
Glucose Transporter Glut1-Dependent Metabolic Reprogramming Regulates Lipopolysaccharide-Induced Inflammation in RAW264.7 Macrophages


Abstract
1. Introduction
2. Materials and Methods
2.1. Macrophage Cell Culture
2.2. Macrophage Treatment with Lipopolysaccharide (LPS)
2.3. Trypan Blue Cell Count
2.4. Protein Extraction and Western Blot
2.5. RNA Extraction and RT-qPCR
2.6. siRNA-Mediated Knockdown of Glut1 Gene
2.7. Low Glucose Treatment
2.8. Flow Cytometry to Measure MHCII, CD40, and CD80 Surface Levels
2.9. Flow Cytometry to Measure Annexin V and 7-AAD
2.10. Glutathione Level Assay
2.11. Autophagy Detection
2.12. Statistical Analysis
3. Results
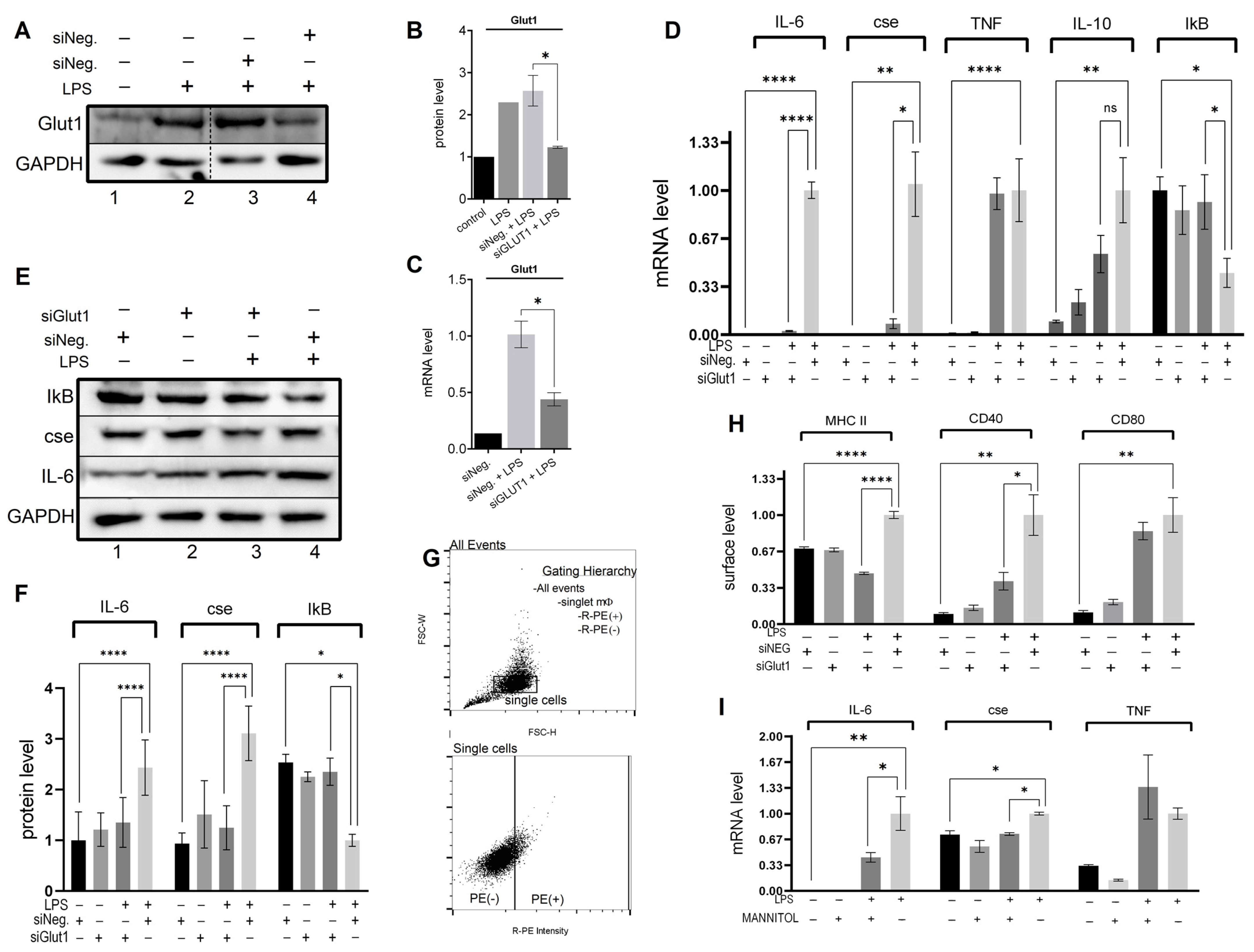
3.1. Glut1-Driven Glucose Uptake Drives NF-κB Mediated Pro-Inflammatory Profile in LPS-Treated Macrophages
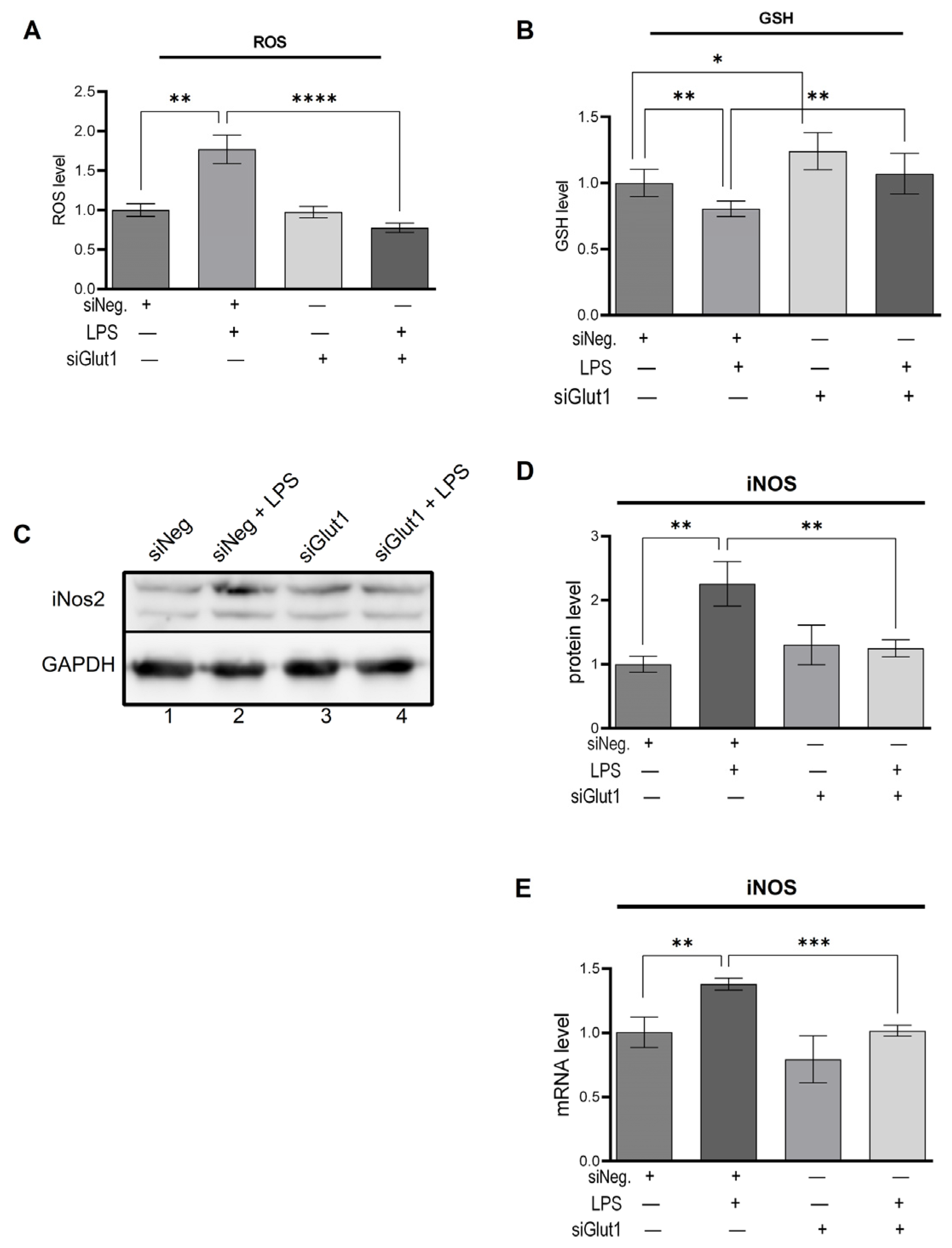
3.2. Glut1-Driven Glucose Uptake Alters Redox in Macrophages
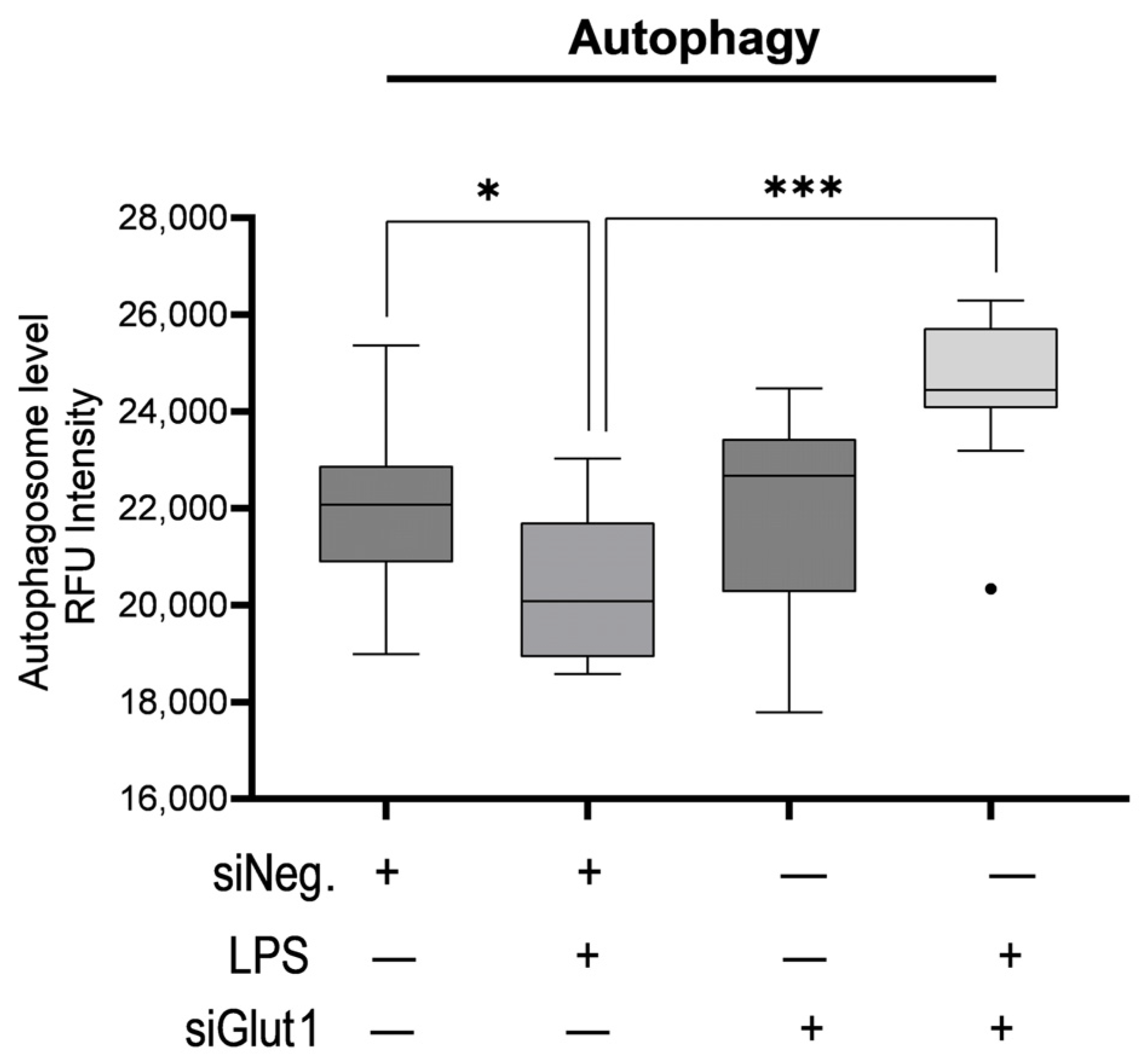
3.3. Glut1 Silencing Increases Autophagy in Macrophages
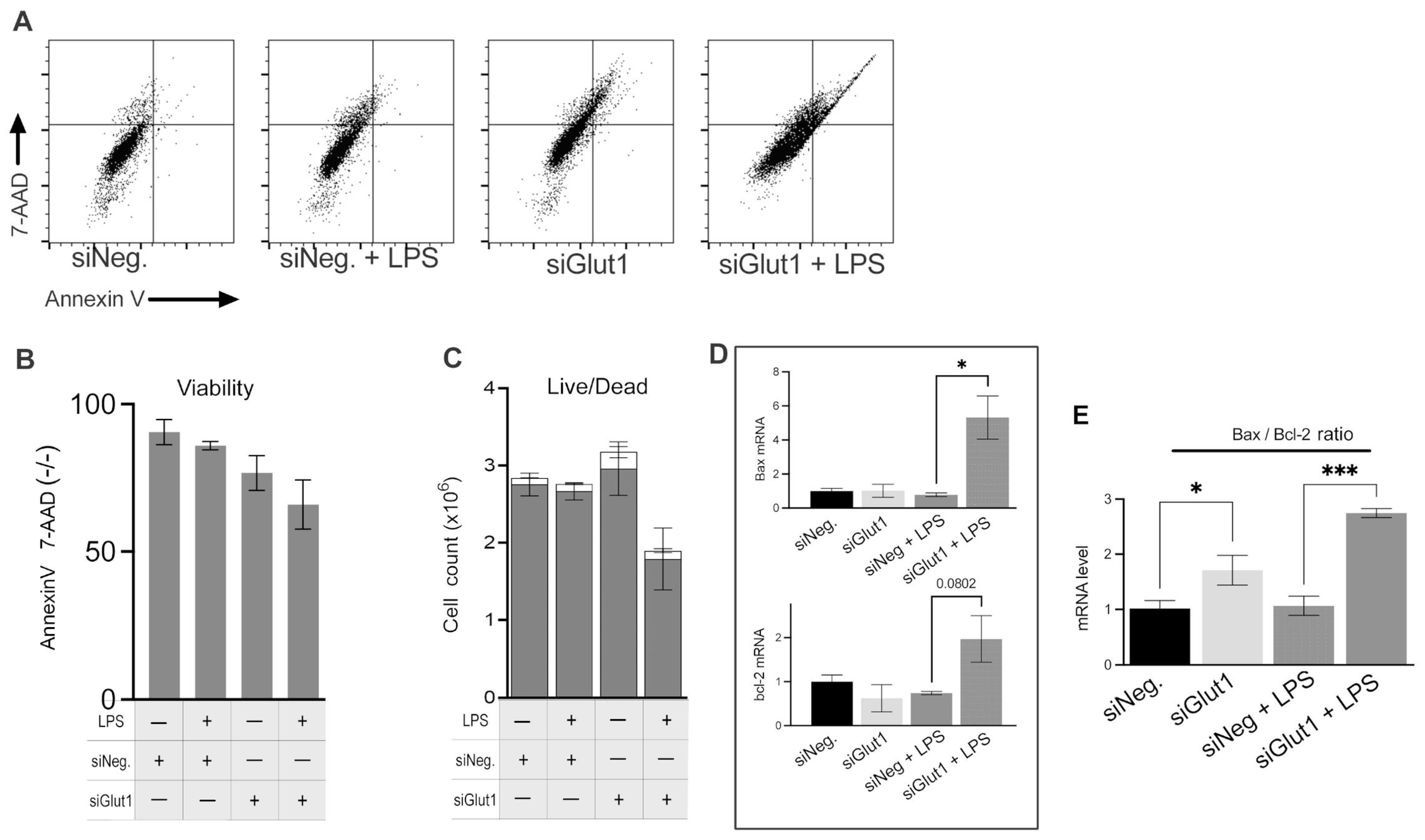
3.4. Silencing of Glut1 Triggers Apoptotic Signaling in Macrophage
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, N.; Liang, H.; Zen, K. Molecular Mechanisms That Influence the Macrophage M1-M2 Polarization Balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic Reprogramming of Macrophages: Glucose Transporter 1 (GLUT1)-Mediated Glucose Metabolism Drives a Proinflammatory Phenotype. J. Biol. Chem. 2014, 289, 7884–7896. [Google Scholar] [CrossRef]
- Jakubzick, C.V.; Randolph, G.J.; Henson, P.M. Monocyte Differentiation and Antigen-Presenting Functions. Nat. Rev. Immunol. 2017, 17, 349–362. [Google Scholar] [CrossRef]
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2018, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.; Münz, C. Innate and Adaptive Immunity through Autophagy. Immunity 2007, 27, 11–21. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-Mcdermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate Is an Inflammatory Signal That Induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Obaid, M.; Udden, S.M.N.; Alluri, P.; Mandal, S.S. LncRNA HOTAIR Regulates Glucose Transporter Glut1 Expression and Glucose Uptake in Macrophages during Inflammation. Sci. Rep. 2021, 11, 232. [Google Scholar] [CrossRef] [PubMed]
- Badiei, A.; Gieseg, S.; Davies, S.; Othman, M.I.; Bhatia, M. LPS Up-Regulates Cystathionine γ -Lyase Gene Expression in Primary Human Macrophages via NF-ΚB/ERK Pathway. Inflamm. Allergy-Drug Targets 2015, 14, 99–104. [Google Scholar] [CrossRef]
- Badiei, A.; Muniraj, N.; Chambers, S.; Bhatia, M. Inhibition of Hydrogen Sulfide Production by Gene Silencing Attenuates Inflammatory Activity by Downregulation of NF-ΚB and MAP Kinase Activity in LPS-Activated RAW 264.7 Cells. Biomed. Res. Int. 2014, 2014, 848570. [Google Scholar] [CrossRef]
- Whiteman, M.; Li, L.; Rose, P.; Tan, C.H.; Parkinson, D.B.; Moore, P.K. The Effect of Hydrogen Sulfide Donors on Lipopolysaccharide-Induced Formation of Inflammatory Mediators in Macrophages. Antioxid. Redox. Signal. 2010, 12, 1147. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Luo, N.; Mu, D.; Jiang, P.; Liu, R.; Sun, H.; Xiong, S.; Liu, X.; Wang, L.; Chu, Y. Lipopolysaccharide Regulates Biosynthesis of Cystathionine γ-Lyase and Hydrogen Sulfide through Toll-like Receptor-4/P38 and Toll-like Receptor-4/NF-ΚB Pathways in Macrophages. Vitr. Cell. Dev. Biol.-Anim. 2013, 49, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of Protein Using Bicinchoninic Acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.C.; Nadeau, K.; Abbasi, M.; Lachance, C.; Nguyen, M.; Fenrich, J. The Ultimate QPCR Experiment: Producing Publication Quality, Reproducible Data the First Time. Trends. Biotechnol. 2019, 37, 761–774. [Google Scholar] [CrossRef]
- Rahman, I.; Kode, A.; Biswas, S.K. Assay for Quantitative Determination of Glutathione and Glutathione Disulfide Levels Using Enzymatic Recycling Method. Nat. Protoc. 2007, 1, 3159–3166. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Kobayashi, K. Macrophages in Inflammation. Curr. Drug Targets-Inflamm. Allergy 2005, 4, 281–286. [Google Scholar] [CrossRef]
- Freemerman, A.J.; Zhao, L.; Pingili, A.K.; Teng, B.; Cozzo, A.J.; Fuller, A.M.; Johnson, A.R.; Milner, J.J.; Lim, M.F.; Galanko, J.A.; et al. Myeloid Slc2a1-Deficient Murine Model Revealed Macrophage Activation and Metabolic Phenotype Are Fueled by GLUT1. J. Immunol. 2019, 202, 1265–1286. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Ben-Neriah, Y. Phosphorylation Meets Ubiquitination: The Control of NF-ΚB Activity. Annu. Rev. Immunol. 2003, 18, 621–663. [Google Scholar] [CrossRef]
- Cornwell, A.; Fedotova, S.; Cowan, S.; Badiei, A. Cystathionine γ-Lyase and Hydrogen Sulfide Modulates Glucose Transporter Glut1 Expression via NF-ΚB and PI3k/Akt in Macrophages during Inflammation. PLoS ONE 2022, 17, e0278910. [Google Scholar] [CrossRef]
- Lee, Z.W.; Low, Y.L.; Huang, S.; Wang, T.; Deng, L.W. The Cystathionine γ-Lyase/Hydrogen Sulfide System Maintains Cellular Glutathione Status. Biochem. J. 2014, 460, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Regulators of the Transsulfuration Pathway. Br. J. Pharmacol. 2019, 176, 583. [Google Scholar] [CrossRef]
- McBean, G.J.; Aslan, M.; Griffiths, H.R.; Torrão, R.C. Thiol Redox Homeostasis in Neurodegenerative Disease. Redox. Biol. 2015, 5, 186–194. [Google Scholar] [CrossRef]
- Paul, B.D.; Sbodio, J.I.; Snyder, S.H. Cysteine Metabolism in Neuronal Redox Homeostasis. Trends Pharmacol. Sci. 2018, 39, 513–524. [Google Scholar] [CrossRef]
- Badiei, A.; Chambers, S.T.; Gaddam, R.R.; Bhatia, M. Cystathionine-γ-Lyase Gene Silencing with SiRNA in Monocytes/Macrophages Attenuates Inflammation in Cecal Ligation and Puncture-Induced Sepsis in the Mouse. J. Biosci. 2016, 41, 87–95. [Google Scholar] [CrossRef]
- Gaddam, R.R.; Fraser, R.; Badiei, A.; Chambers, S.; Cogger, V.C.; Le Couteur, D.G.; Ishii, I.; Bhatia, M. Cystathionine-Gamma-Lyase Gene Deletion Protects Mice against Inflammation and Liver Sieve Injury Following Polymicrobial Sepsis. PLoS ONE 2016, 11, e0160521. [Google Scholar] [CrossRef]
- Muniraj, N.; Stamp, L.K.; Badiei, A.; Hegde, A.; Cameron, V.; Bhatia, M. Hydrogen Sulfide Acts as a Pro-Inflammatory Mediator in Rheumatic Disease. Int. J. Rheum. Dis. 2017, 20, 182–189. [Google Scholar] [CrossRef]
- Lohninger, L.; Tomasova, L.; Praschberger, M.; Hintersteininger, M.; Erker, T.; Gmeiner, B.M.K.; Laggner, H. Hydrogen Sulphide Induces HIF-1α and Nrf2 in THP-1 Macrophages. Biochimie 2015, 112, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Libiad, M.; Vitvitsky, V.; Bostelaar, T.; Bak, D.W.; Lee, H.J.; Sakamoto, N.; Fearon, E.; Lyssiotis, C.A.; Weerapana, E.; Banerjee, R. Hydrogen Sulfide Perturbs Mitochondrial Bioenergetics and Triggers Metabolic Reprogramming in Colon Cells. J. Biol. Chem. 2019, 294, 12077–12090. [Google Scholar] [CrossRef]
- Vitvitsky, V.; Kumar, R.; Libiad, M.; Maebius, A.; Landry, A.P.; Banerjee, R. The Mitochondrial NADH Pool Is Involved in Hydrogen Sulfide Signaling and Stimulation of Aerobic Glycolysis. J. Biol. Chem. 2021, 296, 100736. [Google Scholar] [CrossRef]
- Cornwell, A.; Badiei, A. From Gasotransmitter to Immunomodulator: The Emerging Role of Hydrogen Sulfide in Macrophage Biology. Antioxidants 2023, 12, 935. [Google Scholar] [CrossRef] [PubMed]
- Calvert, J.W.; Jha, S.; Gundewar, S.; Elrod, J.W.; Ramachandran, A.; Pattillo, C.B.; Kevil, C.G.; Lefer, D.J. Hydrogen Sulfide Mediates Cardioprotection through Nrf2 Signaling. Circ. Res. 2009, 105, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Sica, A.; Solinas, G.; Porta, C.; Mantovani, A. The Inflammatory Micro-Environment in Tumor Progression: The Role of Tumor-Associated Macrophages. Crit. Rev. Oncol. Hematol. 2008, 66, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Chen, J.; Ren, G.; Zhang, Y.; Tan, X.; Yang, L. Punicalagin Prevents Inflammation in LPS- Induced RAW264.7 Macrophages by Inhibiting FoxO3a/Autophagy Signaling Pathway. Nutrients 2019, 11, 2794. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.; Pypaert, M.; Münz, C. Antigen-Loading Compartments for Major Histocompatibility Complex Class II Molecules Continuously Receive Input from Autophagosomes. Immunity 2007, 26, 79–92. [Google Scholar] [CrossRef]
- Ying, Y.; Sun, C.-B.; Zhang, S.-Q.; Chen, B.J.; Yu, J.Z.; Liu, F.Y.; Wen, J.; Hou, J.; Han, S.S.; Yan, J.Y.; et al. Induction of Autophagy via the TLR4/NF-ΚB Signaling Pathway by Astragaloside IV Contributes to the Amelioration of Inflammation in RAW264.7 Cells. Biomed. Pharmacother. 2021, 137, 111271. [Google Scholar] [CrossRef]
- Xue, Z.; Zhang, Z.; Liu, H.; Li, W.; Guo, X.; Zhang, Z.; Liu, Y.; Jia, L.; Li, Y.; Ren, Y.; et al. LincRNA-Cox2 Regulates NLRP3 Inflammasome and Autophagy Mediated Neuroinflammation. Cell Death Differ. 2019, 26, 130–145. [Google Scholar] [CrossRef] [PubMed]
- Raisova, M.; Hossini, A.M.; Eberle, J.; Riebeling, C.; Wieder, T.; Sturm, I.; Daniel, P.T.; Orfanos, C.E.; Geilen, C.C. The Bax/Bcl-2 Ratio Determines the Susceptibility of Human Melanoma Cells to CD95/Fas-Mediated Apoptosis. J. Investig. Dermatol. 2001, 117, 333–340. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward (5′–3′) | Reverse (5′–3′) |
|---|---|---|
| Glut1 | GATCTGAGCTACGGGGTCTT | TGTAGAACTCCTCAATAACCTTCTG |
| IL-6 | CGGCCTTCCCTACTTCACAA | TTGCCATTGCACAACTCTTTTC |
| TNFα | CCCACGTCGTAGCAAACCAC | TAGCAAATCGGCTGACGGTG |
| IL-10 | GGTTGCCAAGCCTTATCGGA | CACCTTGGTCTTGGAGCTTATT |
| iNOS2 | GGTGAAGGGACTGAGCTGTT | ACGTTCTCCGTTCTCTTGCAG |
| IkBα | ACAACAGAGATGAGGGCGATG | CTCTGGAGGGTCGGGACTTA |
| CSE | CAAAGCAACACCTCGCACTC | ATGCAAAGGCCAAACTGTGC |
| Bcl-2 | CTGAGTACCTGAACCGGCAT | AGTTCCACAAAGGCATCCCAG |
| Bax | TGGAGCTGCAGAGGATGATT | TCTTGGATCCAGACAAGCAGC |
| GAPDH | CGTCCCGTAGACAAAATGGT | GAGGTCAATGAAGGGGTC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cornwell, A.; Ziółkowski, H.; Badiei, A. Glucose Transporter Glut1-Dependent Metabolic Reprogramming Regulates Lipopolysaccharide-Induced Inflammation in RAW264.7 Macrophages. Biomolecules 2023, 13, 770. https://doi.org/10.3390/biom13050770
Cornwell A, Ziółkowski H, Badiei A. Glucose Transporter Glut1-Dependent Metabolic Reprogramming Regulates Lipopolysaccharide-Induced Inflammation in RAW264.7 Macrophages. Biomolecules. 2023; 13(5):770. https://doi.org/10.3390/biom13050770
Chicago/Turabian StyleCornwell, Alex, Hubert Ziółkowski, and Alireza Badiei. 2023. "Glucose Transporter Glut1-Dependent Metabolic Reprogramming Regulates Lipopolysaccharide-Induced Inflammation in RAW264.7 Macrophages" Biomolecules 13, no. 5: 770. https://doi.org/10.3390/biom13050770
APA StyleCornwell, A., Ziółkowski, H., & Badiei, A. (2023). Glucose Transporter Glut1-Dependent Metabolic Reprogramming Regulates Lipopolysaccharide-Induced Inflammation in RAW264.7 Macrophages. Biomolecules, 13(5), 770. https://doi.org/10.3390/biom13050770