

β-Glucan and Fatty Acid Based Mucoadhesive Carrier for Gastrointestinal Tract Specific Local and Sustained Drug Delivery

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation and Characterization of GADA
2.3. Size and Morphology
2.4. Surface Plasmon Resonance (SPR)
2.5. Drug Loading and In Vitro Release Testing
2.6. In Vivo and Ex Vivo Imaging
2.7. Statistics
3. Results
3.1. Synthesis and Characterization
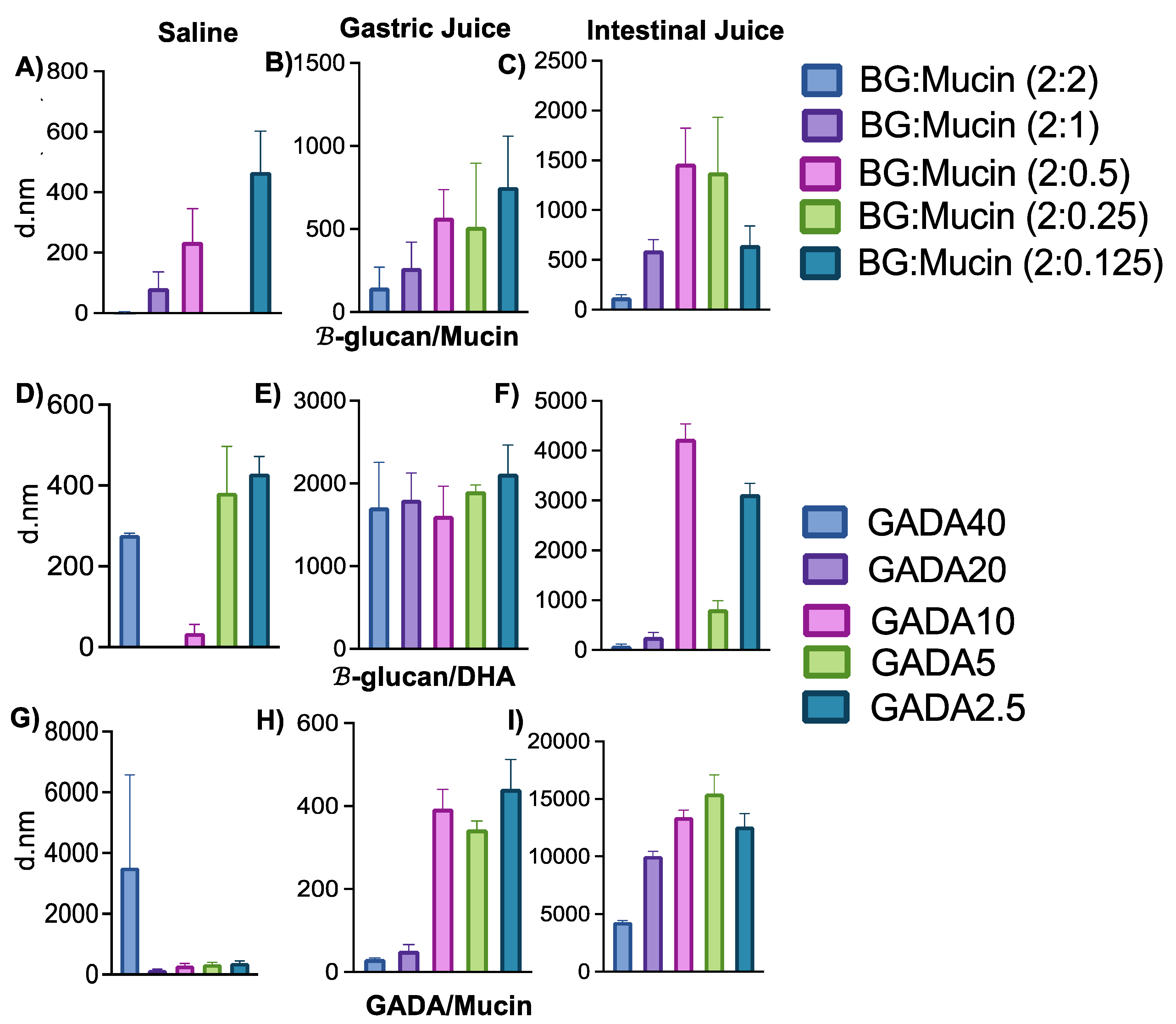
3.2. Interaction between GADA and Mucin
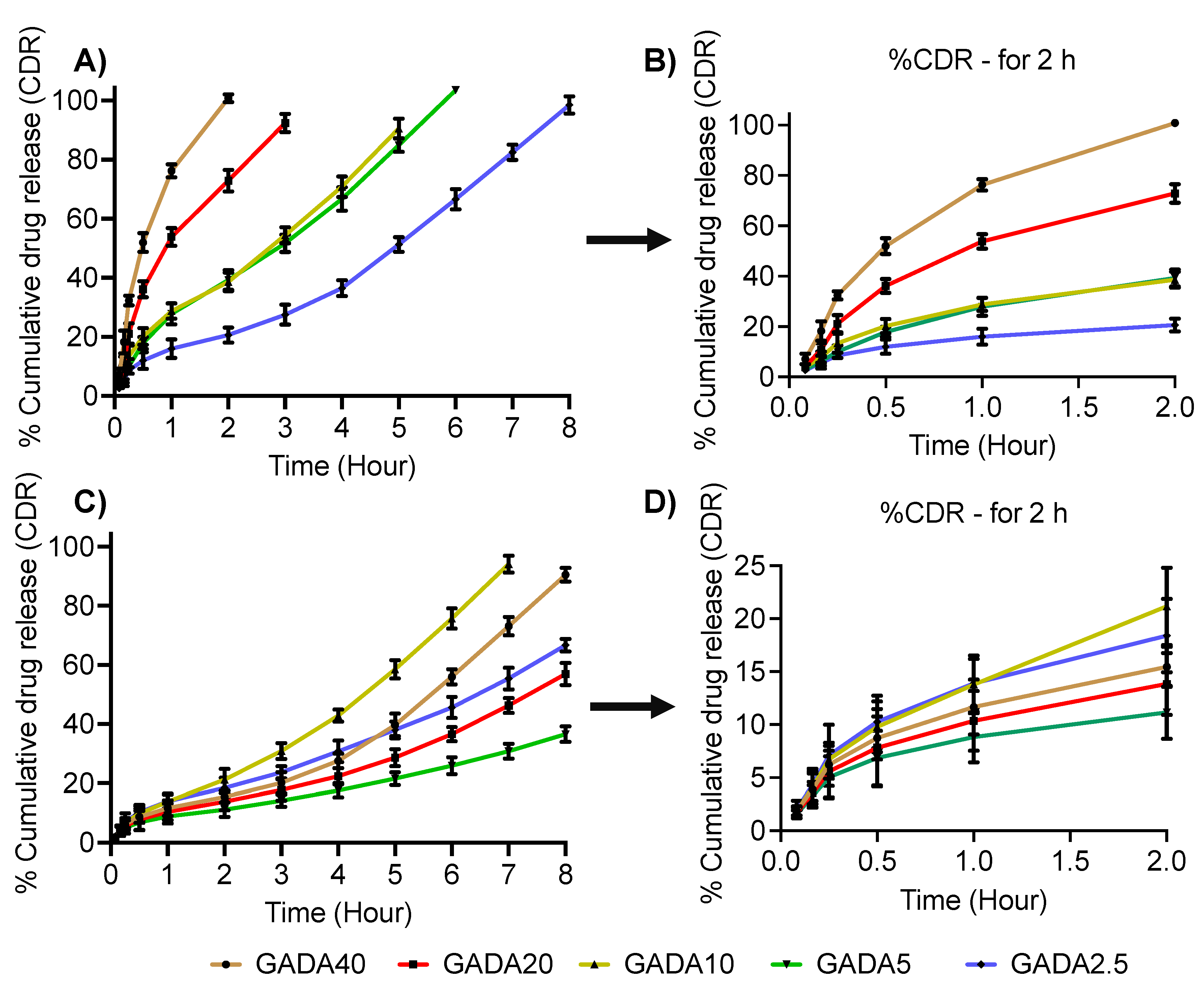
3.3. Drug Loading Efficiency and Release Profile
3.4. In Vivo Imaging and Biodistribution
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saeed, A.F.U.H.; Wang, R.; Ling, S.; Wang, S. Antibody Engineering for Pursuing a Healthier Future. Front. Microbiol. 2017, 8, 495. [Google Scholar] [CrossRef] [PubMed]
- Sasso, J.M.; Ambrose, B.J.B.; Tenchov, R.; Datta, R.S.; Basel, M.T.; Delong, R.K.; Zhou, Q.A. The Progress and Promise of RNA Medicine-An Arsenal of Targeted Treatments. J. Med. Chem. 2022, 65, 6975–7015. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic Peptides: Current Applications and Future Directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Du, A.W.; Stenzel, M.H. Drug Carriers for the Delivery of Therapeutic Peptides. Biomacromolecules 2014, 15, 1097–1114. [Google Scholar] [CrossRef]
- Lou, J.; Duan, H.; Qin, Q.; Teng, Z.; Gan, F.; Zhou, X.; Zhou, X. Advances in Oral Drug Delivery Systems: Challenges and Opportunities. Pharmaceutics 2023, 15, 484. [Google Scholar] [CrossRef]
- Homayun, B.; Lin, X.; Choi, H.J. Challenges and Recent Progress in Oral Drug Delivery Systems for Biopharmaceuticals. Pharmaceutics 2019, 11, 129. [Google Scholar] [CrossRef]
- Hua, S. Advances in Oral Drug Delivery for Regional Targeting in the Gastrointestinal Tract—Influence of Physiological, Pathophysiological and Pharmaceutical Factors. Front. Pharmacol. 2020, 11, 524. [Google Scholar] [CrossRef]
- Kaffash, E.; Shahbazi, M.A.; Hatami, H.; Nokhodchi, A. An Insight into Gastrointestinal Macromolecule Delivery Using Physical Oral Devices. Drug Discovery Today 2022, 27, 2309–2321. [Google Scholar] [CrossRef]
- Patel, V.F.; Liu, F.; Brown, M.B. Advances in Oral Transmucosal Drug Delivery. J. Control. Release 2011, 153, 106–116. [Google Scholar] [CrossRef]
- Mitragotri, S.; Burke, P.A.; Langer, R. Overcoming the Challenges in Administering Biopharmaceuticals: Formulation and Delivery Strategies. Nat. Rev. Drug Discov. 2014, 13, 655–672. [Google Scholar] [CrossRef]
- Vargason, A.M.; Anselmo, A.C.; Mitragotri, S. The Evolution of Commercial Drug Delivery Technologies. Nat. Biomed. Eng. 2021, 5, 951–967. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, D.; Lennernäs, H. Intestinal Permeability and Drug Absorption: Predictive Experimental, Computational and in Vivo Approaches. Pharmaceutics 2019, 11, 411. [Google Scholar] [CrossRef] [PubMed]
- Moroz, E.; Matoori, S.; Leroux, J.C. Oral Delivery of Macromolecular Drugs: Where We Are after Almost 100 Years of Attempts. Adv. Drug Deliv. Rev. 2016, 101, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, M.S.; Kazi, M.; Alsenaidy, M.A.; Ahmad, M.Z. Advances in Oral Drug Delivery. Front. Pharmacol. 2021, 12, 618411. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, D.A.; Langer, R.; Traverso, G. Mucus Interaction to Improve Gastrointestinal Retention and Pharmacokinetics of Orally Administered Nano-Drug Delivery Systems. J. Nanobiotechnology 2022, 20, 1–23. [Google Scholar] [CrossRef]
- Diwan, R.; Khan, S.; Ravi, P.R. Comparative Study of Cilnidipine Loaded PLGA Nanoparticles: Process Optimization by DoE, Physico-Chemical Characterization and in Vivo Evaluation. Drug Deliv. Transl. Res. 2020, 10, 1442–1458. [Google Scholar] [CrossRef]
- Diwan, R.; Ravi, P.R.; Pathare, N.S.; Aggarwal, V. Pharmacodynamic, Pharmacokinetic and Physical Characterization of Cilnidipine Loaded Solid Lipid Nanoparticles for Oral Delivery Optimized Using the Principles of Design of Experiments. Colloids Surf. B Biointerfaces 2020, 193, 111073. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Bowman, K.; Leong, K.W. Chitosan Nanoparticles for Oral Drug and Gene Delivery. Int. J. Nanomed. 2006, 1, 117–128. [Google Scholar] [CrossRef]
- Wang, J.; Chin, D.; Poon, C.; Mancino, V.; Pham, J.; Li, H.; Ho, P.Y.; Hallows, K.R.; Chung, E.J. Oral Delivery of Metformin by Chitosan Nanoparticles for Polycystic Kidney Disease. J. Control. Release 2021, 329, 1198–1209. [Google Scholar] [CrossRef]
- Sung, H.W.; Sonaje, K.; Liao, Z.X.; Hsu, L.W.; Chuang, E.Y. PH-Responsive Nanoparticles Shelled with Chitosan for Oral Delivery of Insulin: From Mechanism to Therapeutic Applications. Acc. Chem. Res. 2012, 45, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Ibsen, K.; Brown, T.; Chen, R.; Agatemor, C.; Mitragotri, S. Ionic Liquids for Oral Insulin Delivery. Proc. Natl. Acad. Sci. USA 2018, 115, 7296–7301. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.D.; Sahbaz, Y.; Ford, L.; Nguyen, T.H.; Scammells, P.J.; Porter, C.J.H. Ionic Liquids Provide Unique Opportunities for Oral Drug Delivery: Structure Optimization and in Vivo Evidence of Utility. Chem. Commun. 2014, 50, 1688–1690. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Sun, Y.; Lu, A.; Wang, X.; Shi, Y. Ionic Liquids: Promising Approach for Oral Drug Delivery. Pharm. Res. 2022, 39, 2353–2365. [Google Scholar] [CrossRef] [PubMed]
- Nyamweya, N.N. Applications of Polymer Blends in Drug Delivery. Future J. Pharm. Sci. 2021, 7, 1–15. [Google Scholar] [CrossRef]
- Jana, P.; Shyam, M.; Singh, S.; Jayaprakash, V.; Dev, A. Biodegradable Polymers in Drug Delivery and Oral Vaccination. Eur. Polym. J. 2021, 142, 110155. [Google Scholar] [CrossRef]
- Ting, J.M.; Porter, W.W.; Mecca, J.M.; Bates, F.S.; Reineke, T.M. Advances in Polymer Design for Enhancing Oral Drug Solubility and Delivery. Bioconjugate Chem. 2018, 29, 939–952. [Google Scholar] [CrossRef]
- Khatun, Z.; Nurunnabi, M.; Reeck, G.R.; Cho, K.J.; Lee, Y.K. Oral Delivery of Taurocholic Acid Linked Heparin–Docetaxel Conjugates for Cancer Therapy. J. Control. Release 2013, 170, 74–82. [Google Scholar] [CrossRef]
- Khatun, Z.; Nurunnabi, M.; Cho, K.J.; Byun, Y.; Bae, Y.H.; Lee, Y.K. Oral Absorption Mechanism and Anti-Angiogenesis Effect of Taurocholic Acid-Linked Heparin-Docetaxel Conjugates. J. Control. Release 2014, 177, 64–73. [Google Scholar] [CrossRef]
- Altreuter, D.H.; Kirtane, A.R.; Grant, T.; Kruger, C.; Traverso, G.; Bellinger, A.M. Changing the Pill: Developments toward the Promise of an Ultra-Long-Acting Gastroretentive Dosage Form. Expert Opin. Drug Deliv. 2018, 15, 1189–1198. [Google Scholar] [CrossRef]
- Singh, B.N.; Kim, K.H. Floating Drug Delivery Systems: An Approach to Oral Controlled Drug Delivery via Gastric Retention. J. Control. Release 2000, 63, 235–259. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Islam, T.; Nurunnabi, M. Mucoadhesive Carriers for Oral Drug Delivery. J. Control. Release 2022, 351, 504–559. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.S.; Geetha Bai, R.; Islam, T.; Abir, M.; Narayan, M.; Khatun, Z.; Nurunnabi, M. Bile Acid Linked β-Glucan Nanoparticles for Liver Specific Oral Delivery of Biologics. Biomater. Sci. 2022, 10, 2929–2939. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.J.; Rezoagli, E.; Major, I.; Rowan, N.J.; Laffey, J.G. β-Glucan Metabolic and Immunomodulatory Properties and Potential for Clinical Application. J. Fungi 2020, 6, 356. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Seviour, R. Medicinal Importance of Fungal β-(1→3), (1→6)-Glucans. Mycol. Res. 2007, 111, 635–652. [Google Scholar] [CrossRef]
- Sima, P.; Vannucci, L.; Vetvicka, V. β-Glucans and Cholesterol (Review). Int. J. Mol. Med. 2018, 41, 1799–1808. [Google Scholar] [CrossRef]
- Lee, D.Y.; Nurunnabi, M.; Kang, S.H.; Nafiujjaman, M.; Huh, K.M.; Lee, Y.K.; Kim, Y.C. Oral Gavage Delivery of PR8 Antigen with β-Glucan-Conjugated GRGDS Carrier to Enhance M-Cell Targeting Ability and Induce Immunity. Biomacromolecules 2017, 18, 1172–1179. [Google Scholar] [CrossRef]
- Islam, T.; Huda, M.N.; Ahsan, M.A.; Afrin, H.; Joseph J Salazar, C.; Nurunnabi, M. Theoretical and Experimental Insights into the Possible Interfacial Interactions between β-Glucan and Fat Molecules in Aqueous Media. J. Phys. Chem. B 2021, 125, 13730–13743. [Google Scholar] [CrossRef]
- Guo, P.; Si, M.; Wu, D.; Xue, H.Y.; Hu, W.; Wong, H.L. Incorporation of Docosahexaenoic Acid (DHA) Enhances Nanodelivery of Antiretroviral across the Blood-Brain Barrier for Treatment of HIV Reservoir in Brain. J. Control. Release 2020, 328, 696–709. [Google Scholar] [CrossRef]
- Chen, J.; Raymond, K.; Curtin, J. Beta-Glucans in the Treatment of Diabetes and Associated Cardiovascular Risks. Vasc. Health Risk Manag. 2008, 4, 1265–1272. [Google Scholar] [CrossRef]
- Caseiro, C.; Dias, J.N.R.; de Andrade Fontes, C.M.G.; Bule, P. From Cancer Therapy to Winemaking: The Molecular Structure and Applications of β-Glucans and β-1, 3-Glucanases. Int. J. Mol. Sci. 2022, 23, 3156. [Google Scholar] [CrossRef] [PubMed]
- Vetvicka, V.; Vannucci, L.; Sima, P.; Richter, J. Beta Glucan: Supplement or Drug? From Laboratory to Clinical Trials. Molecules 2019, 24, 1251. [Google Scholar] [CrossRef]
- Skulas-Ray, A.C.; Wilson, P.W.F.; Harris, W.S.; Brinton, E.A.; Kris-Etherton, P.M.; Richter, C.K.; Jacobson, T.A.; Engler, M.B.; Miller, M.; Robinson, J.G.; et al. Omega-3 Fatty Acids for the Management of Hypertriglyceridemia: A Science Advisory from the American Heart Association. Circulation 2019, 140, e673–e691. [Google Scholar] [CrossRef] [PubMed]
- Horrocks, L.A.; Yeo, Y.K. Health Benefits of Docosahexaenoic Acid (DHA). Pharmacol. Res. 1999, 40, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.U.; Lone, A.N.; Khan, M.S.; Virani, S.S.; Blumenthal, R.S.; Nasir, K.; Miller, M.; Michos, E.D.; Ballantyne, C.M.; Boden, W.E.; et al. Effect of Omega-3 Fatty Acids on Cardiovascular Outcomes: A Systematic Review and Meta-Analysis. EClinicalMedicine 2021, 38, 100997. [Google Scholar] [CrossRef]
- West, L.; Yin, Y.; Pierce, S.R.; Fang, Z.; Fan, Y.; Sun, W.; Tucker, K.; Staley, A.; Zhou, C.; Bae-Jump, V. Original Article Docosahexaenoic Acid (DHA), an Omega-3 Fatty Acid, Inhibits Tumor Growth and Metastatic Potential of Ovarian Cancer. Am. J. Cancer Res. 2020, 10, 1–16. Available online: www.ajcr.us/ (accessed on 6 April 2023).
- Afrin, H.; Huda, M.N.; Islam, T.; Oropeza, B.P.; Alvidrez, E.; Abir, M.I.; Boland, T.; Turbay, D.; Nurunnabi, M. Detection of Anticancer Drug-Induced Cardiotoxicity Using VCAM1-Targeted Nanoprobes. ACS Appl. Mater. Interfaces 2022, 14, 37566–37576. [Google Scholar] [CrossRef]
- Vladár, A.E.; Hodoroaba, V.D. Characterization of Nanoparticles by Scanning Electron Microscopy. In Characterization of Nanoparticles: Measurement Processes for Nanoparticles; Elsevier: Amsterdam, The Netherlands, 2020; pp. 7–27. [Google Scholar] [CrossRef]
- Nurunnabi, M.; Ibsen, K.N.; Tanner, E.E.L.; Mitragotri, S. Oral Ionic Liquid for the Treatment of Diet-Induced Obesity. Proc. Natl. Acad. Sci. USA 2019, 116, 25042–25047. [Google Scholar] [CrossRef]
- Boegh, M.; Nielsen, H.M. Mucus as a Barrier to Drug Delivery—Understanding and Mimicking the Barrier Properties. Basic Clin. Pharmacol. Toxicol. 2015, 116, 179–186. [Google Scholar] [CrossRef]
- Herath, M.; Hosie, S.; Bornstein, J.C.; Franks, A.E.; Hill-Yardin, E.L. The Role of the Gastrointestinal Mucus System in Intestinal Homeostasis: Implications for Neurological Disorders. Front. Cell. Infect. Microbiol. 2020, 10, 248. [Google Scholar] [CrossRef]
- Bandi, S.P.; Bhatnagar, S.; Venuganti, V.V.K. Advanced Materials for Drug Delivery across Mucosal Barriers. Acta Biomater. 2021, 119, 13–29. [Google Scholar] [CrossRef]
- Li, Z.; Chen, G.Y. Current Conjugation Methods for Immunosensors. Nanomaterials 2018, 8, 278. [Google Scholar] [CrossRef] [PubMed]
- Júnior, E.R.; Lopes, M.V.; Bentley, B.; Marchetti, J.M.; Marchetti, J.M. HPLC Assay of Lidocaine in in Vitro Dissolution Test of the Poloxamer 407 Gels. Rev. Bras. Ciências Farm. 2002, 38, 107–111. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esquivel, S.V.; Bhatt, H.N.; Diwan, R.; Habib, A.; Lee, W.-Y.; Khatun, Z.; Nurunnabi, M. β-Glucan and Fatty Acid Based Mucoadhesive Carrier for Gastrointestinal Tract Specific Local and Sustained Drug Delivery. Biomolecules 2023, 13, 768. https://doi.org/10.3390/biom13050768
Esquivel SV, Bhatt HN, Diwan R, Habib A, Lee W-Y, Khatun Z, Nurunnabi M. β-Glucan and Fatty Acid Based Mucoadhesive Carrier for Gastrointestinal Tract Specific Local and Sustained Drug Delivery. Biomolecules. 2023; 13(5):768. https://doi.org/10.3390/biom13050768
Chicago/Turabian StyleEsquivel, Stephanie Vargas, Himanshu N. Bhatt, Rimpy Diwan, Ahsan Habib, Wen-Yee Lee, Zehedina Khatun, and Md Nurunnabi. 2023. "β-Glucan and Fatty Acid Based Mucoadhesive Carrier for Gastrointestinal Tract Specific Local and Sustained Drug Delivery" Biomolecules 13, no. 5: 768. https://doi.org/10.3390/biom13050768
APA StyleEsquivel, S. V., Bhatt, H. N., Diwan, R., Habib, A., Lee, W.-Y., Khatun, Z., & Nurunnabi, M. (2023). β-Glucan and Fatty Acid Based Mucoadhesive Carrier for Gastrointestinal Tract Specific Local and Sustained Drug Delivery. Biomolecules, 13(5), 768. https://doi.org/10.3390/biom13050768