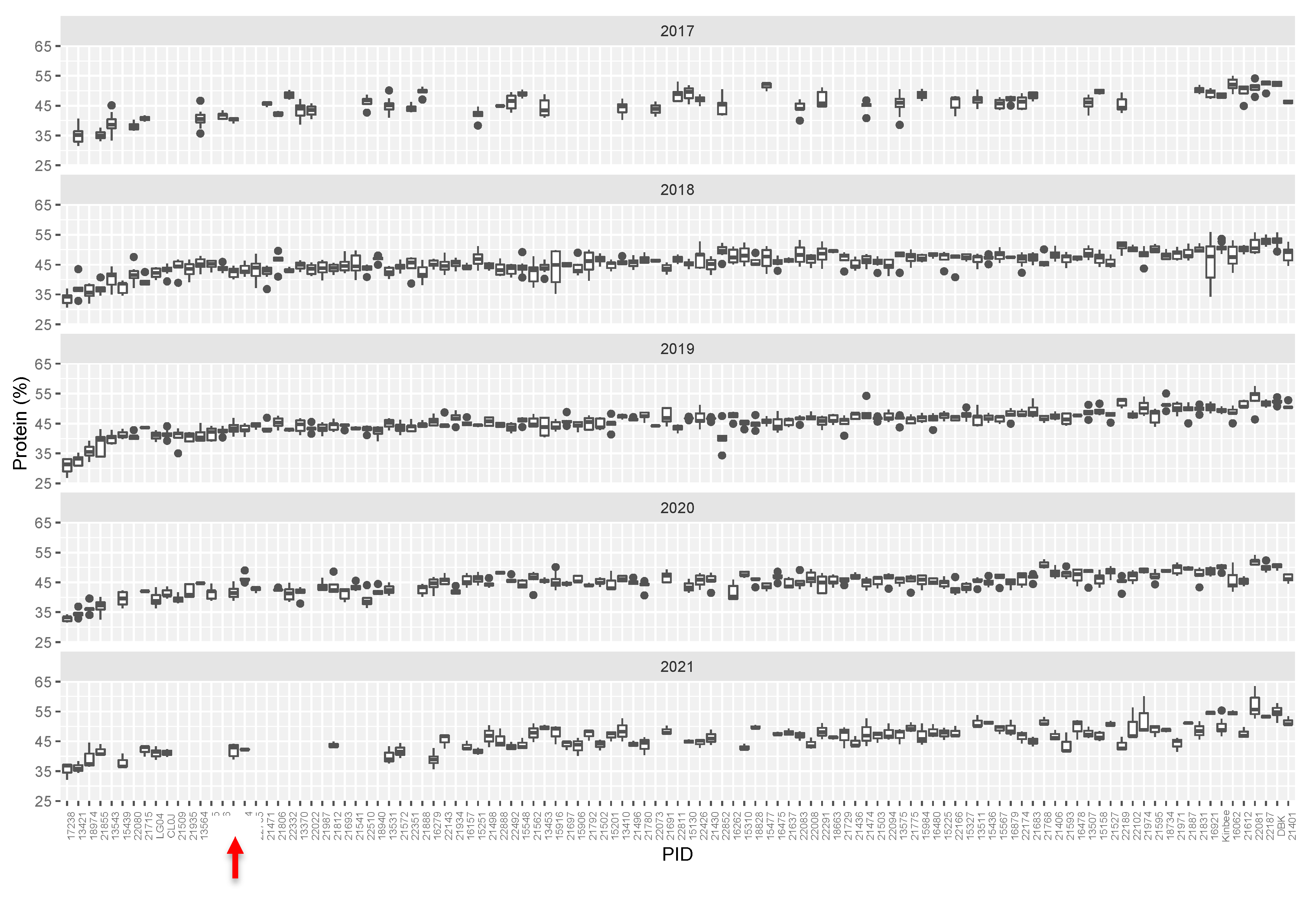
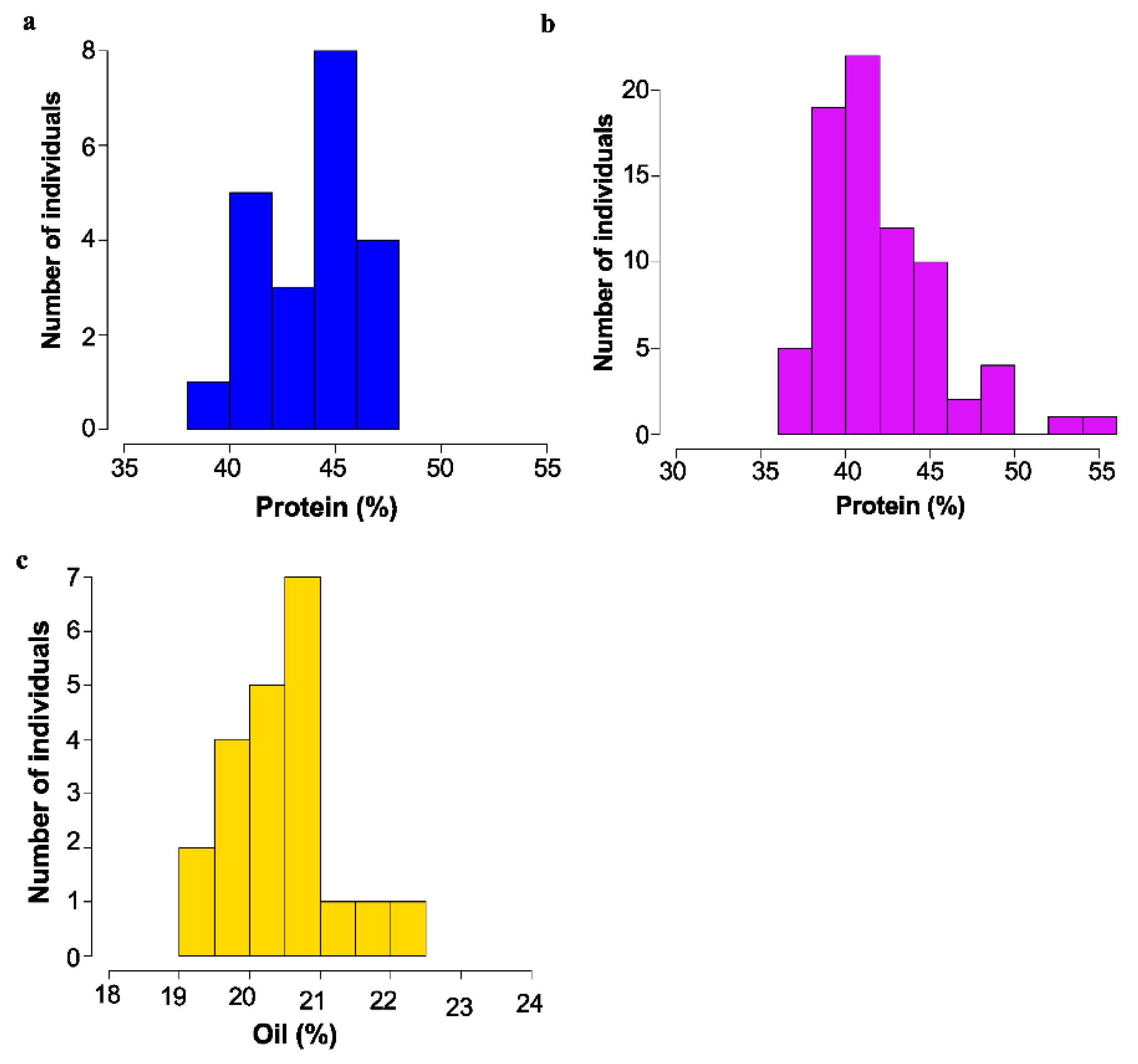
2. Results
2.1. Identification of Mutants
2.2. Genetic Classification of Mutants
3. Discussion
4. Materials and Methods
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jacobs, T.B.; LaFayette, P.R.; Schmitz, R.J.; Parrott, W.A. Targeted genome modifications in soybean with CRISPR/Cas9. BMC Biotechnol. 2015, 15, 16. [Google Scholar] [CrossRef] [PubMed]
- Carrijo, J.; Illa-Berenguer, E.; LaFayette, P.; Torres, N.; Aragão, F.J.L.; Parrott, W.; Vianna, G.R. Two efficient CRISPR/Cas9 systems for gene editing in soybean. Transgenic. Res. 2021, 30, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhang, L.; Zhang, K.; Ran, Y. Progresses, Challenges, and Prospects of Genome Editing in Soybean (Glycine max). Front. Plant Sci. 2020, 11, 571138. [Google Scholar] [CrossRef] [PubMed]
- Espina, M.J.; Ahmed, C.M.S.; Bernardini, A.; Adeleke, E.; Yadegari, Z.; Arelli, P.; Pantalone, V.; Taheri, A. Development and Phenotypic Screening of an Ethyl Methane Sulfonate Mutant Population in Soybean. Front. Plant Sci. 2018, 9, 394. [Google Scholar] [CrossRef] [PubMed]
- Hudson, K. Soybean Oil-Quality Variants Identified by Large-Scale Mutagenesis. Int. J. Agron. 2012, 2012, 7. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, L.; Ma, Y.; Wei, Z.; Hong, H.; Liu, Z.; Lei, J.; Liu, Y.; Guan, R.; Guo, Y.; et al. Development and utilization of a new chemically-induced soybean library with a high mutation density. J. Integr. Plant Biol. 2017, 59, 60–74. [Google Scholar] [CrossRef]
- Clemente, T.E.; Cahoon, E.B. Soybean Oil: Genetic Approaches for Modification of Functionality and Total Content. Plant Physiol. 2009, 151, 1030–1040. [Google Scholar] [CrossRef]
- Wilcox, J.R.; Shibles, R.M. Interrelationships among Seed Quality Attributes in Soybean. Crop Sci. 2001, 41, 11–14. [Google Scholar] [CrossRef]
- Nichols, D.M.; Glover, K.D.; Carlson, S.R.; Specht, J.E.; Diers, B.W. Fine Mapping of a Seed Protein QTL on Soybean Linkage Group I and Its Correlated Effects on Agronomic Traits. Crop Sci. 2006, 46, 834–839. [Google Scholar] [CrossRef]
- Sebolt, A.M.; Shoemaker, R.C.; Diers, B.W. Analysis of a Quantitative Trait Locus Allele from Wild Soybean That Increases Seed Protein Concentration in Soybean. Crop Sci. 2000, 40, 1438–1444. [Google Scholar] [CrossRef]
- Rincker, K.; Nelson, R.; Specht, J.; Sleper, D.; Cary, T.; Cianzio, S.R.; Casteel, S.; Conley, S.; Chen, P.; Davis, V.; et al. Genetic Improvement of U.S. Soybean in Maturity Groups II, III, and IV. Crop Sci. 2014, 54, 1419–1432. [Google Scholar] [CrossRef]
- Patil, G.; Mian, R.; Vuong, T.; Pantalone, V.; Song, Q.; Chen, P.; Shannon, G.J.; Carter, T.C.; Nguyen, H.T. Molecular mapping and genomics of soybean seed protein: A review and perspective for the future. Appl. Genet. 2017, 130, 1975–1991. [Google Scholar] [CrossRef] [PubMed]
- Bandillo, N.; Jarquin, D.; Song, Q.; Nelson, R.; Cregan, P.; Specht, J.; Lorenz, A. A Population Structure and Genome-Wide Association Analysis on the USDA Soybean Germplasm Collection. Plant Genome 2015, 8, plantgenome2015.04.0024. [Google Scholar] [CrossRef] [PubMed]
- Fliege, C.E.; Ward, R.A.; Vogel, P.; Nguyen, H.; Quach, T.; Guo, M.; Viana, J.P.G.; Dos Santos, L.B.; Specht, J.E.; Clemente, T.E.; et al. Fine mapping and cloning of the major seed protein quantitative trait loci on soybean chromosome 20. Plant J. 2022, 110, 114–128. [Google Scholar] [CrossRef]
- Wang, S.; Liu, S.; Wang, J.; Yokosho, K.; Zhou, B.; Yu, Y.C.; Liu, Z.; Frommer, W.B.; Ma, J.F.; Chen, L.Q.; et al. Simultaneous changes in seed size, oil content and protein content driven by selection of SWEET homologues during soybean domestication. Natl. Sci. Rev. 2020, 7, 1776–1786. [Google Scholar] [CrossRef]
- Zhang, H.; Goettel, W.; Song, Q.; Jiang, H.; Hu, Z.; Wang, M.L.; An, Y.C. Selection of GmSWEET39 for oil and protein improvement in soybean. PLoS Genet. 2020, 16, e1009114. [Google Scholar] [CrossRef]
- Manan, S.; Ahmad, M.Z.; Zhang, G.; Chen, B.; Haq, B.U.; Yang, J.; Zhao, J. Soybean LEC2 Regulates Subsets of Genes Involved in Controlling the Biosynthesis and Catabolism of Seed Storage Substances and Seed Development. Front. Plant Sci. 2017, 8, 1604. [Google Scholar] [CrossRef]
- Cooper, J.L.; Till, B.J.; Laport, R.G.; Darlow, M.C.; Kleffner, J.M.; Jamai, A.; El-Mellouki, T.; Liu, S.; Ritchie, R.; Nielsen, N.; et al. TILLING to detect induced mutations in soybean. BMC Plant Biol. 2008, 8, 9. [Google Scholar] [CrossRef]
- Thapa, R.; Carrero-Colon, M.; Rainey, K.M.; Hudson, K. TILLING by Sequencing: A Successful Approach to Identify Rare Alleles in Soybean Populations. Genes 2019, 10, 1003. [Google Scholar] [CrossRef]
- Nelson, R.L.; Johnson, E.O.C. Registration of the High-Yielding Soybean Germplasm Line LG04-6000. J. Plant Regist. 2012, 6, 212–215. [Google Scholar] [CrossRef]
- LeRoy, A.R.; Abney, T.S. Registration of ‘CL0J173-6-2’ and ‘CL0J173-6-8’ Soybeans. J. Plant Regist. 2007, 1, 98–99. [Google Scholar] [CrossRef]
- Song, Q.; Yan, L.; Quigley, C.; Jordan, B.D.; Fickus, E.; Schroeder, S.; Song, B.-H.; Charles An, Y.-Q.; Hyten, D.; Nelson, R.; et al. Genetic Characterization of the Soybean Nested Association Mapping Population. Plant Genome 2017, 10, plantgenome2016.10.0109. [Google Scholar] [CrossRef] [PubMed]
- Manan, S.; Zhao, J. Role of Glycine max ABSCISIC ACID INSENSITIVE 3 (GmABI3) in lipid biosynthesis and stress tolerance in soybean. Funct. Plant Biol. 2021, 48, 171–179. [Google Scholar] [CrossRef]
- Zhang, D.; Sun, L.; Li, S.; Wang, W.; Ding, Y.; Swarm, S.A.; Li, L.; Wang, X.; Tang, X.; Zhang, Z.; et al. Elevation of soybean seed oil content through selection for seed coat shininess. Nat. Plants 2018, 4, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Bolon, Y.T.; Joseph, B.; Cannon, S.B.; Graham, M.A.; Diers, B.W.; Farmer, A.D.; May, G.D.; Muehlbauer, G.J.; Specht, J.E.; Tu, Z.J.; et al. Complementary genetic and genomic approaches help characterize the linkage group I seed protein QTL in soybean. BMC Plant Biol. 2010, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Hyten, D.L.; Song, Q.; Zhu, Y.; Choi, I.-Y.; Nelson, R.L.; Costa, J.M.; Specht, J.E.; Shoemaker, R.C.; Cregan, P.B. Impacts of genetic bottlenecks on soybean genome diversity. Proc. Natl. Acad. Sci. USA 2006, 103, 16666–16671. [Google Scholar] [CrossRef]
- Hudson, K.A.; Hudson, M.E. Genetic Variation for Seed Oil Biosynthesis in Soybean. Plant Mol. Biol. Rep. 2021, 39, 700–709. [Google Scholar] [CrossRef]
- Ritchie, R.; Wiley, H.; Szymanski, D.; Nielsen, N. Targeting Induced Local Lesions in Genomes-TILLING. In Legume Crop Genomics; Wilson, R.F., Stalker, H.T., Brummer, E.C., Eds.; AOCS Press: Champaign, IL, USA, 2004; pp. 194–203. [Google Scholar]
- Shapiro, S.S.; Wilk, M.B. An Analysis of Variance Test for Normality (Complete Samples). Biometrika 1965, 52, 591–611. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Backcross PID | Phenotype | No. of Plants | SW | p |
|---|---|---|---|---|
| 13421 | inconclusive | 39 | 0.914 | 0.006 |
| 13507 | D high protein | 50 | 0.975 | 0.380 |
| 13531 | r high protein | 48 | 0.974 | 0.352 |
| 14015 | r high protein | 55 | 0.939 | 0.008 |
| 15130 | r high protein | 56 | 0.988 | 0.840 |
| 15158 | D high protein | 56 | 0.978 | 0.405 |
| 15201 | inconclusive | 19 | 0.983 | 0.972 |
| 15251 | D high protein | 85 | 0.936 | <0.001 |
| 15310 | r high protein | 107 | 0.992 | 0.763 |
| 15439 | r high oil | 70 | 0.986 | 0.607 |
| 15477 | r high protein | 35 | 0.978 | 0.704 |
| 15548 | r high protein | 40 | 0.930 | 0.017 |
| 15906 | r high protein | 22 | 0.890 | 0.019 |
| 15916 | r high protein | 100 | 0.993 | 0.893 |
| 16062 | D high protein | 96 | 0.971 | 0.032 |
| 16157 | inconclusive | 37 | 0.953 | 0.119 |
| 16262 | r high protein | 38 | 0.980 | 0.734 |
| 16279 | D high protein | 54 | 0.964 | 0.108 |
| 16475 | D high protein | 56 | 0.950 | 0.021 |
| 16480 | r high protein | 38 | 0.951 | 0.094 |
| 16879 | D high protein | 42 | 0.920 | 0.006 |
| 16921 | D high protein | 48 | 0.950 | 0.040 |
| 17238 | r high oil | 44 | 0.872 | <0.001 |
| 18663 | r high protein | 78 | 0.969 | 0.052 |
| 18734 | r high protein | 76 | 0.907 | <0.001 |
| 18940 | inconclusive | 32 | 0.974 | 0.626 |
| 18974 | r high protein | 39 | 0.967 | 0.295 |
| 21401 | D high protein | 49 | 0.972 | 0.288 |
| 21424 | D high protein | 54 | 0.975 | 0.325 |
| 21502 | inconclusive | 41 | 0.980 | 0.687 |
| 21715 | r low oil | 51 | 0.964 | 0.125 |
| 21768 | inconclusive | 42 | 0.991 | 0.982 |
| 21775 | r high protein | 31 | 0.963 | 0.357 |
| 21831 | D high protein | 58 | 0.987 | 0.793 |
| 21855 | r high protein | 48 | 0.959 | 0.091 |
| 21887 | D high protein | 57 | 0.982 | 0.552 |
| 22080 | Inconclusive | 41 | 0.950 | 0.071 |
| 22081 | Inconclusive | 30 | 0.975 | 0.688 |
| 22102 | r high protein | 38 | 0.924 | 0.013 |
| 22143 | D high protein | 40 | 0.939 | 0.031 |
| 22187 | Inconclusive | 56 | 0.988 | 0.841 |
| 22189 | Inconclusive | 53 | 0.970 | 0.199 |
| 22426 | D high protein | 31 | 0.953 | 0.195 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hudson, K. Soybean Protein and Oil Variants Identified through a Forward Genetic Screen for Seed Composition. Plants 2022, 11, 2966. https://doi.org/10.3390/plants11212966
Hudson K. Soybean Protein and Oil Variants Identified through a Forward Genetic Screen for Seed Composition. Plants. 2022; 11(21):2966. https://doi.org/10.3390/plants11212966
Chicago/Turabian StyleHudson, Karen. 2022. "Soybean Protein and Oil Variants Identified through a Forward Genetic Screen for Seed Composition" Plants 11, no. 21: 2966. https://doi.org/10.3390/plants11212966
APA StyleHudson, K. (2022). Soybean Protein and Oil Variants Identified through a Forward Genetic Screen for Seed Composition. Plants, 11(21), 2966. https://doi.org/10.3390/plants11212966