Specific BK Channel Activator NS11021 Protects Rat Renal Proximal Tubular Cells from Cold Storage—Induced Mitochondrial Injury In Vitro


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. NRK Cell Model of Renal Cold Storage Plus Rewarming
2.2. Isolation of NRK Mitochondria
2.3. Protein Lysates and Immunoblotting
2.4. Isolation of NRK Cells and Rat Renal Vascular Smooth Muscle Cells
2.5. Patch-Clamp Measurement of Whole-Cell BK Channel Current
2.6. Measurement of mitoBK Channel-Mediated K+ Uptake in Isolated NRK Mitochondria
2.7. High-Resolution Respirometry
2.8. Detection of Mitochondrial Superoxide Production
2.9. Measurement of Mitochondrial Membrane Potential
2.10. Measurement of NRK Cell Cytotoxicity
2.11. Statistical Analysis
3. Results
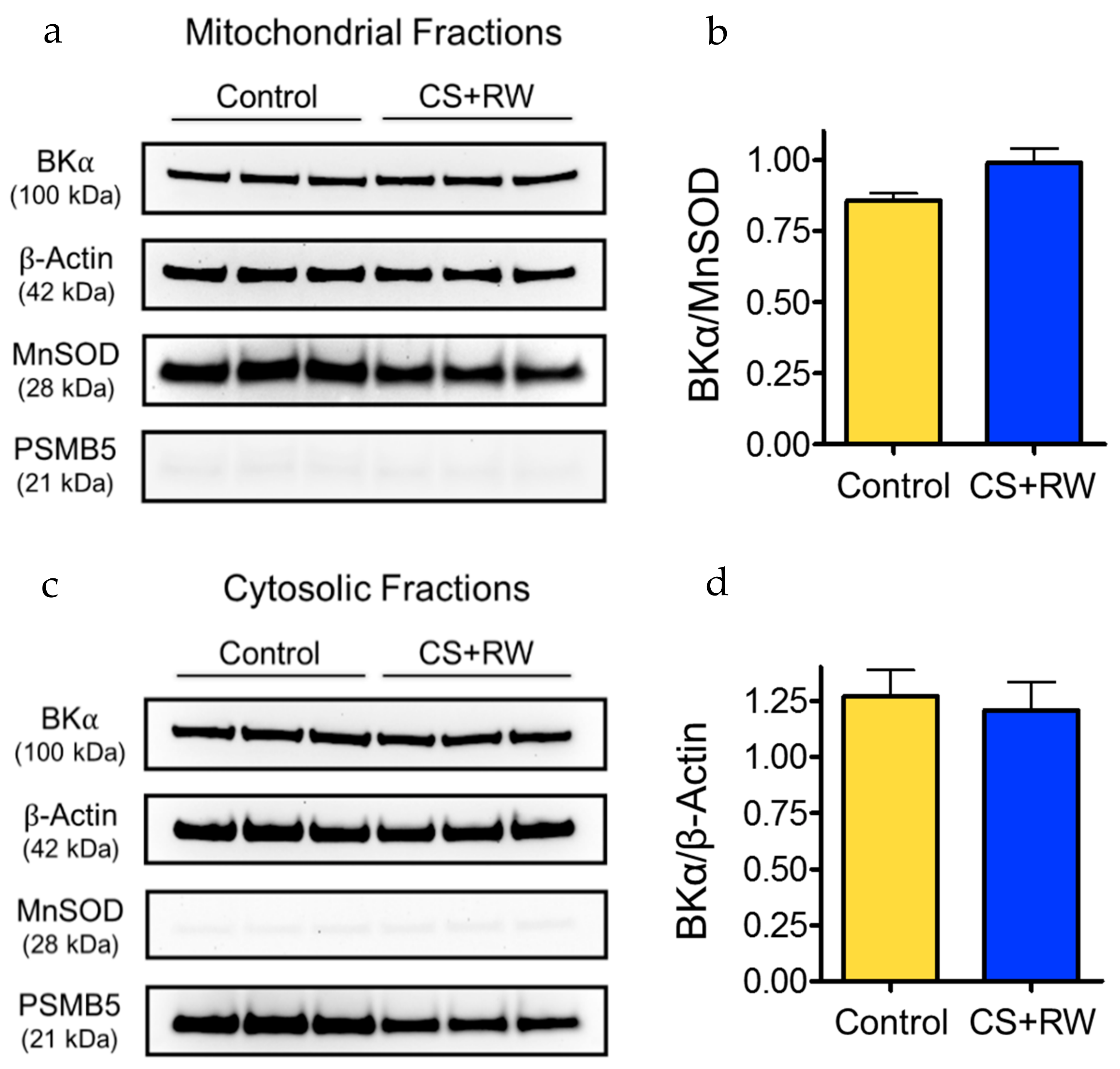
3.1. MitoBK Channels Are Expressed in NRK Cells
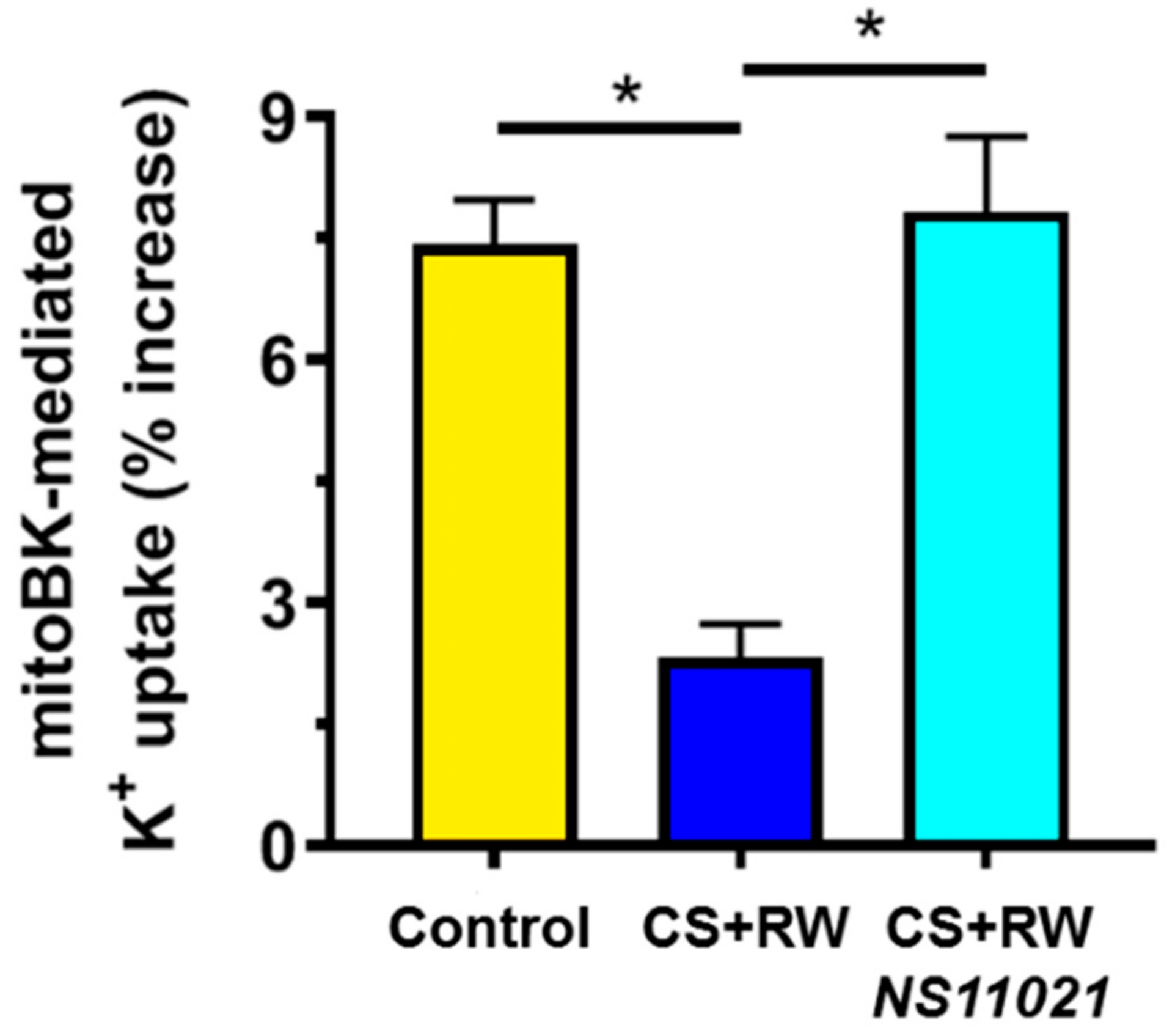
3.2. CS + RW Impairs MitoBK Channel-mediated K+ Uptake in NRK Mitochondria, which is Prevented by NS11021 Treatment During CS
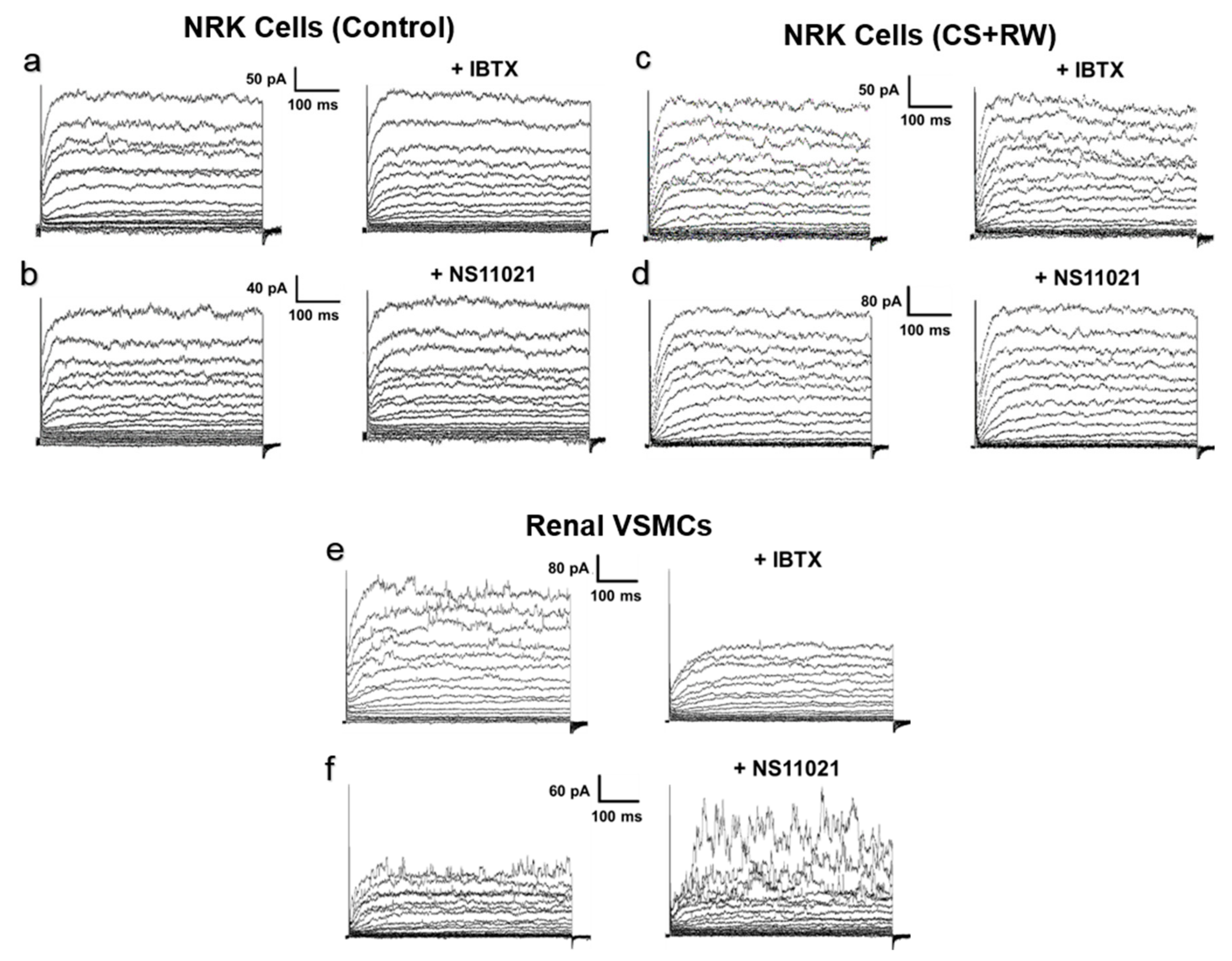
3.3. NRK Cells do not Express Functional BK Channels in Plasma Membrane
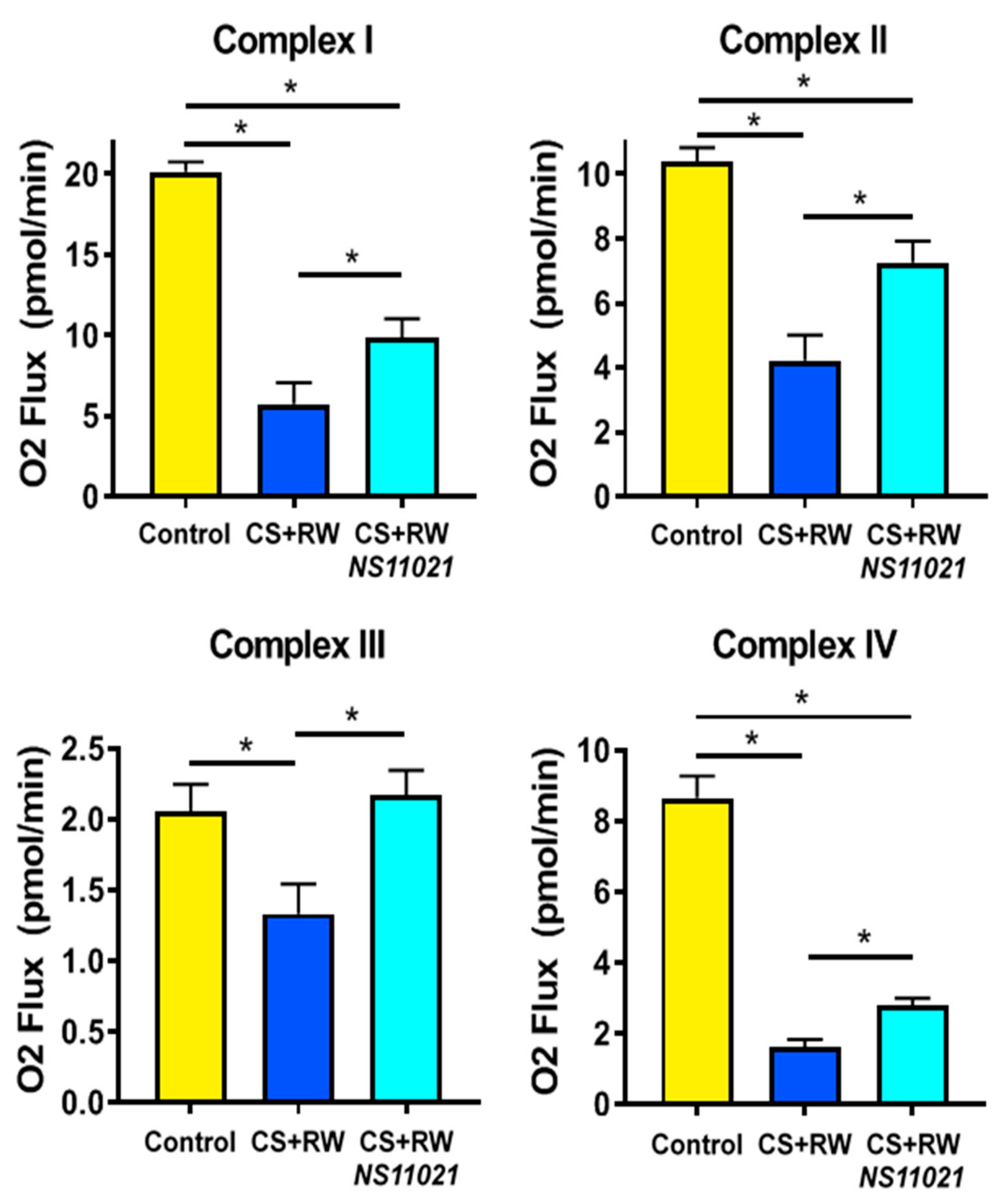
3.4. NS11021 Attenuates CS + RW-Induced Mitochondrial Respiratory Dysfunction
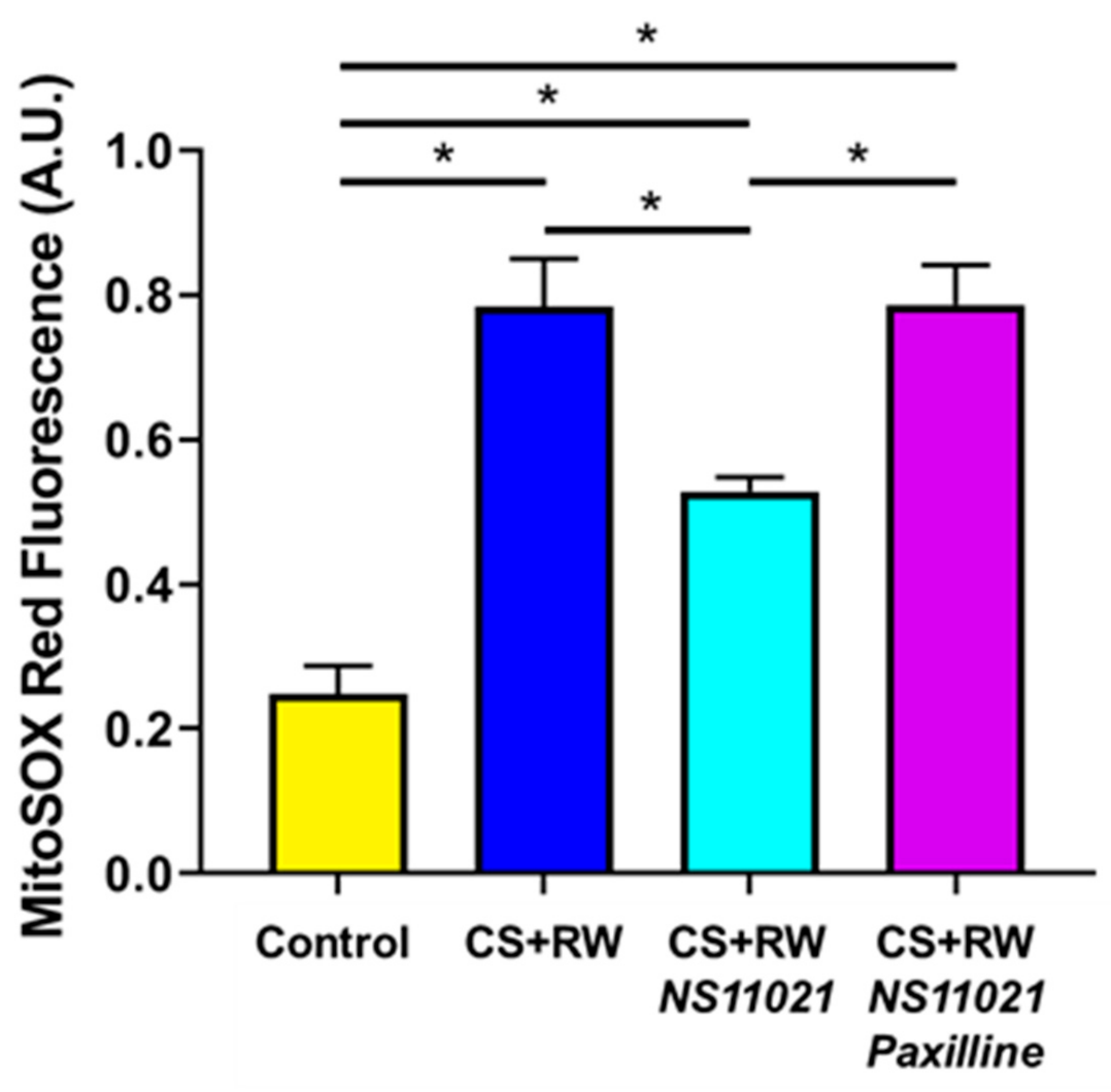
3.5. NS11021 Mitigates CS + RW-Induced Mitochondrial Superoxide Production
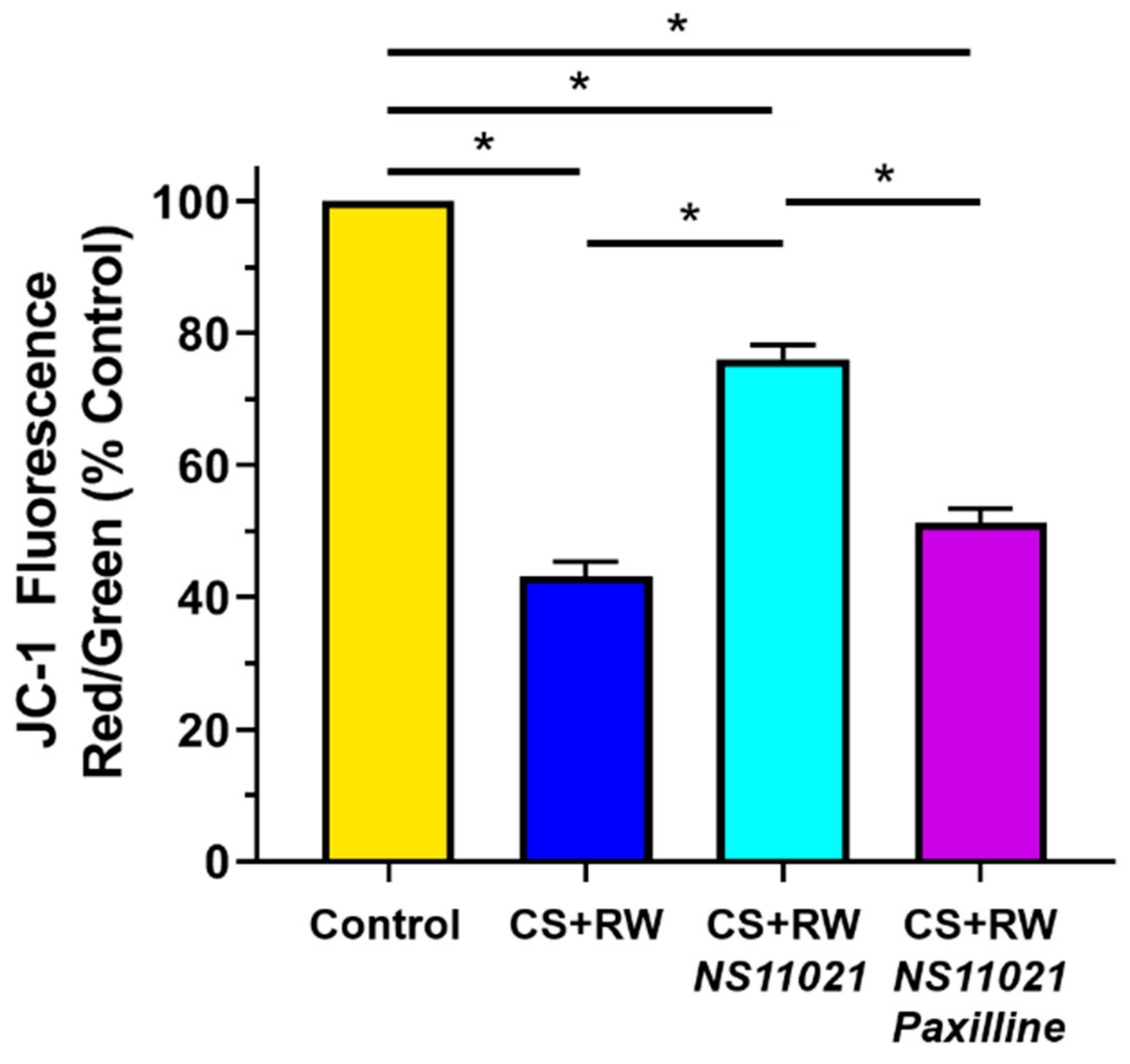
3.6. NS11021 Mitigates CS + RW-Induced Mitochondrial Depolarization
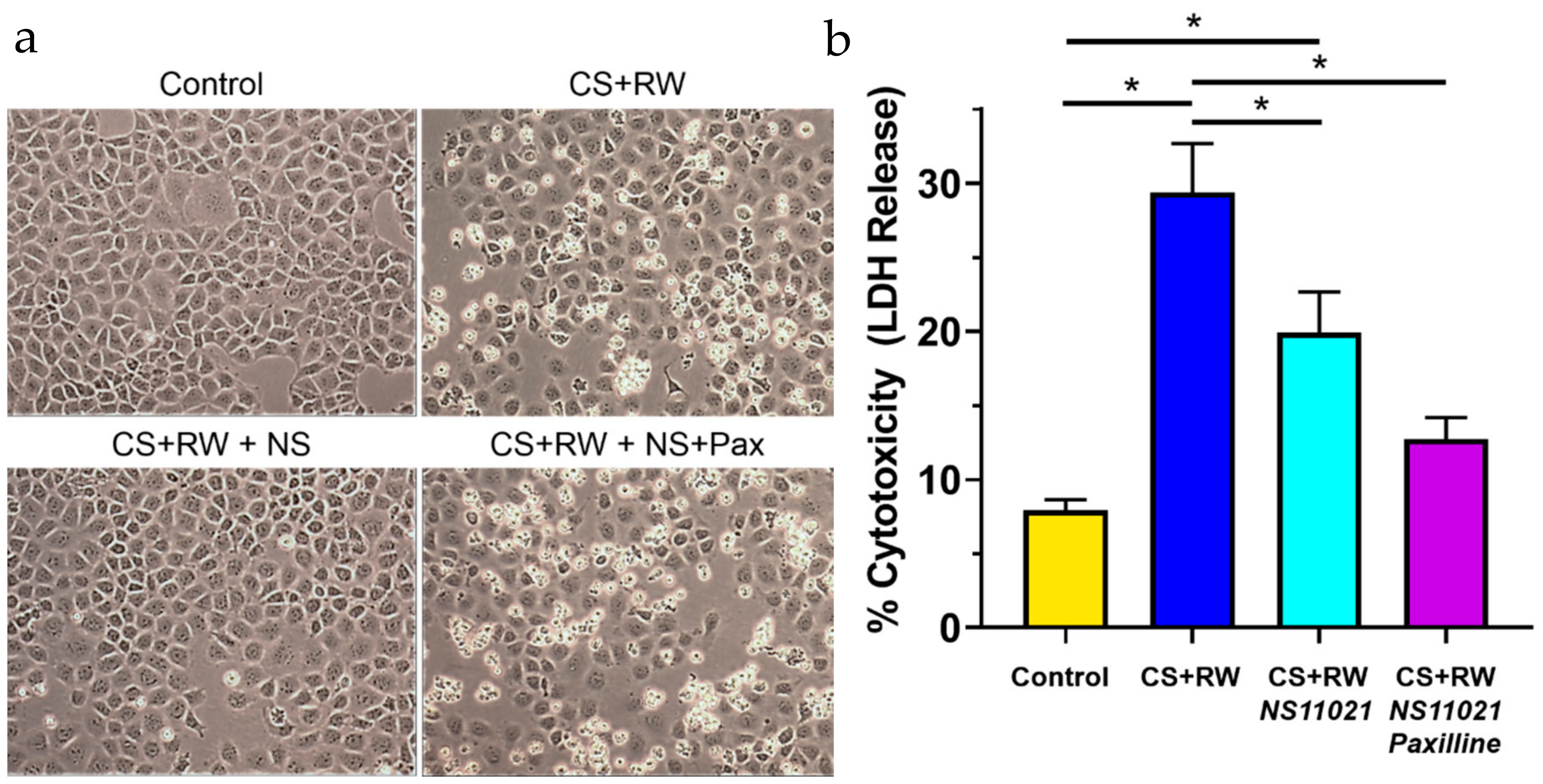
3.7. NS11021 Attenuates CS + RW-Induced Cell Death
4. Discussion
4.1. NRK Cells Express MitoBK Channels
4.2. CS + RW Impairs MitoBK Channel–Mediated K+ Uptake
4.3. NS11021 Restores MitoBK Channel-Mediated K+ Uptake
4.4. NS11021 Protects NRK Cells Against CS + RW-Induced Mitochondrial Injury and Cell Death
4.5. Pharmacological Limitations
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Southard, J.H.; Lutz, M.F.; Ametani, M.S.; Belzer, F.O. Stimulation of ATP synthesis in hypothermically perfused dog kidneys by adenosine and PO4. Cryobiology 1984, 21, 13–19. [Google Scholar] [CrossRef]
- Southard, J.H.; Senzig, K.A.; Belzer, F.O. Effects of hypothermia on canine kidney mitochondria. Cryobiology 1980, 17, 148–153. [Google Scholar] [CrossRef]
- Southard, J.H.; Van der Laan, N.C.; Lutz, M.; Pavlock, G.S.; Belzer, J.P.; Belzer, F.O. Comparison of the effect of temperature on kidney cortex mitochondria from rabbit, dog, pig, and human: Arrhenius plots of ADP-stimulated respiration. Cryobiology 1983, 20, 395–400. [Google Scholar] [CrossRef]
- Belzer, F.O.; Southard, J.H. Principles of solid-organ preservation by cold storage. Transplantation 1988, 45, 673–676. [Google Scholar] [CrossRef]
- Salahudeen, A.K. Cold ischemic injury of transplanted kidneys: New insights from experimental studies. Am. J. Physiol. Ren. Physiol. 2004, 287, F181–F187. [Google Scholar] [CrossRef]
- Salahudeen, A.K.; Haider, N.; May, W. Cold ischemia and the reduced long-term survival of cadaveric renal allografts. Kidney Int. 2004, 65, 713–718. [Google Scholar] [CrossRef]
- Debout, A.; Foucher, Y.; Trebern-Launay, K.; Legendre, C.; Kreis, H.; Mourad, G.; Garrigue, V.; Morelon, E.; Buron, F.; Rostaing, L.; et al. Each additional hour of cold ischemia time significantly increases the risk of graft failure and mortality following renal transplantation. Kidney Int. 2015, 87, 343–349. [Google Scholar] [CrossRef]
- Saba, H.; Munusamy, S.; MacMillan-Crow, L.A. Cold preservation mediated renal injury: Involvement of mitochondrial oxidative stress. Ren. Fail. 2008, 30, 125–133. [Google Scholar] [CrossRef]
- Salahudeen, A.K.; Huang, H.; Joshi, M.; Moore, N.A.; Jenkins, J.K. Involvement of the mitochondrial pathway in cold storage and rewarming-associated apoptosis of human renal proximal tubular cells. Am. J. Transpl. 2003, 3, 273–280. [Google Scholar] [CrossRef]
- Mitchell, T.; Saba, H.; Laakman, J.; Parajuli, N.; MacMillan-Crow, L.A. Role of mitochondrial-derived oxidants in renal tubular cell cold-storage injury. Free Radic. Biol. Med. 2010, 49, 1273–1282. [Google Scholar] [CrossRef]
- Parajuli, N.; Shrum, S.; Tobacyk, J.; Harb, A.; Arthur, J.M.; MacMillan-Crow, L.A. Renal cold storage followed by transplantation impairs expression of key mitochondrial fission and fusion proteins. PLoS ONE 2017, 12, e0185542. [Google Scholar] [CrossRef] [PubMed]
- Shrum, S.; MacMillan-Crow, L.A.; Parajuli, N. Cold storage exacerbates renal and mitochondrial dysfunction following transplantation. J. Kidney 2016, 2, 114. [Google Scholar] [PubMed]
- Salahudeen, A.K.; Huang, H.; Patel, P.; Jenkins, J.K. Mechanism and prevention of cold storage-induced human renal tubular cell injury. Transplantation 2000, 70, 1424–1431. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.; Rotaru, D.; Saba, H.; Smith, R.A.; Murphy, M.P.; MacMillan-Crow, L.A. The mitochondria-targeted antioxidant mitoquinone protects against cold storage injury of renal tubular cells and rat kidneys. J. Pharmacol. Exp. Ther. 2011, 336, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Parajuli, N.; Campbell, L.H.; Marine, A.; Brockbank, K.G.; MacMillan-Crow, L.A. MitoQ blunts mitochondrial and renal damage during cold preservation of porcine kidneys. PLoS ONE 2012, 7, e48590. [Google Scholar] [CrossRef] [PubMed]
- Salahudeen, A.K.; Joshi, M.; Jenkins, J.K. Apoptosis versus necrosis during cold storage and rewarming of human renal proximal tubular cells. Transplantation 2001, 72, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Balderas, E.; Zhang, J.; Stefani, E.; Toro, L. Mitochondrial BKCa channel. Front. Physiol. 2015, 6, 104. [Google Scholar] [CrossRef]
- Bentzen, B.H.; Osadchii, O.; Jespersen, T.; Hansen, R.S.; Olesen, S.P.; Grunnet, M. Activation of big conductance Ca2+-activated K+ channels (BK) protects the heart against ischemia-reperfusion injury. Pflug. Arch. 2009, 457, 979–988. [Google Scholar] [CrossRef]
- Soltysinska, E.; Bentzen, B.H.; Barthmes, M.; Hattel, H.; Thrush, A.B.; Harper, M.E.; Qvortrup, K.; Larsen, F.J.; Schiffer, T.A.; Losa-Reyna, J.; et al. KCNMA1 encoded cardiac BK channels afford protection against ischemia-reperfusion injury. PLoS ONE 2014, 9, e103402. [Google Scholar] [CrossRef]
- Goswami, S.K.; Ponnalagu, D.; Hussain, A.T.; Shah, K.; Karekar, P.; Gururaja, R.S.; Meredith, A.L.; Khan, M.; Singh, H. Expression and activation of BKCa channels in mice protects against ischemia-reperfusion injury of isolated hearts by modulating mitochondrial function. Front. Cardiovasc. Med. 2018, 5, 194. [Google Scholar] [CrossRef]
- Singh, H.; Lu, R.; Bopassa, J.C.; Meredith, A.L.; Stefani, E.; Toro, L. MitoBK(Ca) is encoded by the Kcnma1 gene, and a splicing sequence defines its mitochondrial location. Proc. Natl. Acad. Sci. USA 2013, 110, 10836–10841. [Google Scholar] [CrossRef] [PubMed]
- Borchert, G.H.; Hlavackova, M.; Kolar, F. Pharmacological activation of mitochondrial BK(Ca) channels protects isolated cardiomyocytes against simulated reperfusion-induced injury. Exp. Biol. Med. 2013, 238, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Liu, Y.; Wang, S.; McDonald, T.; Van Eyk, J.E.; Sidor, A.; O’Rourke, B. Cytoprotective role of Ca2+- activated K+ channels in the cardiac inner mitochondrial membrane. Science 2002, 298, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Jiang, M.T.; Su, J.; Hutchins, W.; Konorev, E.; Baker, J.E. Mitochondrial big conductance KCa channel and cardioprotection in infant rabbit heart. J. Cardiovasc. Pharmacol. 2007, 50, 497–502. [Google Scholar] [CrossRef]
- Testai, L.; Da, P.E.; Piano, I.; Pistelli, L.; Gargini, C.; Breschi, M.C.; Braca, A.; Martini, C.; Martelli, A.; Calderone, V. The citrus flavanone naringenin produces cardioprotective effects in hearts from 1 year old rat, through activation of mitoBK channels. Front. Pharmacol. 2017, 8, 71. [Google Scholar] [CrossRef]
- Testai, L.; Martelli, A.; Marino, A.; D’Antongiovanni, V.; Ciregia, F.; Giusti, L.; Lucacchini, A.; Chericoni, S.; Breschi, M.C.; Calderone, V. The activation of mitochondrial BK potassium channels contributes to the protective effects of naringenin against myocardial ischemia/reperfusion injury. Biochem. Pharmacol. 2013, 85, 1634–1643. [Google Scholar] [CrossRef]
- Stowe, D.F.; Aldakkak, M.; Camara, A.K.; Riess, M.L.; Heinen, A.; Varadarajan, S.G.; Jiang, M.T. Cardiac mitochondrial preconditioning by Big Ca2+-sensitive K+ channel opening requires superoxide radical generation. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H434–H440. [Google Scholar] [CrossRef]
- Stowe, D.F.; Yang, M.; Heisner, J.S.; Camara, A.K.S. Endogenous and agonist-induced opening of mitochondrial big versus small Ca2+-sensitive K+ channels on cardiac cell and mitochondrial protection. J. Cardiovasc. Pharmacol. 2017, 70, 314–328. [Google Scholar] [CrossRef]
- Bentzen, B.H.; Olesen, S.P.; Ronn, L.C.; Grunnet, M. BK channel activators and their therapeutic perspectives. Front. Physiol. 2014, 5, 389. [Google Scholar] [CrossRef]
- Testai, L.; Rapposelli, S.; Martelli, A.; Breschi, M.C.; Calderone, V. Mitochondrial potassium channels as pharmacological target for cardioprotective drugs. Med. Res. Rev. 2015, 35, 520–553. [Google Scholar] [CrossRef]
- National Institute of Standards and Technology Database 46v8 Calculator. Available online: https://somapp.ucdmc.ucdavis.edu/pharmacology/bers/maxchelator/CaMgATPEGTA-NIST.htm (accessed on 3 December 2019).
- Aon, M.A.; Cortassa, S.; Wei, A.C.; Grunnet, M.; O’Rourke, B. Energetic performance is improved by specific activation of K+ fluxes through K(Ca) channels in heart mitochondria. Biochim. Biophys. Acta 2010, 1797, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.; MacMillan-Crow, L.A.; Parajuli, N. Renal cold storage followed by transplantation impairs proteasome function and mitochondrial protein homeostasis. Am. J. Physiol. Ren. Physiol. 2019, 316, F42–F53. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.M.; Janes, M.S.; Pehar, M.; Monette, J.S.; Ross, M.F.; Hagen, T.M.; Murphy, M.P.; Beckman, J.S. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc. Natl. Acad. Sci. USA 2006, 103, 15038–15043. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Endara-Coll, M.; Mahmood, R.; Jankauskas, R.; Gjorgjieva, T.; Percipalle, P. Mitochondria-localized beta-actin is essential for priming innate antiviral immune signaling by regulating IRF3 protein stability. Cell. Mol. Immunol. 2019, 16, 837–840. [Google Scholar] [CrossRef]
- Xie, X.; Venit, T.; Drou, N.; Percipalle, P. In Mitochondria α-actin regulates mtDNA transcription and is required for nitochondrial quality control. iScience 2018, 3, 226–237. [Google Scholar] [CrossRef]
- Pesta, D.; Gnaiger, E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol. Biol. 2012, 810, 25–58. [Google Scholar]
- Kuznetsov, A.V.; Veksler, V.; Gellerich, F.N.; Saks, V.; Margreiter, R.; Kunz, W.S. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat. Protoc. 2008, 3, 965–976. [Google Scholar] [CrossRef]
- Lee, U.S.; Cui, J. BK channel activation: Structural and functional insights. Trends Neurosci. 2010, 33, 415–423. [Google Scholar] [CrossRef]
- Gonzalez-Perez, V.; Lingle, C.J. Regulation of BK Channels by Beta and Gamma Subunits. Annu. Rev. Physiol. 2019, 81, 113–137. [Google Scholar] [CrossRef]
- Balderas, E.; Torres, N.S.; Rosa-Garrido, M.; Chaudhuri, D.; Toro, L.; Stefani, E.; Olcese, R. MitoBKCa channel is functionally associated with its regulatory beta1 subunit in cardiac mitochondria. J. Physiol. 2019, 597, 3817–3832. [Google Scholar] [CrossRef]
- Bhattarai, Y.; Fernandes, R.; Kadrofske, M.M.; Lockwood, L.R.; Galligan, J.J.; Xu, H. Western blot analysis of BK channel beta1-subunit expression should be interpreted cautiously when using commercially available antibodies. Physiol. Rep. 2014, 2, e12189. [Google Scholar] [CrossRef]
- Kozyreva, T.V.; Evtushenko, A.A.; Voronova, I.P.; Khramova, G.M.; Kozaruk, V.P. Effect of activation of peripheral ion channel TRPM8 on gene expression of thermosensitive TRP ion channels in the hypothalamus. Comparison with the effect of cooling. Bull. Exp. Biol. Med. 2018, 166, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Jackson, T.C.; Manole, M.D.; Kotermanski, S.E.; Jackson, E.K.; Clark, R.S.; Kochanek, P.M. Cold stress protein RBM3 responds to temperature change in an ultra-sensitive manner in young neurons. Neuroscience 2015, 305, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Ebner, A.; Poitz, D.M.; Augstein, A.; Strasser, R.H.; Deussen, A. Functional, morphologic, and molecular characterization of cold storage injury. J. Vasc. Surg. 2012, 56, 189–198. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tang, X.D.; Garcia, M.L.; Heinemann, S.H.; Hoshi, T. Reactive oxygen species impair Slo1 BK channel function by altering cysteine-mediated calcium sensing. Nat. Struct. Mol. Biol. 2004, 11, 171–178. [Google Scholar] [CrossRef]
- Tang, X.D.; Santarelli, L.C.; Heinemann, S.H.; Hoshi, T. Metabolic regulation of potassium channels. Annu. Rev. Physiol. 2004, 66, 131–159. [Google Scholar] [CrossRef]
- Hermann, A.; Sitdikova, G.F.; Weiger, T.M. Oxidative stress and maxi calcium-activated potassium (BK) channels. Biomolecules 2015, 5, 1870–1911. [Google Scholar] [CrossRef]
- Kyle, B.D.; Braun, A.P. The regulation of BK channel activity by pre- and post-translational modifications. Front. Physiol. 2014, 5, 316. [Google Scholar] [CrossRef]
- Heinen, A.; Strothoff, M.; Schmidt, A.; Stracke, N.; Behmenburg, F.; Bauer, I.; Hollmann, M.W.; Huhn, R. Pharmacological options to protect the aged heart from ischemia and reperfusion injury by targeting the PKA-BK(Ca) signaling pathway. Exp. Gerontol. 2014, 56, 99–105. [Google Scholar] [CrossRef]
- Cao, C.M.; Chen, M.; Wong, T.M. The K(Ca) channel as a trigger for the cardioprotection induced by kappa-opioid receptor stimulation—Its relationship with protein kinase C. Br. J. Pharmacol. 2005, 145, 984–991. [Google Scholar] [CrossRef]
- Frankenreiter, S.; Bednarczyk, P.; Kniess, A.; Bork, N.I.; Straubinger, J.; Koprowski, P.; Wrzosek, A.; Mohr, E.; Logan, A.; Murphy, M.P.; et al. cGMP-elevating compounds and ischemic conditioning provide cardioprotection against ischemia and reperfusion injury via cardiomyocyte-specific BK channels. Circulation 2017, 136, 2337–2355. [Google Scholar] [CrossRef] [PubMed]
- Tano, J.Y.; Gollasch, M. Hypoxia and ischemia-reperfusion: A BiK contribution? Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H811–H817. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Siemen, D.; Loupatatzis, C.; Borecky, J.; Gulbins, E.; Lang, F. Ca2+-activated K channel of the BK-type in the inner mitochondrial membrane of a human glioma cell line. Biochem. Biophys. Res. Commun. 1999, 257, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Kicinska, A.; Augustynek, B.; Kulawiak, B.; Jarmuszkiewicz, W.; Szewczyk, A.; Bednarczyk, P. A large-conductance calcium-regulated K+ channel in human dermal fibroblast mitochondria. Biochem. J. 2016, 473, 4457–4471. [Google Scholar] [CrossRef] [PubMed]
- Olszewska, A.; Bednarczyk, P.; Siemen, D.; Szewczyk, A. Modulation of the mitochondrial large-conductance calcium-regulated potassium channel by polyunsaturated fatty acids. Biochim. Biophys. Acta 2014, 1837, 1602–1610. [Google Scholar] [CrossRef][Green Version]
- Cheng, Y.; Gulbins, E.; Siemen, D. Activation of the permeability transition pore by Bax via inhibition of the mitochondrial BK channel. Cell Physiol. Biochem. 2011, 27, 191–200. [Google Scholar] [CrossRef]
- Cheng, Y.; Gu, X.Q.; Bednarczyk, P.; Wiedemann, F.R.; Haddad, G.G.; Siemen, D. Hypoxia increases activity of the BK-channel in the inner mitochondrial membrane and reduces activity of the permeability transition pore. Cell Physiol. Biochem. 2008, 22, 127–136. [Google Scholar] [CrossRef]
- Cheng, Y.; Debska-Vielhaber, G.; Siemen, D. Interaction of mitochondrial potassium channels with the permeability transition pore. FEBS Lett. 2010, 584, 2005–2012. [Google Scholar] [CrossRef]
- Gu, X.Q.; Pamenter, M.E.; Siemen, D.; Sun, X.; Haddad, G.G. Mitochondrial but not plasmalemmal BK channels are hypoxia-sensitive in human glioma. Glia 2014, 62, 504–513. [Google Scholar] [CrossRef]
- Gu, X.Q.; Siemen, D.; Parvez, S.; Cheng, Y.; Xue, J.; Zhou, D.; Sun, X.; Jonas, E.A.; Haddad, G.G. Hypoxia increases BK channel activity in the inner mitochondrial membrane. Biochem. Biophys. Res. Commun. 2007, 358, 311–316. [Google Scholar] [CrossRef]
- Bednarczyk, P.; Wieckowski, M.R.; Broszkiewicz, M.; Skowronek, K.; Siemen, D.; Szewczyk, A. Putative structural and functional coupling of the mitochondrial BK channel to the respiratory chain. PLoS ONE 2013, 8, e68125. [Google Scholar] [CrossRef] [PubMed]
- Bednarczyk, P.; Koziel, A.; Jarmuszkiewicz, W.; Szewczyk, A. Large-conductance Ca2+-activated potassium channel in mitochondria of endothelial EA.hy926 cells. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1415–H1427. [Google Scholar] [CrossRef] [PubMed]
- Ohya, S.; Kuwata, Y.; Sakamoto, K.; Muraki, K.; Imaizumi, Y. Cardioprotective effects of estradiol include the activation of large-conductance Ca2+-activated K+ channels in cardiac mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1635–H1642. [Google Scholar] [CrossRef] [PubMed]
- Bentzen, B.H.; Nardi, A.; Calloe, K.; Madsen, L.S.; Olesen, S.P.; Grunnet, M. The small molecule NS11021 is a potent and specific activator of Ca2+-activated big-conductance K+ channels. Mol. Pharmacol. 2007, 72, 1033–1044. [Google Scholar] [CrossRef]
- Zhou, Y.; Lingle, C.J. Paxilline inhibits BK channels by an almost exclusively closed-channel block mechanism. J. Gen. Physiol. 2014, 144, 415–440. [Google Scholar] [CrossRef]
- Kun, A.; Matchkov, V.V.; Stankevicius, E.; Nardi, A.; Hughes, A.D.; Kirkeby, H.J.; Demnitz, J.; Simonsen, U. NS11021, a novel opener of large-conductance Ca2+-activated K+ channels, enhances erectile responses in rats. Br. J. Pharmacol. 2009, 158, 1465–1476. [Google Scholar] [CrossRef]
- Layne, J.J.; Nausch, B.; Olesen, S.P.; Nelson, M.T. BK channel activation by NS11021 decreases excitability and contractility of urinary bladder smooth muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R378–R384. [Google Scholar] [CrossRef]
- Guaragnella, N.; Coyne, L.P.; Chen, X.J.; Giannattasio, S. Mitochondria-cytosol-nucleus crosstalk: Learning from Saccharomyces cerevisiae. FEMS Yeast Res. 2018, 18, foy088. [Google Scholar] [CrossRef]
- Inoue, T.; Maekawa, H.; Inagi, R. Organelle crosstalk in the kidney. Kidney Int. 2019, 95, 1318–1325. [Google Scholar] [CrossRef]
- Kaasik, A.; Veksler, V.; Boehm, E.; Novotova, M.; Minajeva, A.; Ventura-Clapier, R. Energetic crosstalk between organelles: Architectural integration of energy production and utilization. Circ. Res. 2001, 89, 153–159. [Google Scholar] [CrossRef]
- Sharaf El Dein, O.; Gallerne, C.; Deniaud, A.; Brenner, C.; Lemaire, C. Role of the permeability transition pore complex in lethal inter-organelle crosstalk. Front. Biosci. 2009, 14, 3465–3482. [Google Scholar] [CrossRef] [PubMed]
- Soto-Heredero, G.; Baixauli, F.; Mittelbrunn, M. Interorganelle communication between mitochondria and the endolysosomal system. Front. Cell Dev. Biol. 2017, 5, 95. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Kim, S.; Peng, W.; Krainc, D. Regulation and function of mitochondria-lysosome membrane contact sites in cellular homeostasis. Trends Cell Biol. 2019, 29, 500–513. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Stefani, E.; Toro, L. Intracellular BKCa (iBKCa) channels. J. Physiol. 2012, 590, 5937–5947. [Google Scholar] [CrossRef]
- Cancherini, D.V.; Queliconi, B.B.; Kowaltowski, A.J. Pharmacological and physiological stimuli do not promote Ca2+-sensitive K+ channel activity in isolated heart mitochondria. Cardiovasc. Res. 2007, 73, 720–728. [Google Scholar] [CrossRef]
- Bilmen, J.G.; Wootton, L.L.; Michelangeli, F. The mechanism of inhibition of the sarco/endoplasmic reticulum Ca2+ ATPase by paxilline. Arch. Biochem. Biophys. 2002, 406, 55–64. [Google Scholar] [CrossRef]
- Ramella-Virieux, S.G.; Steghens, J.P.; Barbieux, A.; Zech, P.; Pozet, N.; Hadj-Aissa, A. Nifedipine improves recovery function of kidneys preserved in a high-sodium, low-potassium cold-storage solution: Study with the isolated perfused rat kidney technique. Nephrol. Dial. Transpl. 1997, 12, 449–455. [Google Scholar] [CrossRef]
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current mechanistic concepts in ischemia and reperfusion injury. Cell Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, S.; Hu, S.; Chen, Y.; Ren, J. ER-Mitochondria microdomains in cardiac ischemia-reperfusion injury: A fresh perspective. Front. Physiol. 2018, 9, 755. [Google Scholar] [CrossRef]
- Bednarczyk, P.; Barker, G.D.; Halestrap, A.P. Determination of the rate of K+ movement through potassium channels in isolated rat heart and liver mitochondria. Biochim. Biophys. Acta 2008, 1777, 540–548. [Google Scholar] [CrossRef]
- Cao, C.M.; Xia, Q.; Gao, Q.; Chen, M.; Wong, T.M. Calcium-activated potassium channel triggers cardioprotection of ischemic preconditioning. J. Pharmacol. Exp. Ther. 2005, 312, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, T.; Domoki, F.; Lenti, L.; Katakam, P.V.; Snipes, J.A.; Bari, F.; Busija, D.W. Immediate neuronal preconditioning by NS1619. Brain Res. 2009, 1285, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Heinen, A.; Aldakkak, M.; Stowe, D.F.; Rhodes, S.S.; Riess, M.L.; Varadarajan, S.G.; Camara, A.K. Reverse electron flow-induced ROS production is attenuated by activation of mitochondrial Ca2+-sensitive K+ channels. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1400–H1407. [Google Scholar] [CrossRef] [PubMed]
- Heinen, A.; Camara, A.K.; Aldakkak, M.; Rhodes, S.S.; Riess, M.L.; Stowe, D.F. Mitochondrial Ca2+-induced K+ influx increases respiration and enhances ROS production while maintaining membrane potential. Am. J. Physiol. Cell Physiol. 2007, 292, C148–C156. [Google Scholar] [CrossRef] [PubMed]
- Kulawiak, B.; Kudin, A.P.; Szewczyk, A.; Kunz, W.S. BK channel openers inhibit ROS production of isolated rat brain mitochondria. Exp. Neurol. 2008, 212, 543–547. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shrum, S.; Rusch, N.J.; MacMillan-Crow, L.A. Specific BK Channel Activator NS11021 Protects Rat Renal Proximal Tubular Cells from Cold Storage—Induced Mitochondrial Injury In Vitro. Biomolecules 2019, 9, 825. https://doi.org/10.3390/biom9120825
Shrum S, Rusch NJ, MacMillan-Crow LA. Specific BK Channel Activator NS11021 Protects Rat Renal Proximal Tubular Cells from Cold Storage—Induced Mitochondrial Injury In Vitro. Biomolecules. 2019; 9(12):825. https://doi.org/10.3390/biom9120825
Chicago/Turabian StyleShrum, Stephen, Nancy J. Rusch, and Lee Ann MacMillan-Crow. 2019. "Specific BK Channel Activator NS11021 Protects Rat Renal Proximal Tubular Cells from Cold Storage—Induced Mitochondrial Injury In Vitro" Biomolecules 9, no. 12: 825. https://doi.org/10.3390/biom9120825
APA StyleShrum, S., Rusch, N. J., & MacMillan-Crow, L. A. (2019). Specific BK Channel Activator NS11021 Protects Rat Renal Proximal Tubular Cells from Cold Storage—Induced Mitochondrial Injury In Vitro. Biomolecules, 9(12), 825. https://doi.org/10.3390/biom9120825