
A Multispecific Checkpoint Inhibitor Nanofitin with a Fast Tumor Accumulation Property and Anti-Tumor Activity in Immune Competent Mice


 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmid Construct, Expression, and Purification of Proteins
2.2. Biolayer Interferometry Analyses
2.3. Cell Culture
2.4. Cell Surface Binding by Flow Cytometry
2.5. Agilent X-Celligence Real-Time Cell Analysis
2.6. Animal Experiments
2.7. Immunohistochemistry Analyses
3. Results
3.1. The Bispecific B10–B11 Nanofitin Binds to A431 Cells and Induces a Cytotoxic Effect
3.2. Tumor Accumulation of the Bispecific B10–B11 Nanofitin in an A431 Xenograft Model
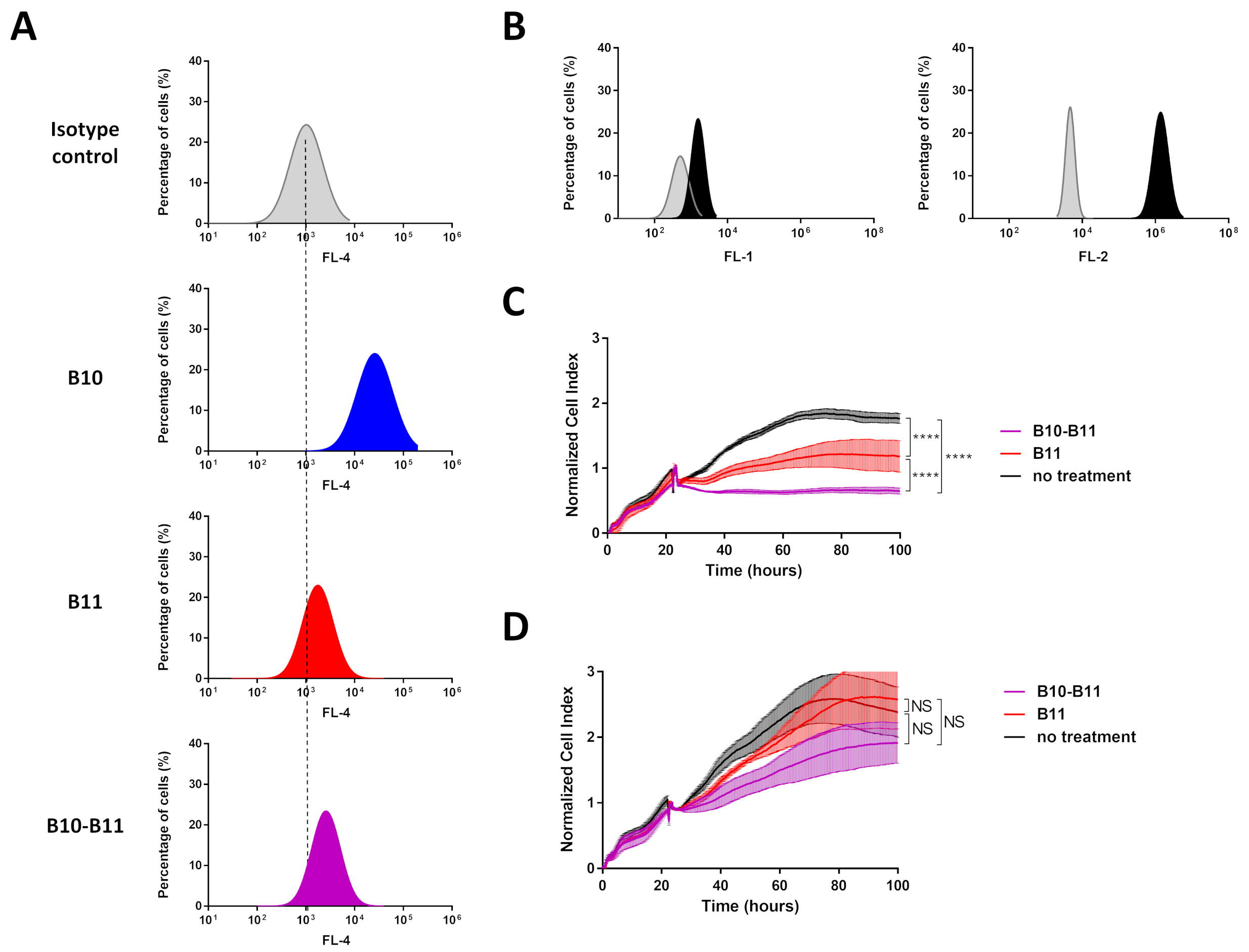
3.3. The Bispecific B10–B11 Nanofitin Is Cross-Reactive with Mouse EGFR and PD-L1 and Binds to CT26 Cells
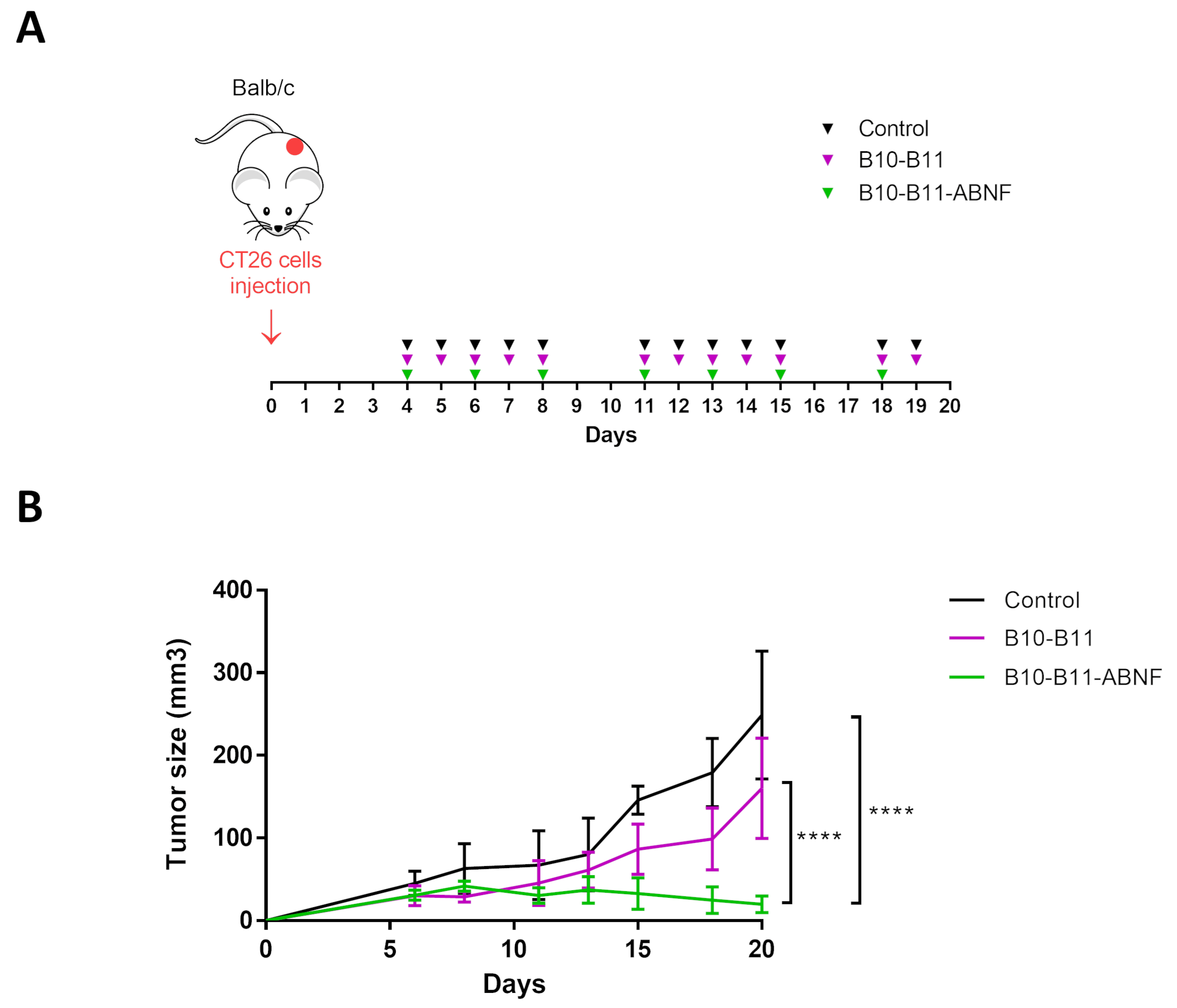
3.4. The Bispecific B10–B11 Nanofitin Reduces Tumor Proliferation in an Immunocompetent Murine Model
3.5. The Bispecific B10–B11 Nanofitin Promotes Immune Cell Recruiting
4. Discussion
4.1. Bispecific B10–B11 Nanofitin Simultaneously Engages EGFR and PD-L1 on Human A431 and Murine CT26 Cell Lines
4.2. Nanofitin Accumulation in Tumor Is Driven by Its Size
4.3. Bispecific B10–B11 Nanofitin Demonstrates a Potent Anti-PD-L1 Activity in an Immunocompetent Mice Model
4.4. Taking Advantages of Small and Bispecific PD-L1 Immune Checkpoint Inhibitor
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FDA | Food and Drug Administration |
| ICIs | Immune checkpoint inhibitors |
| CTLA-4 | Cytotoxic T-Lymphocyte-Associated protein 4 |
| PD-1 | Programmed cell Death protein 1 |
| PD-L1 | Programmed death-ligand 1 |
| irAEs | Immune-Related Adverse Events |
| PD-L2 | Programmed cell death-ligand 2 |
| EGFR | Epithelial Growth Factor Receptor |
| ABNF | Albumin-binding Nanofitin |
| HSA | Human Serum Albumin |
| EpCAM | Epithelial Cell Adhesion Molecule |
| CD3 | Cluster of Differentiation 3 |
| BiTE | Bispecific T-cell Engager |
| RTCA | Real-Time Cell Analysis |
| IHC | Immunohistochemistry |
| scFv | Single-Chain Variable Fragment |
| TAMs | Tumor-Associated Macrophages |
References
- Zhao, B.; Zhao, H.; Zhao, J. Efficacy of PD-1/PD-L1 blockade monotherapy in clinical trials. Ther. Adv. Med. Oncol. 2020, 12, 175883592093761. [Google Scholar] [CrossRef]
- Hiltbrunner, S.; Cords, L.; Kasser, S.; Freiberger, S.N.; Kreutzer, S.; Toussaint, N.C.; Grob, L.; Opitz, I.; Messerli, M.; Zoche, M.; et al. Acquired resistance to anti-PD1 therapy in patients with NSCLC associates with immunosuppressive T cell phenotype. Nat. Commun. 2023, 14, 5154. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, S.; Togashi, Y. Resistance to immune checkpoint inhibitors and the tumor microenvironment. Exp. Dermatol. 2023, 32, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Wu, L.; Han, L.; Zheng, X.; Tong, R.; Li, L.; Bai, L.; Bian, Y. Immune-related adverse events of immune checkpoint inhibitors: A review. Front. Immunol. 2023, 14, 1167975. [Google Scholar] [CrossRef] [PubMed]
- El Osta, B.; Hu, F.; Sadek, R.; Chintalapally, R.; Tang, S.-C. Not all immune-checkpoint inhibitors are created equal: Meta-analysis and systematic review of immune-related adverse events in cancer trials. Crit. Rev. Oncol./Hematol. 2017, 119,, 1–12. [Google Scholar] [CrossRef]
- Khunger, M.; Rakshit, S.; Pasupuleti, V.; Hernandez, A.V.; Mazzone, P.; Stevenson, J.; Pennell, N.A.; Velcheti, V. Incidence of Pneumonitis With Use of Programmed Death 1 and Programmed Death-Ligand 1 Inhibitors in Non-Small Cell Lung Cancer. Chest 2017, 152, 271–281. [Google Scholar] [CrossRef]
- Shklovskaya, E.; Rizos, H. Spatial and Temporal Changes in PD-L1 Expression in Cancer: The Role of Genetic Drivers, Tumor Microenvironment and Resistance to Therapy. Int. J. Mol. Sci. 2020, 21, 7139. [Google Scholar] [CrossRef]
- Kurino, T.; Matsuda, R.; Terui, A.; Suzuki, H.; Kokubo, T.; Uehara, T.; Arano, Y.; Hisaka, A.; Hatakeyama, H. Poor outcome with anti-programmed death-ligand 1 (PD-L1) antibody due to poor pharmacokinetic properties in PD-1/PD-L1 blockade-sensitive mouse models. J. Immunother. Cancer 2020, 8, e000400. [Google Scholar] [CrossRef]
- Koopmans, I.; Hendriks, D.; Samplonius, D.F.; Van Ginkel, R.J.; Heskamp, S.; Wierstra, P.J.; Bremer, E.; Helfrich, W. A novel bispecific antibody for EGFR-directed blockade of the PD-1/PD-L1 immune checkpoint. OncoImmunology 2018, 7, e1466016. [Google Scholar] [CrossRef]
- Jacquot, P.; Muñoz-Garcia, J.; Fleury, M.; Cochonneau, D.; Gaussin, R.; Enouf, E.; Roze, C.; Ollivier, E.; Cinier, M.; Heymann, D. Engineering of a Bispecific Nanofitin with Immune Checkpoint Inhibitory Activity Conditioned by the Cross-Arm Binding to EGFR and PDL1. Biomolecules 2023, 13, 636. [Google Scholar] [CrossRef]
- Dewhirst, M.W.; Secomb, T.W. Transport of drugs from blood vessels to tumour tissue. Nat. Rev. Cancer 2017, 17, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Tannock, I.F. The distribution of the therapeutic monoclonal antibodies cetuximab and trastuzumab within solid tumors. BMC Cancer 2010, 10, 255. [Google Scholar] [CrossRef]
- Huet, S.; Zeisser Labouebe, M.; Castro, R.; Jacquot, P.; Pedrault, J.; Viollet, S.; Van Simaeys, G.; Doumont, G.; Larbanoix, L.; Zindy, E.; et al. Targeted Nanofitin-drug Conjugates Achieve Efficient Tumor Delivery and Therapeutic Effect in an EGFRpos Mouse Xenograft Model. Mol. Cancer Ther. 2023, 22, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Nedrow, J.R.; Josefsson, A.; Park, S.; Ranka, S.; Roy, S.; Sgouros, G. Imaging of Programmed Cell Death Ligand 1: Impact of Protein Concentration on Distribution of Anti-PD-L1 SPECT Agents in an Immunocompetent Murine Model of Melanoma. J Nucl. Med. 2017, 58, 1560–1566. [Google Scholar] [CrossRef]
- Michot, N.; Guyochin, A.; Cinier, M.; Savignard, C.; Kitten, O.; Pascual, M.H.; Pouzieux, S.; Ozoux, M.L.; Verdier, P.; Vicat, P.; et al. Albumin binding Nanofitins, a new scaffold to extend half-life of biologics—a case study with exenatide peptide. Peptide 2022, 152, 170760. [Google Scholar] [CrossRef]
- Goux, M.; Becker, G.; Gorré, H.; Dammicco, S.; Desselle, A.; Egrise, D.; Leroi, N.; Lallemand, F.; Bahri, M.A.; Doumont, G.; et al. Nanofitin as a New Molecular-Imaging Agent for the Diagnosis of Epidermal Growth Factor Receptor Over-Expressing Tumors. Bioconjugate Chem. 2017, 28, 2361–2371. [Google Scholar] [CrossRef] [PubMed]
- Marcion, G.; Hermetet, F.; Neiers, F.; Uyanik, B.; Dondaine, L.; Dias, A.M.M.; Da Costa, L.; Moreau, M.; Bellaye, P.; Collin, B.; et al. Nanofitins targeting heat shock protein 110: An innovative immunotherapeutic modality in cancer. Int. J. Cancer 2021, 148, 3019–3031. [Google Scholar] [CrossRef]
- Heskamp, S.; Wierstra, P.J.; Molkenboer-Kuenen, J.D.; Sandker, G.W.; Thordardottir, S.; Cany, J.; Olive, D.; Bussink, J.; Boerman, O.C.; Dolstra, H.; et al. PD-L1 microSPECT/CT Imaging for Longitudinal Monitoring of PD-L1 Expression in Syngeneic and Humanized Mouse Models for Cancer. Cancer Immunol. Research 2019, 7, 150–161. [Google Scholar] [CrossRef]
- Zahnd, C.; Kawe, M.; Stumpp, M.T.; de Pasquale, C.; Tamaskovic, R.; Nagy-Davidescu, G.; Dreier, B.; Schibli, R.; Binz, H.K.; Waibel, R.; et al. Efficient Tumor Targeting with High-Affinity Designed Ankyrin Repeat Proteins: Effects of Affinity and Molecular Size. Cancer Research. 2010, 70, 1595–1605. [Google Scholar] [CrossRef]
- Debie, P.; Lafont, C.; Defrise, M.; Hansen, I.; van Willigen, D.M.; van Leeuwen, F.W.; Gijsbers, R.; D’Huyvetter, M.; Devoogdt, N.; Lahoutte, T.; et al. Size and affinity kinetics of nanobodies influence targeting and penetration of solid tumours. J. Control. Release 2020, 317, 34–42. [Google Scholar] [CrossRef]
- Wittrup, K.D.; Thurber, G.M.; Schmidt, M.M.; Rhoden, J.J. Practical Theoretic Guidance for the Design of Tumor-Targeting Agents. Methods Enzymol. 2012, 503, 255–268. [Google Scholar] [PubMed]
- Fiser, A.; Do, R.K.G.; Šali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [PubMed]
- Modak, M.; Mattes, A.-K.; Reiss, D.; Skronska-Wasek, W.; Langlois, R.; Sabarth, N.; Konopitzky, R.; Ramirez, F.; Lehr, K.; Mayr, T.; et al. CD206+ tumor-associated macrophages cross-present tumor antigen and drive antitumor immunity. JCI Insight 2022, 7, e155022. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Tijink, B.M.; Laeremans, T.; Budde, M.; Walsum, M.S.-V.; Dreier, T.; de Haard, H.J.; Leemans, C.R.; van Dongen, G.A. Improved tumor targeting of anti–epidermal growth factor receptor Nanobodies through albumin binding: Taking advantage of modular Nanobody technology. Mol. Cancer Ther. 2008, 7, 2288–2297. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | PBD | Molecular Weight (kDa) | Stokes Radius (Å) | Rh (nm) |
|---|---|---|---|---|
| Nanofitin | 1AZP | 7.60 | 15.81 | 1.581 |
| Dimeric Nanofitin | * | 16.14 | 23.90 | 2.390 |
| DARPin | 5KNH | 18.21 | 19.44 | 1.944 |
| scFv | 8DGR | 29.59 | 23.46 | 2.346 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacquot, P.; Muñoz-Garcia, J.; Léger, A.; Babuty, A.; Taupin, M.; Fradet, L.; Dupont, F.; Heymann, M.-F.; Cinier, M.; Heymann, D. A Multispecific Checkpoint Inhibitor Nanofitin with a Fast Tumor Accumulation Property and Anti-Tumor Activity in Immune Competent Mice. Biomolecules 2025, 15, 471. https://doi.org/10.3390/biom15040471
Jacquot P, Muñoz-Garcia J, Léger A, Babuty A, Taupin M, Fradet L, Dupont F, Heymann M-F, Cinier M, Heymann D. A Multispecific Checkpoint Inhibitor Nanofitin with a Fast Tumor Accumulation Property and Anti-Tumor Activity in Immune Competent Mice. Biomolecules. 2025; 15(4):471. https://doi.org/10.3390/biom15040471
Chicago/Turabian StyleJacquot, Perrine, Javier Muñoz-Garcia, Antoine Léger, Antoine Babuty, Manon Taupin, Laurie Fradet, Fabio Dupont, Marie-Françoise Heymann, Mathieu Cinier, and Dominique Heymann. 2025. "A Multispecific Checkpoint Inhibitor Nanofitin with a Fast Tumor Accumulation Property and Anti-Tumor Activity in Immune Competent Mice" Biomolecules 15, no. 4: 471. https://doi.org/10.3390/biom15040471
APA StyleJacquot, P., Muñoz-Garcia, J., Léger, A., Babuty, A., Taupin, M., Fradet, L., Dupont, F., Heymann, M.-F., Cinier, M., & Heymann, D. (2025). A Multispecific Checkpoint Inhibitor Nanofitin with a Fast Tumor Accumulation Property and Anti-Tumor Activity in Immune Competent Mice. Biomolecules, 15(4), 471. https://doi.org/10.3390/biom15040471