

The Implications of Microglial Regulation in Neuroplasticity-Dependent Stroke Recovery

Abstract
1. Introduction
2. Pathophysiology and Therapeutic Target of Stroke Recovery
2.1. Pathophysiology of Stroke in Different Phases
2.2. The Basis of Functional Recovery: Neuroplasticity
2.2.1. Remodeling of Dendrites and Dendritic Spines
2.2.2. Axonal Sprouting
2.2.3. Myelin Regeneration
2.2.4. Neurogenesis
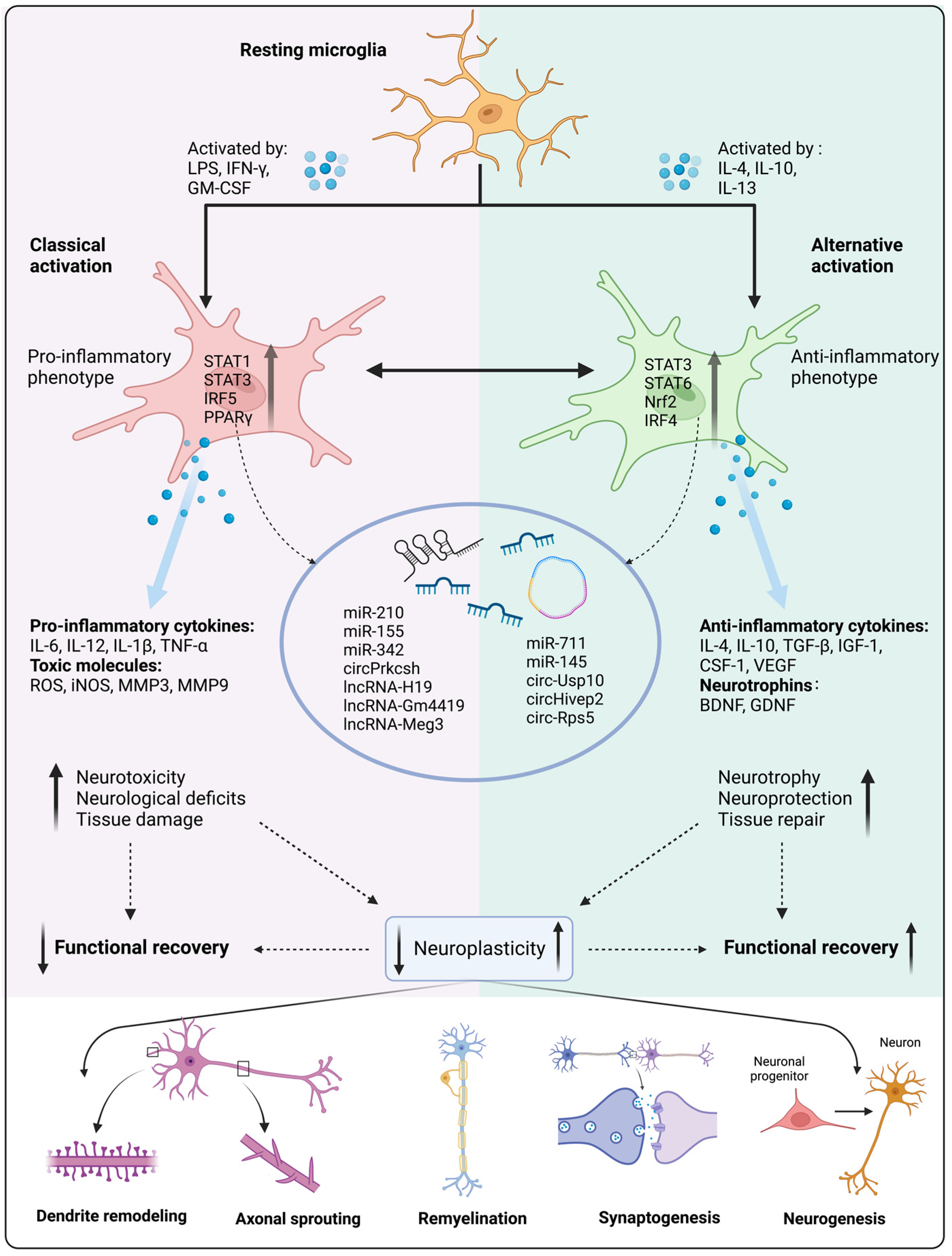
3. Activation and Polarization of Microglia following Stroke
3.1. Microglia in CNS
3.2. Spatiotemporal Distribution of Microglia and Colocalization between Microglia Activation and Neuroplasticity after Stroke
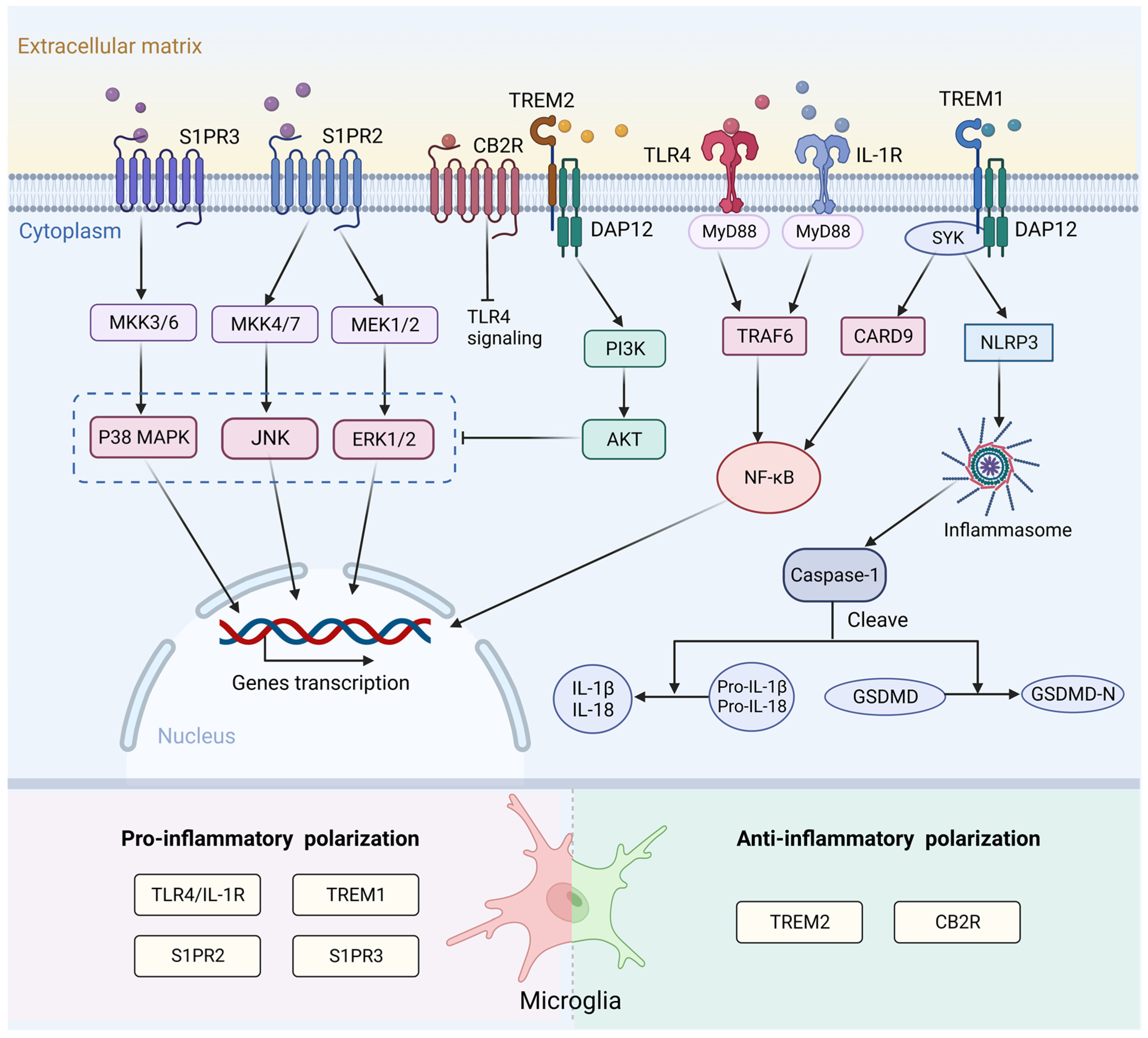
3.3. Molecular Mechanism of Microglial Activation and Polarization Following Stroke
3.3.1. Surface Receptor
3.3.2. Transcription Factor
3.3.3. Non-Coding RNA
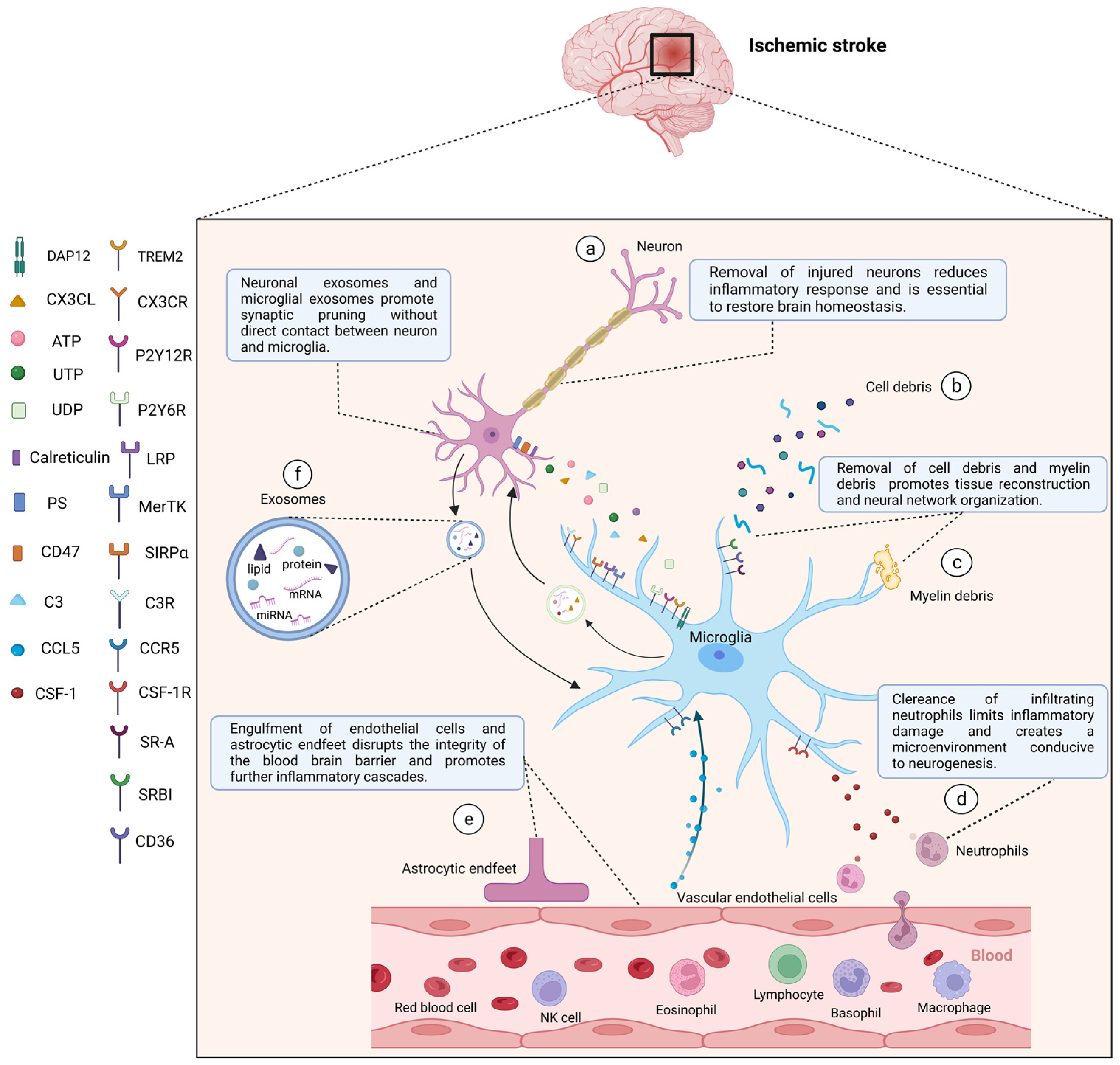
4. Microglia Phagocytic Function following Stroke
4.1. Microglia Phagocytotic Function during Physiological State
4.2. Functional Changes of Microglia Phagocytosis after Stroke
4.3. Phagocytic Signaling
4.3.1. Find Me Signal
4.3.2. Eat Me Signal
4.3.3. Do Not Eat Me Signal
5. Therapeutic Intervention Targeting Microglia in Stroke Rehabilitation
5.1. Pharmacotherapy

{kind=link}
{kind=link}
{kind=link}
| Drug | In Vivo | In Vitro | Effect on Microglia | Signal | Reference | ||
|---|---|---|---|---|---|---|---|
| Animal Model | Treatment | Cell Culture | Treatment | ||||
| Minocycline | Mouse (tMCAO) | Intraperitoneal injections after MCAO induction; 10, 25, and 50 mg/kg/day for 3 consecutive days | BV2 microglial cells oxygen–glucose deprivation/reoxygenation (OGD/R) cell model | Minocycline at doses of 0.01, 0.1, 1, 10, and 100 μM; preincubated 1 h before OGD/R injury | IL-1β↓, IL-18↓, NLRP3↓ | [239] | |
| Rats (tMCAO) | Intravenous injection after reperfusion onset; a single dose (3 mg/kg) | IL-1β↓, TNF-α↓, IL-10↑, TGF-β↑, Ym1↑ | [240] | ||||
| Mouse (MCAO/R) | Intraperitoneal injections after reperfusion; 10, 25, or 50 mg/kg/day for 2 weeks | Primary microglia from cerebral cortex of newborn mice | LPS (100 ng/mL) + IFN-γ (20 ng/mL) + minocycline (50 μM); incubation for 24 h | IL-1β↓, IL-6↓, iNOS↓, TNF-α↓, Arg-1↑, IL-10↑, TGF-β↑, Ym1↑ | STAT1/STAT6 pathways ↓ | [241] | |
| Wnt-3a | Mouse (tMCAO) | Intranasally delivered at the time of reperfusion and next 2 days; 2 μg/kg/day | iNOS↓, TNF-α↓, Arg-1↑, CD206 ↑ | [242] | |||
| Gal-3 | Mouse (MCAO) | Intracortical injection at 24 h following MCAO; 100 ng/mouse (20–25 g) | Primary cell cultures from the brains of the adult, 8–9-week-old C57BL/6 wild-type mice | Incubation with Gal-3 (5 μM) for 24 h | TNF-α↓, IL-1β↓, IFN-γ↓, IL-17↓, iNOS↓, Ym1↑, IL-4 ↑, IL-6 ↑ | [243] | |
| Atorvastatin | Mouse (pMCAO) | Oral gavage after MCAO induction; 20 mg/kg/day | IL-6↓, TNF-α↓, MCP-1↓, IL-10 ↑ | [244] | |||
| Exendin-4 | Mouse (MCAO) | Intraperitoneal injection; 50 mg/kg at 1.5 h after MCAO induction; 0.2 mg/kg daily for 3 days until sacrifice | Primary microglia-enriched cultures were prepared from whole brains of 2- to 3- day-old mice | LPS (10 ng/mL) + Ex-4 (40 ng/mL); incubation for 24 h | CD206↑, Arg-1↑, Ym1/2↑ | [245] | |
| Bendavia | Mouse (tMCAO) | Intraperitoneal injection immediately after reperfusion and 4 h later; 5 mg/kg | MMP9↓, TNF-α ↓ | [246] | |||
| Vx-765 | Mouse (MCAO) | intraperitoneal injection starting immediately after MCAO induction; 50 mg/kg for 3 consecutive days | IL-1β↓, TNF-α↓, iNOS↓, TGF-β↑, Ym1↑ | NF-κB signaling↓ | [247] | ||
| Baicalein | Mouse (MCAO) | Intragastrical administration after reperfusion; 100 mg/kg/day for 3 days | BV2 microglial cells |
| Ym1/2↑, Arg-1↑, CD206↑, TNF-α↓, IL-1β↓, IL-6↓, NO↓ | TLR4/NF-κB ↓, phosphorylated STAT1 ↓ | [248] |
| Cottonseed oil | Rats (MCAO/R) | Subcutaneous injection before MCAO; 1.3 mL/kg/day for 3 weeks | IL-1β↓, IL-6↓, TNF-α↓ | TLR4/NF-κB ↓ | [249] | ||
| GJ-4 | Rats (MCAO/R) | Oral administration after MCAO induction; 10, 25, 50 mg/kg/day for 12 days | iNOS↓, COX-2↓, MMP9↓ | JAK2/STAT1 ↓ | [251] | ||
| Melatonin | Mouse (dMCAO) | 20 mg/kg at 0 and 24 h after reperfusion | Co-culture of BV2 cells (growing on culture inserts) and OGD neuron | Melatonin (100, 200, or 400 mM); incubation for 12 h | pro-inflammatory markers ↓, anti-inflammatory markers ↑ | p-STAT3/STAT3↑ | [252] |
| Ki20227 | Mouse (PT) | Oral gavage before modeling; 0.002 mg/kg/day for 7 consecutive days | TNF-α↓, iNOS ↓, IL-10↑, Arg-1 ↑, NLRP3↓, Active caspase 1 ↓ | NF-κB signaling↓ | [253] | ||
| Curcumin | Mouse (tMCAO) | Intraperitoneal injection; 150 mg/kg at 0 h and 24 h after reperfusion | BV2 microglial cells | LPS (100 ng/mL) + IFN-γ (20 ng/mL) + curcumin (12.5 and 25 µmol/L); incubation for 48 h | TNF-α↓, IL-12p70↓, IL-6 ↓ | [255] | |
| Mouse (MCAO/R) | Intraperitoneal injection; 150 mg/kg/day for 7 days after ischemic stroke | Primary microglia were isolated from the whole brains of neonatal C57BL/6J mice | LPS (100 ng/mL) + curcumin (12.5 μM); incubation for 24 h | NLRP3/NF-κB pathway ↓ | [256] | ||
5.2. Exercise
| Type | Treatment (Intensity, Time, Frequency, Duration) | Model | Effect on Microglia | Outcome | Reference | |
|---|---|---|---|---|---|---|
| Exercise | Treadmill exercise | 12 m/min; 30 min/day; 3 or 6 consecutive days | Rats (MCAO) | IL-4↑ M1-like markers↓ M2-like markers↑ | Improving neurobehavioral outcomes | [270] |
| 5–6 m/min; 5 min/day; 3 consecutive days | Mouse (MCAO) | NLRP3↓ | Showing better improvements at functional levels | [268] | ||
| HIT program: 10 days (>25 m/min) MOD program: 2 days (<20 m/min) | Rats (MCAO) | IL-10↑, p75NTR↑, BDNF↑ | Promoting cerebral plasticity | [269] | ||
| 30 min/day; 5 days/week; 4 weeks | Mouse (MCAO) | Iba1+↑ (hippocampal CA1 region) | Alleviating increased neuroinflammation | [271] | ||
| 25 cm/s; 30 min/day; 3 days/week 4.5 weeks | Mouse (MCAO) | IL-10↑, NLRP3↑, IFN-γ↑, Gal-3↓ (caused by stress) | Having beneficial neuro-inflammatory effects; inducing detrimental stress response by forced running | [280] | ||
| 10 m/min; 60 min/day; 5 weeks | Mouse (microinjection of collagenase into the striatum region) | CD36/Iba1-double positive cells↑ | Contributing to neuroprotection | [278] | ||
| 12 m/min; 30 min/day; 5 times/week; 4 weeks | Rats (MCAO) (exosomes injection) | Excessive microglial activation↓, Syn↑, PSD-95↑ | Regulating synaptic plasticity and protecting neural function | [273] | ||
| Skilled reaching training of the impaired forelimb | 5 days/week; 14 or 42 days | Rats (PT) | Excessive microglial activation↓ | Modulating perilesional cellular plasticity and contributing to a better functional recovery | [276] | |
| 10 or 42 days | Rats (PT) | Excessive microglial activation↓ | Improving functional recovery | [277] | ||
| rTMS | Continuous TBS | 5 min (3 pulses of 50 Hz repeated every 200 ms); 5 days | Rats (PT) | Pro-inflammatory cytokines↓ | Improving the local neuronal microenvironment | [281] |
| 5 min (3 pulses of 50 Hz, repeated every 200 ms); 6 days | Rats (PT) | TGF-β↑, VEGF↑, HIF-1α↑ | Presenting protective effects in the context of ischemic stroke; contributing to vascular repair and protection | [282] | ||
| Intermittent TBS | Ten 50 Hz bursts with 3 pulses each repeated 20 times at 5 Hz intervals; twice per day; 7 continuous days | Mouse (MCAO) | TLR4/NF-κB/NLRP3 signaling pathway↓ | Alleviating locomotor deficits and neuronal pyroptosis | [283] | |
| High frequency | 10 Hz rTMS with a total of 60 trains; 20 pulses per train (1200 pulses); 10 s intertrain interval; for 11 min 44 s | Rats (MCAO) | NF-κB↓, STAT6↓ | Promoting neurogenesis and improving neural function recovery | [284] | |
| tDCS | Cathodal | 500 µA, 15 min; once per day; 10 days | Rats (MCAO) | Iba1+↓ Pro-inflammatory factors↓ Anti-inflammatory factor↑ | Accelerating recovery from neurologic deficit and brain damage | [285] |
| 250 µA; 40 min; 1 day | Mouse (PT) | CD206↑ CD68↓ | Being effective from a functional point of view | [286] | ||
| 250 µA; 40 min | Mouse (MCAO) | Iba1+↓ GABA and glutamate↓ | Exerting a measurable neuroprotective effect | [287] | ||
| Anodal | 250 µA; 15 min; 10 days | Mouse (MCAO) | Iba1+↓ | Inducing regeneration and promoting functional recovery | [288] | |
| Cathodal or anodal | 250 µA (110.13 A/m2) or 500 µA (220.3 A/m2); 15 days | Mouse (PT) | CD16/32↓, Iba1+↓ | Impacting neurogenesis and influencing functional recovery | [289] | |
| TUS/ tFUS | Low intensity | 528 mW/cm2; 5 days; 15 min/day; 5 days before MCAO | Mouse (MCAO) | VEGF↑, BDNF↑, Caspase-3↓ | Ameliorating brain damage | [290] |
| 86 mW/cm2; 60 min | Rats (dMCAO) | Inflammatory factors↓ | Increasing cerebral blood flow and supporting neuroprotection | [291] | ||
| 0.5 MHz; 120 mW/cm2; 7 consecutive days; | Mouse (MCAO) | M2 microglia↑ IL-10 and IL-10R↑ | Promoting neurorehabilitation | [292] |
5.3. Cell-Based Therapy
5.4. Noninvasive Brain Stimulation
6. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iadecola, C.; Buckwalter, M.S.; Anrather, J. Immune responses to stroke: Mechanisms, modulation, and therapeutic potential. J. Clin. Investig. 2020, 130, 2777–2788. [Google Scholar] [CrossRef] [PubMed]
- Maida, C.D.; Norrito, R.L.; Daidone, M.; Tuttolomondo, A.; Pinto, A. Neuroinflammatory Mechanisms in Ischemic Stroke: Focus on Cardioembolic Stroke, Background, and Therapeutic Approaches. Int. J. Mol. Sci. 2020, 21, 6454. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhao, J.; Li, X.; Bai, Y.; Wang, B.; Qu, Y.; Li, B.; Zhao, S.; Committee Chinese Stroke Association Stroke Council Guideline Writing. Chinese Stroke Association guidelines for clinical management of cerebrovascular disorders: Executive summary and 2019 update of clinical management of stroke rehabilitation. Stroke Vasc. Neurol. 2020, 5, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Hankey, G.J. Stroke. Lancet 2017, 389, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Stark, B.A.; Johnson, C.O.; Roth, G.A.; Bisignano, C.; Abady, G.G.; Abbasifard, M.; Abbasi-Kangevari, M.; Abd-Allah, F.; Abedi, V.; et al. Global, regional, and national burden of stroke and its risk factors, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021, 20, 795–820. [Google Scholar] [CrossRef]
- Desai, S.M.; Jha, R.M.; Linfante, I. Collateral Circulation Augmentation and Neuroprotection as Adjuvant to Mechanical Thrombectomy in Acute Ischemic Stroke. Neurology 2021, 97 (Suppl. 2), S178–S184. [Google Scholar] [CrossRef]
- Yu, F.; Huang, T.; Ran, Y.; Li, D.; Ye, L.; Tian, G.; Xi, J.; Liu, Z. New Insights Into the Roles of Microglial Regulation in Brain Plasticity-Dependent Stroke Recovery. Front. Cell. Neurosci. 2021, 15, 727899. [Google Scholar] [CrossRef]
- Wolf, S.A.; Boddeke, H.W.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef]
- Wang, Y.; Leak, R.K.; Cao, G. Microglia-mediated neuroinflammation and neuroplasticity after stroke. Front. Cell. Neurosci. 2022, 16, 980722. [Google Scholar] [CrossRef]
- Panatier, A.; Robitaille, R. The soothing touch: Microglial contact influences neuronal excitability. Dev. Cell 2012, 23, 1125–1126. [Google Scholar] [CrossRef]
- Ji, K.; Akgul, G.; Wollmuth, L.P.; Tsirka, S.E. Microglia actively regulate the number of functional synapses. PLoS ONE 2013, 8, e56293. [Google Scholar] [CrossRef]
- Hayashi, Y.; Nakanishi, H. Synaptic plasticity and synaptic reorganization regulated by microglia. Nihon Shinkei Seishin Yakurigaku Zasshi 2013, 33, 211–216. [Google Scholar]
- Kuriakose, D.; Xiao, Z. Pathophysiology and Treatment of Stroke: Present Status and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 7609. [Google Scholar] [CrossRef]
- Jiang, C.; Guo, H.; Zhang, Z.; Wang, Y.; Liu, S.; Lai, J.; Wang, T.J.; Li, S.; Zhang, J.; Zhu, L.; et al. Molecular, Pathological, Clinical, and Therapeutic Aspects of Perihematomal Edema in Different Stages of Intracerebral Hemorrhage. Oxid. Med. Cell. Longev. 2022, 2022, 3948921. [Google Scholar] [CrossRef]
- Lan, X.; Han, X.; Li, Q.; Yang, Q.W.; Wang, J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat. Rev. Neurol. 2017, 13, 420–433. [Google Scholar] [CrossRef]
- Chen, Y. Disturbed cerebral circulation and metabolism matters: A preface to the special issue “Stroke and Energy Metabolism”: A preface to the special issue “Stroke and Energy Metabolism”. J. Neurochem. 2022, 160, 10–12. [Google Scholar] [CrossRef]
- Bir, S.C.; Kelley, R.E. Carotid atherosclerotic disease: A systematic review of pathogenesis and management. Brain Circ. 2022, 8, 127–136. [Google Scholar] [CrossRef]
- Feske, S.K. Ischemic Stroke. Am. J. Med. 2021, 134, 1457–1464. [Google Scholar] [CrossRef]
- Gauberti, M.; De Lizarrondo, S.M.; Vivien, D. The “inflammatory penumbra” in ischemic stroke: From clinical data to experimental evidence. Eur. Stroke J. 2016, 1, 20–27. [Google Scholar] [CrossRef]
- Wolf, V.L.; Ergul, A. Progress and challenges in preclinical stroke recovery research. Brain Circ. 2021, 7, 230–240. [Google Scholar] [CrossRef]
- Sun, M.S.; Jin, H.; Sun, X.; Huang, S.; Zhang, F.L.; Guo, Z.N.; Yang, Y. Free Radical Damage in Ischemia-Reperfusion Injury: An Obstacle in Acute Ischemic Stroke after Revascularization Therapy. Oxid. Med. Cell. Longev. 2018, 2018, 3804979. [Google Scholar] [CrossRef] [PubMed]
- Clark, B.; Whitall, J.; Kwakkel, G.; Mehrholz, J.; Ewings, S.; Burridge, J. The effect of time spent in rehabilitation on activity limitation and impairment after stroke. Cochrane Database Syst. Rev. 2021, 10, CD012612. [Google Scholar] [CrossRef] [PubMed]
- Coleman, E.R.; Moudgal, R.; Lang, K.; Hyacinth, H.I.; Awosika, O.O.; Kissela, B.M.; Feng, W. Early Rehabilitation after Stroke: A Narrative Review. Curr. Atheroscler. Rep. 2017, 19, 59. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Yun, H.J.; Ding, Y. Timing is everything: Exercise therapy and remote ischemic conditioning for acute ischemic stroke patients. Brain Circ. 2021, 7, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Nave, A.H.; Rackoll, T.; Grittner, U.; Blasing, H.; Gorsler, A.; Nabavi, D.G.; Audebert, H.J.; Klostermann, F.; Muller-Werdan, U.; Steinhagen-Thiessen, E.; et al. Physical Fitness Training in Patients with Subacute Stroke (PHYS-STROKE): Multicentre, randomised controlled, endpoint blinded trial. BMJ 2019, 366, l5101. [Google Scholar] [CrossRef]
- Jin, R.; Yang, G.; Li, G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J. Leukoc. Biol. 2010, 87, 779–789. [Google Scholar] [CrossRef]
- Zhang, B.; Saatman, K.E.; Chen, L. Therapeutic potential of natural compounds from Chinese medicine in acute and subacute phases of ischemic stroke. Neural Regen. Res. 2020, 15, 416–424. [Google Scholar] [CrossRef]
- Jin, R.; Liu, L.; Zhang, S.; Nanda, A.; Li, G. Role of inflammation and its mediators in acute ischemic stroke. J. Cardiovasc. Transl. Res. 2013, 6, 834–851. [Google Scholar] [CrossRef]
- Paul, S.; Candelario-Jalil, E. Emerging neuroprotective strategies for the treatment of ischemic stroke: An overview of clinical and preclinical studies. Exp. Neurol. 2021, 335, 113518. [Google Scholar] [CrossRef]
- Chamorro, A.; Lo, E.H.; Renu, A.; van Leyen, K.; Lyden, P.D. The future of neuroprotection in stroke. J. Neurol. Neurosurg. Psychiatry 2021, 92, 129–135. [Google Scholar] [CrossRef]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef]
- Jurcau, A.; Simion, A. Neuroinflammation in Cerebral Ischemia and Ischemia/Reperfusion Injuries: From Pathophysiology to Therapeutic Strategies. Int. J. Mol. Sci. 2021, 23, 14. [Google Scholar] [CrossRef]
- Xiong, X.Y.; Liu, L.; Yang, Q.W. Refocusing Neuroprotection in Cerebral Reperfusion Era: New Challenges and Strategies. Front. Neurol. 2018, 9, 249. [Google Scholar] [CrossRef]
- Strinitz, M.; Pham, M.; Marz, A.G.; Feick, J.; Weidner, F.; Vogt, M.L.; Essig, F.; Neugebauer, H.; Stoll, G.; Schuhmann, M.K.; et al. Immune Cells Invade the Collateral Circulation during Human Stroke: Prospective Replication and Extension. Int. J. Mol. Sci. 2021, 22, 9161. [Google Scholar] [CrossRef]
- Kumar, A.; Kitago, T. Pharmacological Enhancement of Stroke Recovery. Curr. Neurol. Neurosci. Rep. 2019, 19, 43. [Google Scholar] [CrossRef]
- Amantea, D.; Nappi, G.; Bernardi, G.; Bagetta, G.; Corasaniti, M.T. Post-ischemic brain damage: Pathophysiology and role of inflammatory mediators. FEBS J. 2009, 276, 13–26. [Google Scholar] [CrossRef]
- Alia, C.; Spalletti, C.; Lai, S.; Panarese, A.; Lamola, G.; Bertolucci, F.; Vallone, F.; Di Garbo, A.; Chisari, C.; Micera, S.; et al. Neuroplastic Changes Following Brain Ischemia and their Contribution to Stroke Recovery: Novel Approaches in Neurorehabilitation. Front. Cell. Neurosci. 2017, 11, 76. [Google Scholar] [CrossRef]
- Brown, C.E.; Li, P.; Boyd, J.D.; Delaney, K.R.; Murphy, T.H. Extensive turnover of dendritic spines and vascular remodeling in cortical tissues recovering from stroke. J. Neurosci. 2007, 27, 4101–4109. [Google Scholar] [CrossRef]
- Stroemer, R.P.; Kent, T.A.; Hulsebosch, C.E. Neocortical neural sprouting, synaptogenesis, and behavioral recovery after neocortical infarction in rats. Stroke 1995, 26, 2135–2144. [Google Scholar] [CrossRef]
- Grutzendler, J.; Kasthuri, N.; Gan, W.B. Long-term dendritic spine stability in the adult cortex. Nature 2002, 420, 812–816. [Google Scholar] [CrossRef]
- Harris, K.M. Structure, development, and plasticity of dendritic spines. Curr. Opin. Neurobiol. 1999, 9, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Trachtenberg, J.T.; Chen, B.E.; Knott, G.W.; Feng, G.; Sanes, J.R.; Welker, E.; Svoboda, K. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 2002, 420, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Holtmaat, A.; Wilbrecht, L.; Knott, G.W.; Welker, E.; Svoboda, K. Experience-dependent and cell-type-specific spine growth in the neocortex. Nature 2006, 441, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wang, L.; Ju, F.; Ran, Y.; Wang, C.; Zhang, S. Transient global cerebral ischemia induces rapid and sustained reorganization of synaptic structures. J. Cereb. Blood Flow Metab. 2017, 37, 2756–2767. [Google Scholar] [CrossRef]
- Keck, T.; Mrsic-Flogel, T.D.; Vaz Afonso, M.; Eysel, U.T.; Bonhoeffer, T.; Hubener, M. Massive restructuring of neuronal circuits during functional reorganization of adult visual cortex. Nat. Neurosci. 2008, 11, 1162–1167. [Google Scholar] [CrossRef]
- Mostany, R.; Chowdhury, T.G.; Johnston, D.G.; Portonovo, S.A.; Carmichael, S.T.; Portera-Cailliau, C. Local hemodynamics dictate long-term dendritic plasticity in peri-infarct cortex. J. Neurosci. 2010, 30, 14116–14126. [Google Scholar] [CrossRef]
- Benowitz, L.I.; Carmichael, S.T. Promoting axonal rewiring to improve outcome after stroke. Neurobiol. Dis. 2010, 37, 259–266. [Google Scholar] [CrossRef]
- Weidner, N.; Ner, A.; Salimi, N.; Tuszynski, M.H. Spontaneous Corticospinal Axonal Plasticity and Functional Recovery after Adult Central Nervous System Injury. Proc. Natl. Acad. Sci. USA 2001, 98, 3513–3518. [Google Scholar] [CrossRef]
- Overman, J.J.; Clarkson, A.N.; Wanner, I.B.; Overman, W.T.; Eckstein, I.; Maguire, J.L.; Dinov, I.D.; Toga, A.W.; Carmichael, S.T. A role for ephrin-A5 in axonal sprouting, recovery, and activity-dependent plasticity after stroke. Proc. Natl. Acad. Sci. USA 2012, 109, E2230–E2239. [Google Scholar] [CrossRef]
- Li, S.; Overman, J.J.; Katsman, D.; Kozlov, S.V.; Donnelly, C.J.; Twiss, J.L.; Giger, R.J.; Coppola, G.; Geschwind, D.H.; Carmichael, S.T. An age-related sprouting transcriptome provides molecular control of axonal sprouting after stroke. Nat. Neurosci. 2010, 13, 1496–1504. [Google Scholar] [CrossRef]
- Li, S.; Nie, E.H.; Yin, Y.; Benowitz, L.I.; Tung, S.; Vinters, H.V.; Bahjat, F.R.; Stenzel-Poore, M.P.; Kawaguchi, R.; Coppola, G.; et al. GDF10 is a signal for axonal sprouting and functional recovery after stroke. Nat. Neurosci. 2015, 18, 1737–1745. [Google Scholar] [CrossRef]
- Dancause, N.; Barbay, S.; Frost, S.B.; Plautz, E.J.; Chen, D.; Zoubina, E.V.; Stowe, A.M.; Nudo, R.J. Extensive cortical rewiring after brain injury. J. Neurosci. 2005, 25, 10167–10179. [Google Scholar] [CrossRef]
- Morecraft, R.J.; Ge, J.; Stilwell-Morecraft, K.S.; McNeal, D.W.; Hynes, S.M.; Pizzimenti, M.A.; Rotella, D.L.; Darling, W.G. Vulnerability of the medial frontal corticospinal projection accompanies combined lateral frontal and parietal cortex injury in rhesus monkey. J. Comp. Neurol. 2015, 523, 669–697. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Y.; Zhang, Z.G.; Cui, X.; Cui, Y.; Lu, M.; Savant-Bhonsale, S.; Chopp, M. Bone marrow stromal cells enhance inter- and intracortical axonal connections after ischemic stroke in adult rats. J. Cereb. Blood Flow Metab. 2010, 30, 1288–1295. [Google Scholar] [CrossRef]
- Carmichael, S.T.; Kathirvelu, B.; Schweppe, C.A.; Nie, E.H. Molecular, cellular and functional events in axonal sprouting after stroke. Exp. Neurol. 2017, 287, 384–394. [Google Scholar] [CrossRef]
- Wahl, A.S.; Schwab, M.E. Finding an optimal rehabilitation paradigm after stroke: Enhancing fiber growth and training of the brain at the right moment. Front. Hum. Neurosci. 2014, 8, 381. [Google Scholar] [CrossRef]
- Jia, W.; Kamen, Y.; Pivonkova, H.; Karadottir, R.T. Neuronal activity-dependent myelin repair after stroke. Neurosci. Lett. 2019, 703, 139–144. [Google Scholar] [CrossRef]
- Shi, H.; Hu, X.; Leak, R.K.; Shi, Y.; An, C.; Suenaga, J.; Chen, J.; Gao, Y. Demyelination as a rational therapeutic target for ischemic or traumatic brain injury. Exp. Neurol. 2015, 272, 17–25. [Google Scholar] [CrossRef]
- Mifsud, G.; Zammit, C.; Muscat, R.; Di Giovanni, G.; Valentino, M. Oligodendrocyte pathophysiology and treatment strategies in cerebral ischemia. CNS Neurosci. Ther. 2014, 20, 603–612. [Google Scholar] [CrossRef]
- Raffaele, S.; Fumagalli, M. Dynamics of Microglia Activation in the Ischemic Brain: Implications for Myelin Repair and Functional Recovery. Front. Cell. Neurosci. 2022, 16, 950819. [Google Scholar] [CrossRef]
- Moyon, S.; Dubessy, A.L.; Aigrot, M.S.; Trotter, M.; Huang, J.K.; Dauphinot, L.; Potier, M.C.; Kerninon, C.; Melik Parsadaniantz, S.; Franklin, R.J.; et al. Demyelination causes adult CNS progenitors to revert to an immature state and express immune cues that support their migration. J. Neurosci. 2015, 35, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.M.; Ffrench-Constant, C. Regenerating CNS myelin—From mechanisms to experimental medicines. Nat. Rev. Neurosci. 2017, 18, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Lampron, A.; Larochelle, A.; Laflamme, N.; Prefontaine, P.; Plante, M.M.; Sanchez, M.G.; Yong, V.W.; Stys, P.K.; Tremblay, M.E.; Rivest, S. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J. Exp. Med. 2015, 212, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E. Microglia-driven regulation of oligodendrocyte lineage cells, myelination, and remyelination. J. Leukoc. Biol. 2017, 101, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Plemel, J.R.; Keough, M.B.; Duncan, G.J.; Sparling, J.S.; Yong, V.W.; Stys, P.K.; Tetzlaff, W. Remyelination after spinal cord injury: Is it a target for repair? Prog. Neurobiol. 2014, 117, 54–72. [Google Scholar] [CrossRef]
- Qi, C.; Luo, L.D.; Feng, I.; Ma, S. Molecular mechanisms of synaptogenesis. Front. Synaptic Neurosci. 2022, 14, 939793. [Google Scholar] [CrossRef]
- Jin, Y. Synaptogenesis. In WormBook; The C. elegans Research Community, Ed.; National Library of Medicine: Pasadena, CA, USA, 2005; pp. 1–11. [Google Scholar] [CrossRef]
- Petzoldt, A.G.; Sigrist, S.J. Synaptogenesis. Curr. Biol. 2014, 24, R1076–R1080. [Google Scholar] [CrossRef]
- DeBoer, S.R.; Hubbard, R.; Mersha, M.; Pinilla Monsalve, G.; Winter, S.; Zeiler, S.R. Enhanced Spontaneous Motor Recovery after Stroke in Mice Treated with Cerebrolysin. Neurorehabil. Neural Repair 2021, 35, 525–533. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Chopp, M. Neurorestorative therapies for stroke: Underlying mechanisms and translation to the clinic. Lancet Neurol. 2009, 8, 491–500. [Google Scholar] [CrossRef]
- Mine, Y.; Tatarishvili, J.; Oki, K.; Monni, E.; Kokaia, Z.; Lindvall, O. Grafted human neural stem cells enhance several steps of endogenous neurogenesis and improve behavioral recovery after middle cerebral artery occlusion in rats. Neurobiol. Dis. 2013, 52, 191–203. [Google Scholar] [CrossRef]
- Vonderwalde, I.; Azimi, A.; Rolvink, G.; Ahlfors, J.E.; Shoichet, M.S.; Morshead, C.M. Transplantation of Directly Reprogrammed Human Neural Precursor Cells Following Stroke Promotes Synaptogenesis and Functional Recovery. Transl. Stroke Res. 2020, 11, 93–107. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, G.; Gu, Y.; Guo, X. Meta-Analysis and Systematic Review of Neural Stem Cells therapy for experimental ischemia stroke in preclinical studies. Sci. Rep. 2016, 6, 32291. [Google Scholar] [CrossRef]
- Zhao, T.; Zhu, T.; Xie, L.; Li, Y.; Xie, R.; Xu, F.; Tang, H.; Zhu, J. Neural Stem Cells Therapy for Ischemic Stroke: Progress and Challenges. Transl. Stroke Res. 2022, 13, 665–675. [Google Scholar] [CrossRef]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef]
- Nayak, D.; Roth, T.L.; McGavern, D.B. Microglia development and function. Annu. Rev. Immunol. 2014, 32, 367–402. [Google Scholar] [CrossRef]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Ueno, M.; Fujita, Y.; Tanaka, T.; Nakamura, Y.; Kikuta, J.; Ishii, M.; Yamashita, T. Layer V cortical neurons require microglial support for survival during postnatal development. Nat. Neurosci. 2013, 16, 543–551. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Stence, N.; Waite, M.; Dailey, M.E. Dynamics of microglial activation: A confocal time-lapse analysis in hippocampal slices. Glia 2001, 33, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Nolte, C.; Moller, T.; Walter, T.; Kettenmann, H. Complement 5a controls motility of murine microglial cells in vitro via activation of an inhibitory G-protein and the rearrangement of the actin cytoskeleton. Neuroscience 1996, 73, 1091–1107. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.Y.; Liu, L.; Yang, Q.W. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog. Neurobiol. 2016, 142, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, M.; Ninomiya, I.; Hatakeyama, M.; Takahashi, T.; Shimohata, T. Microglia and Monocytes/Macrophages Polarization Reveal Novel Therapeutic Mechanism against Stroke. Int. J. Mol. Sci. 2017, 18, 2135. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.Q.; Ma, X.T.; Hu, Z.W.; Yang, S.; Chen, M.; Bosco, D.B.; Wu, L.J.; Tian, D.S. Dual Functions of Microglia in Ischemic Stroke. Neurosci. Bull. 2019, 35, 921–933. [Google Scholar] [CrossRef]
- Dikmen, H.O.; Hemmerich, M.; Lewen, A.; Hollnagel, J.O.; Chausse, B.; Kann, O. GM-CSF induces noninflammatory proliferation of microglia and disturbs electrical neuronal network rhythms in situ. J. Neuroinflamm. 2020, 17, 235. [Google Scholar] [CrossRef]
- Han, C.; Sheng, Y.; Wang, J.; Zhou, X.; Li, W.; Zhang, C.; Guo, L.; Yang, Y. Double-negative T cells mediate M1 polarization of microglial cells via TNF-alpha-NLRP3 to aggravate neuroinflammation and cognitive impairment in Alzheimer’s disease mice. J. Cell. Physiol. 2022, 237, 3860–3871. [Google Scholar] [CrossRef]
- Meng, H.; Zhao, H.; Cao, X.; Hao, J.; Zhang, H.; Liu, Y.; Zhu, M.S.; Fan, L.; Weng, L.; Qian, L.; et al. Double-negative T cells remarkably promote neuroinflammation after ischemic stroke. Proc. Natl. Acad. Sci. USA 2019, 116, 5558–5563. [Google Scholar] [CrossRef]
- Jia, J.; Yang, L.; Chen, Y.; Zheng, L.; Chen, Y.; Xu, Y.; Zhang, M. The Role of Microglial Phagocytosis in Ischemic Stroke. Front. Immunol. 2021, 12, 790201. [Google Scholar] [CrossRef]
- Wang, J.; Xing, H.; Wan, L.; Jiang, X.; Wang, C.; Wu, Y. Treatment targets for M2 microglia polarization in ischemic stroke. Biomed. Pharmacther. 2018, 105, 518–525. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, J.; Wang, Y.; Yang, G.Y. The biphasic function of microglia in ischemic stroke. Prog. Neurobiol. 2017, 157, 247–272. [Google Scholar] [CrossRef]
- Latta, C.H.; Brothers, H.M.; Wilcock, D.M. Neuroinflammation in Alzheimer’s disease; A source of heterogeneity and target for personalized therapy. Neuroscience 2015, 302, 103–111. [Google Scholar] [CrossRef]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Ore, M.V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Savman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 2013, 32, 70–85. [Google Scholar] [CrossRef]
- Mecha, M.; Feliu, A.; Carrillo-Salinas, F.J.; Rueda-Zubiaurre, A.; Ortega-Gutierrez, S.; de Sola, R.G.; Guaza, C. Endocannabinoids drive the acquisition of an alternative phenotype in microglia. Brain Behav. Immun. 2015, 49, 233–245. [Google Scholar] [CrossRef]
- Pont-Lezica, L.; Beumer, W.; Colasse, S.; Drexhage, H.; Versnel, M.; Bessis, A. Microglia shape corpus callosum axon tract fasciculation: Functional impact of prenatal inflammation. Eur. J. Neurosci. 2014, 39, 1551–1557. [Google Scholar] [CrossRef]
- Mittelbronn, M. The M1/M2 immune polarization concept in microglia: A fair transfer? Neuroimmunol. Neuroinflamm. 2014, 1, 6–7. [Google Scholar] [CrossRef]
- Zeisel, A.; Munoz-Manchado, A.B.; Codeluppi, S.; Lonnerberg, P.; La Manno, G.; Jureus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C.; et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Bottcher, C.; Amann, L.; Sagar; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e6. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef] [PubMed]
- Smajic, S.; Prada-Medina, C.A.; Landoulsi, Z.; Ghelfi, J.; Delcambre, S.; Dietrich, C.; Jarazo, J.; Henck, J.; Balachandran, S.; Pachchek, S.; et al. Single-cell sequencing of human midbrain reveals glial activation and a Parkinson-specific neuronal state. Brain 2022, 145, 964–978. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Tian, D.-C.; Li, Z.-G.; Ducruet, A.F.; Lawton, M.T.; Shi, F.-D. Global brain inflammation in stroke. Lancet Neurol. 2019, 18, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Chopp, M.; Chen, J. Experimental animal models and inflammatory cellular changes in cerebral ischemic and hemorrhagic stroke. Neurosci. Bull. 2015, 31, 717–734. [Google Scholar] [CrossRef]
- Rupalla, K.; Allegrini, P.R.; Sauer, D.; Wiessner, C. Time course of microglia activation and apoptosis in various brain regions after permanent focal cerebral ischemia in mice. Acta Neuropathol. 1998, 96, 172–178. [Google Scholar] [CrossRef]
- Kanazawa, M.; Kawamura, K.; Takahashi, T.; Miura, M.; Tanaka, Y.; Koyama, M.; Toriyabe, M.; Igarashi, H.; Nakada, T.; Nishihara, M.; et al. Multiple therapeutic effects of progranulin on experimental acute ischaemic stroke. Brain 2015, 138 Pt 7, 1932–1948. [Google Scholar] [CrossRef]
- Walberer, M.; Jantzen, S.U.; Backes, H.; Rueger, M.A.; Keuters, M.H.; Neumaier, B.; Hoehn, M.; Fink, G.R.; Graf, R.; Schroeter, M. In-vivo detection of inflammation and neurodegeneration in the chronic phase after permanent embolic stroke in rats. Brain Res. 2014, 1581, 80–88. [Google Scholar] [CrossRef]
- Cao, Z.; Harvey, S.S.; Chiang, T.; Foltz, A.G.; Lee, A.G.; Cheng, M.Y.; Steinberg, G.K. Unique Subtype of Microglia in Degenerative Thalamus after Cortical Stroke. Stroke 2021, 52, 687–698. [Google Scholar] [CrossRef]
- Yu, T.B.; Cheng, Y.S.; Zhao, P.; Kou, D.W.; Sun, K.; Chen, B.H.; Wang, A.M. Immune therapy with cultured microglia grafting into the injured spinal cord promoting the recovery of rat’s hind limb motor function. Chin. J. Traumatol. 2009, 12, 291–295. [Google Scholar]
- Taylor, R.A.; Chang, C.F.; Goods, B.A.; Hammond, M.D.; Mac Grory, B.; Ai, Y.; Steinschneider, A.F.; Renfroe, S.C.; Askenase, M.H.; McCullough, L.D.; et al. TGF-beta1 modulates microglial phenotype and promotes recovery after intracerebral hemorrhage. J. Clin. Investig. 2017, 127, 280–292. [Google Scholar] [CrossRef]
- Anttila, J.E.; Whitaker, K.W.; Wires, E.S.; Harvey, B.K.; Airavaara, M. Role of microglia in ischemic focal stroke and recovery: Focus on Toll-like receptors. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 79 Pt A, 3–14. [Google Scholar] [CrossRef]
- Thored, P.; Heldmann, U.; Gomes-Leal, W.; Gisler, R.; Darsalia, V.; Taneera, J.; Nygren, J.M.; Jacobsen, S.E.; Ekdahl, C.T.; Kokaia, Z.; et al. Long-term accumulation of microglia with proneurogenic phenotype concomitant with persistent neurogenesis in adult subventricular zone after stroke. Glia 2009, 57, 835–849. [Google Scholar] [CrossRef]
- Otxoa-de-Amezaga, A.; Miro-Mur, F.; Pedragosa, J.; Gallizioli, M.; Justicia, C.; Gaja-Capdevila, N.; Ruiz-Jaen, F.; Salas-Perdomo, A.; Bosch, A.; Calvo, M.; et al. Microglial cell loss after ischemic stroke favors brain neutrophil accumulation. Acta Neuropathol. 2019, 137, 321–341. [Google Scholar] [CrossRef]
- Gelosa, P.; Lecca, D.; Fumagalli, M.; Wypych, D.; Pignieri, A.; Cimino, M.; Verderio, C.; Enerback, M.; Nikookhesal, E.; Tremoli, E.; et al. Microglia is a key player in the reduction of stroke damage promoted by the new antithrombotic agent ticagrelor. J. Cereb. Blood Flow Metab. 2014, 34, 979–988. [Google Scholar] [CrossRef]
- Sandvig, I.; Augestad, I.L.; Haberg, A.K.; Sandvig, A. Neuroplasticity in stroke recovery. The role of microglia in engaging and modifying synapses and networks. Eur. J. Neurosci. 2018, 47, 1414–1428. [Google Scholar] [CrossRef]
- Yao, X.; Liu, S.; Ding, W.; Yue, P.; Jiang, Q.; Zhao, M.; Hu, F.; Zhang, H. TLR4 signal ablation attenuated neurological deficits by regulating microglial M1/M2 phenotype after traumatic brain injury in mice. J. Neuroimmunol. 2017, 310, 38–45. [Google Scholar] [CrossRef]
- Beutler, B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature 2004, 430, 257–263. [Google Scholar] [CrossRef]
- Zhao, S.C.; Ma, L.S.; Chu, Z.H.; Xu, H.; Wu, W.Q.; Liu, F. Regulation of microglial activation in stroke. Acta Pharmacol. Sin. 2017, 38, 445–458. [Google Scholar] [CrossRef]
- Biancardi, V.C.; Stranahan, A.M.; Krause, E.G.; de Kloet, A.D.; Stern, J.E. Cross talk between AT1 receptors and Toll-like receptor 4 in microglia contributes to angiotensin II-derived ROS production in the hypothalamic paraventricular nucleus. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H404–H415. [Google Scholar] [CrossRef]
- Hyakkoku, K.; Hamanaka, J.; Tsuruma, K.; Shimazawa, M.; Tanaka, H.; Uematsu, S.; Akira, S.; Inagaki, N.; Nagai, H.; Hara, H. Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience 2010, 171, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, J.; Li, Q.; Mi, Y.; Zhou, D.; Wang, J.; Chen, G.; Liang, D.; Li, N.; Hou, Y. Loureirin C ameliorates ischemia and reperfusion injury in rats by inhibiting the activation of the TLR4/NF-kappaB pathway and promoting TLR4 degradation. Phytother. Res. 2022, 36, 4527–4541. [Google Scholar] [CrossRef] [PubMed]
- Cartier, A.; Hla, T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science 2019, 366, eaar5551. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, I.; Alam, S.; Jayagopi, S.; Frohberger, S.J.; Hansen, J.N.; Kuehlwein, J.; Holbling, B.V.; Schumak, B.; Hubner, M.P.; Graler, M.H.; et al. Neural sphingosine 1-phosphate accumulation activates microglia and links impaired autophagy and inflammation. Glia 2019, 67, 1859–1872. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 2003, 4, 397–407. [Google Scholar] [CrossRef]
- Zhong, L.; Jiang, X.; Zhu, Z.; Qin, H.; Dinkins, M.B.; Kong, J.N.; Leanhart, S.; Wang, R.; Elsherbini, A.; Bieberich, E.; et al. Lipid transporter Spns2 promotes microglia pro-inflammatory activation in response to amyloid-beta peptide. Glia 2019, 67, 498–511. [Google Scholar] [CrossRef]
- Lv, M.; Zhang, D.; Dai, D.; Zhang, W.; Zhang, L. Sphingosine kinase 1/sphingosine-1-phosphate regulates the expression of interleukin-17A in activated microglia in cerebral ischemia/reperfusion. Inflamm. Res. 2016, 65, 551–562. [Google Scholar] [CrossRef]
- Sapkota, A.; Gaire, B.P.; Kang, M.G.; Choi, J.W. S1P2 contributes to microglial activation and M1 polarization following cerebral ischemia through ERK1/2 and JNK. Sci. Rep. 2019, 9, 12106. [Google Scholar] [CrossRef]
- Xu, D.; Gao, Q.; Wang, F.; Peng, Q.; Wang, G.; Wei, Q.; Lei, S.; Zhao, S.; Zhang, L.; Guo, F. Sphingosine-1-phosphate receptor 3 is implicated in BBB injury via the CCL2-CCR2 axis following acute intracerebral hemorrhage. CNS Neurosci. Ther. 2021, 27, 674–686. [Google Scholar] [CrossRef]
- Xu, P.; Zhang, X.; Liu, Q.; Xie, Y.; Shi, X.; Chen, J.; Li, Y.; Guo, H.; Sun, R.; Hong, Y.; et al. Microglial TREM-1 receptor mediates neuroinflammatory injury via interaction with SYK in experimental ischemic stroke. Cell Death Dis. 2019, 10, 555. [Google Scholar] [CrossRef]
- Gervois, P.; Lambrichts, I. The Emerging Role of Triggering Receptor Expressed on Myeloid Cells 2 as a Target for Immunomodulation in Ischemic Stroke. Front. Immunol. 2019, 10, 1668. [Google Scholar] [CrossRef]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Microglia and Aging: The Role of the TREM2-DAP12 and CX3CL1-CX3CR1 Axes. Int. J. Mol. Sci. 2018, 19, 318. [Google Scholar] [CrossRef]
- Ma, W.Y.; Wang, S.S.; Wu, Q.L.; Zhou, X.; Chu, S.F.; Chen, N.H. The versatile role of TREM2 in regulating of microglia fate in the ischemic stroke. Int. Immunopharmacol. 2022, 109, 108733. [Google Scholar] [CrossRef]
- Turnbull, I.R.; Colonna, M. Activating and inhibitory functions of DAP12. Nat. Rev. Immunol. 2007, 7, 155–161. [Google Scholar] [CrossRef]
- Kawabori, M.; Kacimi, R.; Kauppinen, T.; Calosing, C.; Kim, J.Y.; Hsieh, C.L.; Nakamura, M.C.; Yenari, M.A. Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J. Neurosci. 2015, 35, 3384–3396. [Google Scholar] [CrossRef]
- Tanaka, M.; Sackett, S.; Zhang, Y. Endocannabinoid Modulation of Microglial Phenotypes in Neuropathology. Front. Neurol. 2020, 11, 87. [Google Scholar] [CrossRef]
- Komorowska-Muller, J.A.; Schmole, A.C. CB2 Receptor in Microglia: The Guardian of Self-Control. Int. J. Mol. Sci. 2020, 22, 19. [Google Scholar] [CrossRef]
- Zarruk, J.G.; Fernandez-Lopez, D.; Garcia-Yebenes, I.; Garcia-Gutierrez, M.S.; Vivancos, J.; Nombela, F.; Torres, M.; Burguete, M.C.; Manzanares, J.; Lizasoain, I.; et al. Cannabinoid type 2 receptor activation downregulates stroke-induced classic and alternative brain macrophage/microglial activation concomitant to neuroprotection. Stroke 2012, 43, 211–219. [Google Scholar] [CrossRef]
- Karamyan, V.; Yu, S.-J.; Reiner, D.; Shen, H.; Wu, K.-J.; Liu, Q.-R.; Wang, Y. Time-Dependent Protection of CB2 Receptor Agonist in Stroke. PLoS ONE 2015, 10, e0132487. [Google Scholar] [CrossRef]
- Kolb, B.; Saber, H.; Fadel, H.; Rajah, G. The endocannabinoid system and stroke: A focused review. Brain Circ. 2019, 5, 1–7. [Google Scholar] [CrossRef]
- Zhang, W.; Tian, T.; Gong, S.X.; Huang, W.Q.; Zhou, Q.Y.; Wang, A.P.; Tian, Y. Microglia-associated neuroinflammation is a potential therapeutic target for ischemic stroke. Neural Regen. Res. 2021, 16, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Benekli, M.; Baumann, H.; Wetzler, M. Targeting signal transducer and activator of transcription signaling pathway in leukemias. J. Clin. Oncol. 2009, 27, 4422–4432. [Google Scholar] [CrossRef] [PubMed]
- Butturini, E.; Boriero, D.; Carcereri de Prati, A.; Mariotto, S. STAT1 drives M1 microglia activation and neuroinflammation under hypoxia. Arch. Biochem. Biophys. 2019, 669, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Fan, W.H.; Liu, Q.; Shang, K.; Murugan, M.; Wu, L.J.; Wang, W.; Tian, D.S. Fingolimod Protects Against Ischemic White Matter Damage by Modulating Microglia Toward M2 Polarization via STAT3 Pathway. Stroke 2017, 48, 3336–3346. [Google Scholar] [CrossRef]
- Ding, Y.; Qian, J.; Li, H.; Shen, H.; Li, X.; Kong, Y.; Xu, Z.; Chen, G. Effects of SC99 on cerebral ischemia-perfusion injury in rats: Selective modulation of microglia polarization to M2 phenotype via inhibiting JAK2-STAT3 pathway. Neurosci. Res. 2019, 142, 58–68. [Google Scholar] [CrossRef]
- Li, F.; Zhao, H.; Han, Z.; Wang, R.; Tao, Z.; Fan, Z.; Zhang, S.; Li, G.; Chen, Z.; Luo, Y. Xuesaitong May Protect Against Ischemic Stroke by Modulating Microglial Phenotypes and Inhibiting Neuronal Cell Apoptosis via the STAT3 Signaling Pathway. CNS Neurol. Disord. Drug Targets 2019, 18, 115–123. [Google Scholar] [CrossRef]
- Cai, W.; Dai, X.; Chen, J.; Zhao, J.; Xu, M.; Zhang, L.; Yang, B.; Zhang, W.; Rocha, M.; Nakao, T.; et al. STAT6/Arg1 promotes microglia/macrophage efferocytosis and inflammation resolution in stroke mice. JCI Insight 2019, 4, e131355. [Google Scholar] [CrossRef]
- Sigfridsson, E.; Marangoni, M.; Hardingham, G.E.; Horsburgh, K.; Fowler, J.H. Deficiency of Nrf2 exacerbates white matter damage and microglia/macrophage levels in a mouse model of vascular cognitive impairment. J. Neuroinflamm. 2020, 17, 367. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Liao, S.; Wu, J.; Liu, R.; Wang, S.; Luo, J.; Yang, Y.; Qin, Y.; Li, T.; Zheng, X.; Song, J.; et al. A novel compound DBZ ameliorates neuroinflammation in LPS-stimulated microglia and ischemic stroke rats: Role of Akt(Ser473)/GSK3beta(Ser9)-mediated Nrf2 activation. Redox Biol. 2020, 36, 101644. [Google Scholar] [CrossRef]
- Bellezza, I.; Mierla, A.L.; Minelli, A. Nrf2 and NF-kappaB and Their Concerted Modulation in Cancer Pathogenesis and Progression. Cancers 2010, 2, 483. [Google Scholar] [CrossRef]
- Al Mamun, A.; Chauhan, A.; Qi, S.; Ngwa, C.; Xu, Y.; Sharmeen, R.; Hazen, A.L.; Li, J.; Aronowski, J.A.; McCullough, L.D.; et al. Microglial IRF5-IRF4 regulatory axis regulates neuroinflammation after cerebral ischemia and impacts stroke outcomes. Proc. Natl. Acad. Sci. USA 2020, 117, 1742–1752. [Google Scholar] [CrossRef]
- Negishi, H.; Fujita, Y.; Yanai, H.; Sakaguchi, S.; Ouyang, X.; Shinohara, M.; Takayanagi, H.; Ohba, Y.; Taniguchi, T.; Honda, K. Evidence for Licensing of Ifn-Gamma-Induced Ifn Regulatory Factor 1 Transcription Factor by Myd88 in Toll-Like Receptor-Dependent Gene Induction Program. Proc. Natl. Acad. Sci. USA 2006, 103, 15136–15141. [Google Scholar] [CrossRef]
- Al Mamun, A.; Chauhan, A.; Yu, H.; Xu, Y.; Sharmeen, R.; Liu, F. Interferon regulatory factor 4/5 signaling impacts on microglial activation after ischemic stroke in mice. Eur. J. Neurosci. 2018, 47, 140–149. [Google Scholar] [CrossRef]
- Masuda, T.; Tsuda, M.; Yoshinaga, R.; Tozaki-Saitoh, H.; Ozato, K.; Tamura, T.; Inoue, K. IRF8 is a critical transcription factor for transforming microglia into a reactive phenotype. Cell Rep. 2012, 1, 334–340. [Google Scholar] [CrossRef]
- Horiuchi, M.; Wakayama, K.; Itoh, A.; Kawai, K.; Pleasure, D.; Ozato, K.; Itoh, T. Interferon regulatory factor 8/interferon consensus sequence binding protein is a critical transcription factor for the physiological phenotype of microglia. J. Neuroinflamm. 2012, 9, 227. [Google Scholar] [CrossRef]
- Jiang, C.T.; Wu, W.F.; Deng, Y.H.; Ge, J.W. Modulators of microglia activation and polarization in ischemic stroke (Review). Mol. Med. Rep. 2020, 21, 2006–2018. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Utreras, E.; Cabezas-Opazo, F.A. Role of PPAR gamma in the Differentiation and Function of Neurons. PPAR Res. 2014, 2014, 768594. [Google Scholar] [CrossRef]
- Ji, J.; Xue, T.F.; Guo, X.D.; Yang, J.; Guo, R.B.; Wang, J.; Huang, J.Y.; Zhao, X.J.; Sun, X.L. Antagonizing peroxisome proliferator-activated receptor gamma facilitates M1-to-M2 shift of microglia by enhancing autophagy via the LKB1-AMPK signaling pathway. Aging Cell 2018, 17, e12774. [Google Scholar] [CrossRef]
- Wang, S.W.; Liu, Z.; Shi, Z.S. Non-Coding RNA in Acute Ischemic Stroke: Mechanisms, Biomarkers and Therapeutic Targets. Cell Transplant. 2018, 27, 1763–1777. [Google Scholar] [CrossRef]
- Lian, L.; Zhang, Y.; Liu, L.; Yang, L.; Cai, Y.; Zhang, J.; Xu, S. Neuroinflammation in Ischemic Stroke: Focus on MicroRNA-mediated Polarization of Microglia. Front. Mol. Neurosci. 2021, 13, 612439. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dasgupta, C.; Huang, L.; Meng, X.; Zhang, L. MiRNA-210 induces microglial activation and regulates microglia-mediated neuroinflammation in neonatal hypoxic-ischemic encephalopathy. Cell. Mol. Immunol. 2020, 17, 976–991. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef]
- Bras, J.P.; Bravo, J.; Freitas, J.; Barbosa, M.A.; Santos, S.G.; Summavielle, T.; Almeida, M.I. TNF-alpha-induced microglia activation requires miR-342: Impact on NF-kB signaling and neurotoxicity. Cell Death Dis. 2020, 11, 415. [Google Scholar] [CrossRef] [PubMed]
- Freilich, R.W.; Woodbury, M.E.; Ikezu, T. Integrated expression profiles of mRNA and miRNA in polarized primary murine microglia. PLoS ONE 2013, 8, e79416. [Google Scholar] [CrossRef]
- Zhang, D.; Cai, G.; Liu, K.; Zhuang, Z.; Jia, K.; Pei, S.; Wang, X.; Wang, H.; Xu, S.; Cui, C.; et al. Microglia exosomal miRNA-137 attenuates ischemic brain injury through targeting Notch1. Aging 2021, 13, 4079–4095. [Google Scholar] [CrossRef]
- Huang, S.; Ge, X.; Yu, J.; Han, Z.; Yin, Z.; Li, Y.; Chen, F.; Wang, H.; Zhang, J.; Lei, P. Increased miR-124-3p in microglial exosomes following traumatic brain injury inhibits neuronal inflammation and contributes to neurite outgrowth via their transfer into neurons. FASEB J. 2018, 32, 512–528. [Google Scholar] [CrossRef]
- You, X.; Vlatkovic, I.; Babic, A.; Will, T.; Epstein, I.; Tushev, G.; Akbalik, G.; Wang, M.; Glock, C.; Quedenau, C.; et al. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat. Neurosci. 2015, 18, 603–610. [Google Scholar] [CrossRef]
- Mehta, S.L.; Chokkalla, A.K.; Vemuganti, R. Noncoding RNA crosstalk in brain health and diseases. Neurochem. Int. 2021, 149, 105139. [Google Scholar] [CrossRef]
- Tong, D.; Zhao, Y.; Tang, Y.; Ma, J.; Wang, Z.; Li, C. Circ-Usp10 promotes microglial activation and induces neuronal death by targeting miRNA-152-5p/CD84. Bioengineered 2021, 12, 10812–10822. [Google Scholar] [CrossRef] [PubMed]
- Xiaoying, G.; Guo, M.; Jie, L.; Yanmei, Z.; Ying, C.; Shengjie, S.; Haiyan, G.; Feixiang, S.; Sihua, Q.; Jiahang, S. CircHivep2 contributes to microglia activation and inflammation via miR-181a-5p/SOCS2 signalling in mice with kainic acid-induced epileptic seizures. J. Cell. Mol. Med. 2020, 24, 12980–12993. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kang, J.; Lv, H.; Liu, R.; Chen, J.; Zhang, Y.; Zhang, Y.; Yu, G.; Zhang, X.; Ning, B. CircPrkcsh, a circular RNA, contributes to the polarization of microglia towards the M1 phenotype induced by spinal cord injury and acts via the JNK/p38 MAPK pathway. FASEB J. 2021, 35, e22014. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Tu, Z.; Yang, D.; Hu, M.; Zhou, L.; Li, Q.; Yu, B.; Hou, S. Exosomes from hypoxic pre-treated ADSCs attenuate acute ischemic stroke-induced brain injury via delivery of circ-Rps5 and promote M2 microglia/macrophage polarization. Neurosci. Lett. 2022, 769, 136389. [Google Scholar] [CrossRef] [PubMed]
- Han, C.L.; Liu, Y.P.; Guo, C.J.; Du, T.T.; Jiang, Y.; Wang, K.L.; Shao, X.Q.; Meng, F.G.; Zhang, J.G. The lncRNA H19 binding to let-7b promotes hippocampal glial cell activation and epileptic seizures by targeting Stat3 in a rat model of temporal lobe epilepsy. Cell Prolif. 2020, 53, e12856. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, H.; Fan, Z.; Li, G.; Ma, Q.; Tao, Z.; Wang, R.; Feng, J.; Luo, Y. Long Noncoding RNA H19 Promotes Neuroinflammation in Ischemic Stroke by Driving Histone Deacetylase 1-Dependent M1 Microglial Polarization. Stroke 2017, 48, 2211–2221. [Google Scholar] [CrossRef]
- Wen, Y.; Yu, Y.; Fu, X. LncRNA Gm4419 contributes to OGD/R injury of cerebral microglial cells via IkappaB phosphorylation and NF-kappaB activation. Biochem. Biophys. Res. Commun. 2017, 487, 923–929. [Google Scholar] [CrossRef]
- Meng, J.; Ding, T.; Chen, Y.; Long, T.; Xu, Q.; Lian, W.; Liu, W. LncRNA-Meg3 promotes Nlrp3-mediated microglial inflammation by targeting miR-7a-5p. Int. Immunopharmacol. 2021, 90, 107141. [Google Scholar] [CrossRef]
- Qi, X.; Shao, M.; Sun, H.; Shen, Y.; Meng, D.; Huo, W. Long non-coding RNA SNHG14 promotes microglia activation by regulating miR-145-5p/PLA2G4A in cerebral infarction. Neuroscience 2017, 348, 98–106. [Google Scholar] [CrossRef]
- Vilalta, A.; Brown, G.C. Neurophagy, the phagocytosis of live neurons and synapses by glia, contributes to brain development and disease. FEBS J. 2018, 285, 3566–3575. [Google Scholar] [CrossRef]
- Sierra, A.; Abiega, O.; Shahraz, A.; Neumann, H. Janus-faced microglia: Beneficial and detrimental consequences of microglial phagocytosis. Front. Cell. Neurosci. 2013, 7, 6. [Google Scholar] [CrossRef]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 2009, 29, 3974–3980. [Google Scholar] [CrossRef]
- Riccomagno, M.M.; Kolodkin, A.L. Sculpting neural circuits by axon and dendrite pruning. Annu. Rev. Cell Dev. Biol. 2015, 31, 779–805. [Google Scholar] [CrossRef]
- Schuldiner, O.; Yaron, A. Mechanisms of developmental neurite pruning. Cell. Mol. Life Sci. 2015, 72, 101–119. [Google Scholar] [CrossRef]
- Cunningham, C.L.; Martinez-Cerdeno, V.; Noctor, S.C. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J. Neurosci. 2013, 33, 4216–4233. [Google Scholar] [CrossRef]
- Fricker, M.; Neher, J.J.; Zhao, J.W.; Thery, C.; Tolkovsky, A.M.; Brown, G.C. MFG-E8 mediates primary phagocytosis of viable neurons during neuroinflammation. J. Neurosci. 2012, 32, 2657–2666. [Google Scholar] [CrossRef]
- Fricker, M.; Oliva-Martin, M.J.; Brown, G.C. Primary phagocytosis of viable neurons by microglia activated with LPS or Abeta is dependent on calreticulin/LRP phagocytic signalling. J. Neuroinflamm. 2012, 9, 196. [Google Scholar] [CrossRef]
- Hornik, T.C.; Vilalta, A.; Brown, G.C. Activated microglia cause reversible apoptosis of pheochromocytoma cells, inducing their cell death by phagocytosis. J. Cell Sci. 2016, 129, 65–79. [Google Scholar] [CrossRef]
- Neher, J.J.; Neniskyte, U.; Zhao, J.W.; Bal-Price, A.; Tolkovsky, A.M.; Brown, G.C. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J. Immunol. 2011, 186, 4973–4983. [Google Scholar] [CrossRef]
- Andoh, M.; Koyama, R. Comparative Review of Microglia and Monocytes in CNS Phagocytosis. Cells 2021, 10, 2555. [Google Scholar] [CrossRef]
- Tanaka, R.; Komine-Kobayashi, M.; Mochizuki, H.; Yamada, M.; Furuya, T.; Migita, M.; Shimada, T.; Mizuno, Y.; Urabe, T. Migration of enhanced green fluorescent protein expressing bone marrow-derived microglia/macrophage into the mouse brain following permanent focal ischemia. Neuroscience 2003, 117, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Neher, J.J. Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 2014, 15, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Sauerzweig, S.; Ronicke, R.; Gunzer, F.; Dinkel, K.; Ullrich, O.; Gunzer, M.; Reymann, K.G. Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: A new mechanism of CNS immune privilege. J. Neurosci. 2008, 28, 5965–5975. [Google Scholar] [CrossRef] [PubMed]
- Denes, A.; Vidyasagar, R.; Feng, J.; Narvainen, J.; McColl, B.W.; Kauppinen, R.A.; Allan, S.M. Proliferating resident microglia after focal cerebral ischaemia in mice. J. Cereb. Blood Flow Metab. 2007, 27, 1941–1953. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Morrison, H.W.; Filosa, J.A. Stroke and the neurovascular unit: Glial cells, sex differences, and hypertension. Am. J. Physiol. Cell Physiol. 2019, 316, C325–C339. [Google Scholar] [CrossRef]
- Kang, R.; Gamdzyk, M.; Lenahan, C.; Tang, J.; Tan, S.; Zhang, J.H. The Dual Role of Microglia in Blood-Brain Barrier Dysfunction after Stroke. Curr. Neuropharmacol. 2020, 18, 1237–1249. [Google Scholar] [CrossRef]
- Wilton, D.K.; Dissing-Olesen, L.; Stevens, B. Neuron-Glia Signaling in Synapse Elimination. Annu. Rev. Neurosci. 2019, 42, 107–127. [Google Scholar] [CrossRef]
- Prada, I.; Gabrielli, M.; Turola, E.; Iorio, A.; D’Arrigo, G.; Parolisi, R.; De Luca, M.; Pacifici, M.; Bastoni, M.; Lombardi, M.; et al. Glia-to-neuron transfer of miRNAs via extracellular vesicles: A new mechanism underlying inflammation-induced synaptic alterations. Acta Neuropathol. 2018, 135, 529–550. [Google Scholar] [CrossRef]
- Bahrini, I.; Song, J.H.; Diez, D.; Hanayama, R. Neuronal exosomes facilitate synaptic pruning by up-regulating complement factors in microglia. Sci. Rep. 2015, 5, 7989. [Google Scholar] [CrossRef]
- Yanguas-Casas, N.; Crespo-Castrillo, A.; Arevalo, M.A.; Garcia-Segura, L.M. Aging and sex: Impact on microglia phagocytosis. Aging Cell 2020, 19, e13182. [Google Scholar] [CrossRef] [PubMed]
- Butler, C.A.; Popescu, A.S.; Kitchener, E.J.A.; Allendorf, D.H.; Puigdellivol, M.; Brown, G.C. Microglial phagocytosis of neurons in neurodegeneration, and its regulation. J. Neurochem. 2021, 158, 621–639. [Google Scholar] [CrossRef]
- Lyons, A.; Lynch, A.M.; Downer, E.J.; Hanley, R.; O’Sullivan, J.B.; Smith, A.; Lynch, M.A. Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attentuates microglial activation in vivo and in vitro. J. Neurochem. 2009, 110, 1547–1556. [Google Scholar] [CrossRef]
- Pagani, F.; Paolicelli, R.C.; Murana, E.; Cortese, B.; Di Angelantonio, S.; Zurolo, E.; Guiducci, E.; Ferreira, T.A.; Garofalo, S.; Catalano, M.; et al. Defective microglial development in the hippocampus of Cx3cr1 deficient mice. Front. Cell. Neurosci. 2015, 9, 111. [Google Scholar] [CrossRef]
- Sipe, G.O.; Lowery, R.L.; Tremblay, M.E.; Kelly, E.A.; Lamantia, C.E.; Majewska, A.K. Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat. Commun. 2016, 7, 10905. [Google Scholar] [CrossRef]
- Kurpius, D.; Nolley, E.P.; Dailey, M.E. Purines induce directed migration and rapid homing of microglia to injured pyramidal neurons in developing hippocampus. Glia 2007, 55, 873–884. [Google Scholar] [CrossRef]
- Jiang, P.; Xing, F.; Guo, B.; Yang, J.; Li, Z.; Wei, W.; Hu, F.; Lee, I.; Zhang, X.; Pan, L.; et al. Nucleotide transmitters ATP and ADP mediate intercellular calcium wave communication via P2Y12/13 receptors among BV-2 microglia. PLoS ONE 2017, 12, e0183114. [Google Scholar] [CrossRef]
- Koizumi, S.; Shigemoto-Mogami, Y.; Nasu-Tada, K.; Shinozaki, Y.; Ohsawa, K.; Tsuda, M.; Joshi, B.V.; Jacobson, K.A.; Kohsaka, S.; Inoue, K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 2007, 446, 1091–1095. [Google Scholar] [CrossRef]
- Neher, J.J.; Neniskyte, U.; Hornik, T.; Brown, G.C. Inhibition of UDP/P2Y6 purinergic signaling prevents phagocytosis of viable neurons by activated microglia in vitro and in vivo. Glia 2014, 62, 1463–1475. [Google Scholar] [CrossRef]
- Emmrich, J.V.; Hornik, T.C.; Neher, J.J.; Brown, G.C. Rotenone induces neuronal death by microglial phagocytosis of neurons. FEBS J. 2013, 280, 5030–5038. [Google Scholar] [CrossRef]
- Fu, R.; Shen, Q.; Xu, P.; Luo, J.J.; Tang, Y. Phagocytosis of microglia in the central nervous system diseases. Mol. Neurobiol. 2014, 49, 1422–1434. [Google Scholar] [CrossRef] [PubMed]
- Kansal, S.; Tandon, R.; Dwivedi, P.; Misra, P.; Verma, P.R.; Dube, A.; Mishra, P.R. Development of nanocapsules bearing doxorubicin for macrophage targeting through the phosphatidylserine ligand: A system for intervention in visceral leishmaniasis. J. Antimicrob. Chemother. 2012, 67, 2650–2660. [Google Scholar] [CrossRef] [PubMed]
- Bevers, E.M.; Comfurius, P.; Dekkers, D.W.; Zwaal, R.F. Lipid translocation across the plasma membrane of mammalian cells. Biochim. Biophys. Acta 1999, 1439, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Kasikara, C.; Kumar, S.; Kimani, S.; Tsou, W.I.; Geng, K.; Davra, V.; Sriram, G.; Devoe, C.; Nguyen, K.N.; Antes, A.; et al. Phosphatidylserine Sensing by TAM Receptors Regulates AKT-Dependent Chemoresistance and PD-L1 Expression. Mol. Cancer Res. 2017, 15, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chiou, B.; Gilman, C.K.; Luo, R.; Koshi, T.; Yu, D.; Oak, H.C.; Giera, S.; Johnson-Venkatesh, E.; Muthukumar, A.K.; et al. A splicing isoform of GPR56 mediates microglial synaptic refinement via phosphatidylserine binding. EMBO J. 2020, 39, e104136. [Google Scholar] [CrossRef]
- Wijeyesakere, S.J.; Bedi, S.K.; Huynh, D.; Raghavan, M. The C-Terminal Acidic Region of Calreticulin Mediates Phosphatidylserine Binding and Apoptotic Cell Phagocytosis. J. Immunol. 2016, 196, 3896–3909. [Google Scholar] [CrossRef]
- Caberoy, N.B.; Alvarado, G.; Li, W. Tubby regulates microglial phagocytosis through MerTK. J. Neuroimmunol. 2012, 252, 40–48. [Google Scholar] [CrossRef]
- Caberoy, N.B.; Alvarado, G.; Bigcas, J.L.; Li, W. Galectin-3 is a new MerTK-specific eat-me signal. J. Cell. Physiol. 2012, 227, 401–407. [Google Scholar] [CrossRef]
- Neher, J.J.; Emmrich, J.V.; Fricker, M.; Mander, P.K.; Thery, C.; Brown, G.C. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, E4098–E4107. [Google Scholar] [CrossRef]
- Reid, K.M.; Kitchener, E.J.A.; Butler, C.A.; Cockram, T.O.J.; Brown, G.C. Brain Cells Release Calreticulin That Attracts and Activates Microglia, and Inhibits Amyloid Beta Aggregation and Neurotoxicity. Front. Immunol. 2022, 13, 859686. [Google Scholar] [CrossRef]
- Cockram, T.O.J.; Puigdellivol, M.; Brown, G.C. Calreticulin and Galectin-3 Opsonise Bacteria for Phagocytosis by Microglia. Front. Immunol. 2019, 10, 2647. [Google Scholar] [CrossRef]
- Andoh, M.; Koyama, R. Microglia regulate synaptic development and plasticity. Dev. Neurobiol. 2021, 81, 568–590. [Google Scholar] [CrossRef]
- Lehrman, E.K.; Wilton, D.K.; Litvina, E.Y.; Welsh, C.A.; Chang, S.T.; Frouin, A.; Walker, A.J.; Heller, M.D.; Umemori, H.; Chen, C.; et al. CD47 Protects Synapses from Excess Microglia-Mediated Pruning during Development. Neuron 2018, 100, 120–134.e6. [Google Scholar] [CrossRef]
- Elberg, G.; Liraz-Zaltsman, S.; Reichert, F.; Matozaki, T.; Tal, M.; Rotshenker, S. Deletion of SIRPalpha (signal regulatory protein-alpha) promotes phagocytic clearance of myelin debris in Wallerian degeneration, axon regeneration, and recovery from nerve injury. J. Neuroinflamm. 2019, 16, 277. [Google Scholar] [CrossRef]
- Puigdellivol, M.; Allendorf, D.H.; Brown, G.C. Sialylation and Galectin-3 in Microglia-Mediated Neuroinflammation and Neurodegeneration. Front. Cell. Neurosci. 2020, 14, 162. [Google Scholar] [CrossRef]
- Wang, Y.; Neumann, H. Alleviation of neurotoxicity by microglial human Siglec-11. J. Neurosci. 2010, 30, 3482–3488. [Google Scholar] [CrossRef]
- Klaus, C.; Hansen, J.N.; Ginolhac, A.; Gerard, D.; Gnanapragassam, V.S.; Horstkorte, R.; Rossdam, C.; Buettner, F.F.R.; Sauter, T.; Sinkkonen, L.; et al. Reduced sialylation triggers homeostatic synapse and neuronal loss in middle-aged mice. Neurobiol. Aging 2020, 88, 91–107. [Google Scholar] [CrossRef]
- Allendorf, D.H.; Franssen, E.H.; Brown, G.C. Lipopolysaccharide activates microglia via neuraminidase 1 desialylation of Toll-like Receptor 4. J. Neurochem. 2020, 155, 403–416. [Google Scholar] [CrossRef]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef]
- Sampaio, N.G.; Yu, W.; Cox, D.; Wyckoff, J.; Condeelis, J.; Stanley, E.R.; Pixley, F.J. Phosphorylation of CSF-1R Y721 mediates its association with PI3K to regulate macrophage motility and enhancement of tumor cell invasion. J. Cell Sci. 2011, 124 Pt 12, 2021–2031. [Google Scholar] [CrossRef] [PubMed]
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J. Immunol. 2017, 198, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Chen, Z.; Pathak, J.L.; Carneiro, A.M.D.; Chung, C.Y. Differential Regulation of Adhesion and Phagocytosis of Resting and Activated Microglia by Dopamine. Front. Cell. Neurosci. 2018, 12, 309. [Google Scholar] [CrossRef] [PubMed]
- Husemann, J.; Loike, J.D.; Anankov, R.; Febbraio, M.; Silverstein, S.C. Scavenger receptors in neurobiology and neuropathology: Their role on microglia and other cells of the nervous system. Glia 2002, 40, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Fadok, V.A.; Savill, J.S.; Haslett, C.; Bratton, D.L.; Doherty, D.E.; Campbell, P.A.; Henson, P.M. Different populations of macrophages use either the vitronectin receptor or the phosphatidylserine receptor to recognize and remove apoptotic cells. J. Immunol. 1992, 149, 4029–4035. [Google Scholar] [CrossRef]
- Eugenin, J.; Vecchiola, A.; Murgas, P.; Arroyo, P.; Cornejo, F.; von Bernhardi, R. Expression Pattern of Scavenger Receptors and Amyloid-beta Phagocytosis of Astrocytes and Microglia in Culture are Modified by Acidosis: Implications for Alzheimer’s Disease. J. Alzheimers Dis. 2016, 53, 857–873. [Google Scholar] [CrossRef]
- Grajchen, E.; Wouters, E.; van de Haterd, B.; Haidar, M.; Hardonniere, K.; Dierckx, T.; Van Broeckhoven, J.; Erens, C.; Hendrix, S.; Kerdine-Romer, S.; et al. CD36-mediated uptake of myelin debris by macrophages and microglia reduces neuroinflammation. J. Neuroinflamm. 2020, 17, 224. [Google Scholar] [CrossRef]
- Lu, Y.; Xiao, G.; Luo, W. Minocycline Suppresses NLRP3 Inflammasome Activation in Experimental Ischemic Stroke. Neuroimmunomodulation 2016, 23, 230–238. [Google Scholar] [CrossRef]
- Yang, Y.; Salayandia, V.M.; Thompson, J.F.; Yang, L.Y.; Estrada, E.Y.; Yang, Y. Attenuation of acute stroke injury in rat brain by minocycline promotes blood-brain barrier remodeling and alternative microglia/macrophage activation during recovery. J. Neuroinflamm. 2015, 12, 26. [Google Scholar] [CrossRef]
- Lu, Y.; Zhou, M.; Li, Y.; Li, Y.; Hua, Y.; Fan, Y. Minocycline promotes functional recovery in ischemic stroke by modulating microglia polarization through STAT1/STAT6 pathways. Biochem. Pharmacol. 2021, 186, 114464. [Google Scholar] [CrossRef]
- Zhang, D.; Lu, Z.; Man, J.; Cui, K.; Fu, X.; Yu, L.; Gao, Y.; Liao, L.; Xiao, Q.; Guo, R.; et al. Wnt-3a alleviates neuroinflammation after ischemic stroke by modulating the responses of microglia/macrophages and astrocytes. Int. Immunopharmacol. 2019, 75, 105760. [Google Scholar] [CrossRef]
- Rahimian, R.; Lively, S.; Abdelhamid, E.; Lalancette-Hebert, M.; Schlichter, L.; Sato, S.; Kriz, J. Delayed Galectin-3-Mediated Reprogramming of Microglia after Stroke Is Protective. Mol. Neurobiol. 2019, 56, 6371–6385. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, X.; Huang, Y.; Chen, J.; Shang, W.; Shi, G.; Zhang, L.; Zhang, C.; Chen, R. Atorvastatin alleviates microglia-mediated neuroinflammation via modulating the microbial composition and the intestinal barrier function in ischemic stroke mice. Free Radic. Biol. Med. 2021, 162, 104–117. [Google Scholar] [CrossRef]
- Darsalia, V.; Hua, S.; Larsson, M.; Mallard, C.; Nathanson, D.; Nystrom, T.; Sjoholm, A.; Johansson, M.E.; Patrone, C. Exendin-4 reduces ischemic brain injury in normal and aged type 2 diabetic mice and promotes microglial M2 polarization. PLoS ONE 2014, 9, e103114. [Google Scholar] [CrossRef]
- Imai, T.; Matsubara, H.; Nakamura, S.; Hara, H.; Shimazawa, M. The Mitochondria-targeted Peptide, Bendavia, Attenuated Ischemia/Reperfusion-induced Stroke Damage. Neuroscience 2020, 443, 110–119. [Google Scholar] [CrossRef]
- Li, Q.; Dai, Z.; Cao, Y.; Wang, L. Caspase-1 inhibition mediates neuroprotection in experimental stroke by polarizing M2 microglia/macrophage and suppressing NF-kappaB activation. Biochem. Biophys. Res. Commun. 2019, 513, 479–485. [Google Scholar] [CrossRef]
- Ran, Y.; Qie, S.; Gao, F.; Ding, Z.; Yang, S.; Tian, G.; Liu, Z.; Xi, J. Baicalein ameliorates ischemic brain damage through suppressing proinflammatory microglia polarization via inhibiting the TLR4/NF-kappaB and STAT1 pathway. Brain Res. 2021, 1770, 147626. [Google Scholar] [CrossRef]
- Liu, M.; Xu, Z.; Wang, L.; Zhang, L.; Liu, Y.; Cao, J.; Fu, Q.; Liu, Y.; Li, H.; Lou, J.; et al. Cottonseed oil alleviates ischemic stroke injury by inhibiting the inflammatory activation of microglia and astrocyte. J. Neuroinflamm. 2020, 17, 270. [Google Scholar] [CrossRef]
- Qie, S.; Ran, Y.; Lu, X.; Su, W.; Li, W.; Xi, J.; Gong, W.; Liu, Z. Candesartan modulates microglia activation and polarization via NF-kappaB signaling pathway. Int. J. Immunopathol. Pharmacol. 2020, 34, 2058738420974900. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Z.; Zang, C.; Wang, L.; Yang, H.; Sheng, C.; Shang, J.; Zhao, Z.; Yuan, F.; Yu, Y.; et al. GJ-4 ameliorates memory impairment in focal cerebral ischemia/reperfusion of rats via inhibiting JAK2/STAT1-mediated neuroinflammation. J. Ethnopharmacol. 2021, 267, 113491. [Google Scholar] [CrossRef]
- Liu, Z.J.; Ran, Y.Y.; Qie, S.Y.; Gong, W.J.; Gao, F.H.; Ding, Z.T.; Xi, J.N. Melatonin protects against ischemic stroke by modulating microglia/macrophage polarization toward anti-inflammatory phenotype through STAT3 pathway. CNS Neurosci. Ther. 2019, 25, 1353–1362. [Google Scholar] [CrossRef]
- Du, X.; Xu, Y.; Chen, S.; Fang, M. Inhibited CSF1R Alleviates Ischemia Injury via Inhibition of Microglia M1 Polarization and NLRP3 Pathway. Neural Plast. 2020, 2020, 8825954. [Google Scholar] [CrossRef] [PubMed]
- Hou, B.; Jiang, C.; Wang, D.; Wang, G.; Wang, Z.; Zhu, M.; Kang, Y.; Su, J.; Wei, P.; Ren, H.; et al. Pharmacological Targeting of CSF1R Inhibits Microglial Proliferation and Aggravates the Progression of Cerebral Ischemic Pathology. Front. Cell. Neurosci. 2020, 14, 267. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ran, Y.; Huang, S.; Wen, S.; Zhang, W.; Liu, X.; Ji, Z.; Geng, X.; Ji, X.; Du, H.; et al. Curcumin Protects against Ischemic Stroke by Titrating Microglia/Macrophage Polarization. Front. Aging Neurosci. 2017, 9, 233. [Google Scholar] [CrossRef] [PubMed]
- Ran, Y.; Su, W.; Gao, F.; Ding, Z.; Yang, S.; Ye, L.; Chen, X.; Tian, G.; Xi, J.; Liu, Z. Curcumin Ameliorates White Matter Injury after Ischemic Stroke by Inhibiting Microglia/Macrophage Pyroptosis through NF-kappaB Suppression and NLRP3 Inflammasome Inhibition. Oxid. Med. Cell. Longev. 2021, 2021, 1552127. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wen, S.; Yan, F.; Liu, K.; Liu, L.; Wang, L.; Zhao, S.; Ji, X. Salidroside provides neuroprotection by modulating microglial polarization after cerebral ischemia. J. Neuroinflamm. 2018, 15, 39. [Google Scholar] [CrossRef]
- Jiang, M.; Liu, X.; Zhang, D.; Wang, Y.; Hu, X.; Xu, F.; Jin, M.; Cao, F.; Xu, L. Celastrol treatment protects against acute ischemic stroke-induced brain injury by promoting an IL-33/ST2 axis-mediated microglia/macrophage M2 polarization. J. Neuroinflamm. 2018, 15, 78. [Google Scholar] [CrossRef]
- Pan, J.; Jin, J.L.; Ge, H.M.; Yin, K.L.; Chen, X.; Han, L.J.; Chen, Y.; Qian, L.; Li, X.X.; Xu, Y. Malibatol A regulates microglia M1/M2 polarization in experimental stroke in a PPARgamma-dependent manner. J. Neuroinflamm. 2015, 12, 51. [Google Scholar] [CrossRef]
- Fan, X.; Elkin, K.; Shi, Y.; Zhang, Z.; Cheng, Y.; Gu, J.; Liang, J.; Wang, C.; Ji, X. Schisandrin B improves cerebral ischemia and reduces reperfusion injury in rats through TLR4/NF-kappaB signaling pathway inhibition. Neurol. Res. 2020, 42, 693–702. [Google Scholar] [CrossRef]
- Bobinski, F.; Teixeira, J.M.; Sluka, K.A.; Santos, A.R.S. Interleukin-4 mediates the analgesia produced by low-intensity exercise in mice with neuropathic pain. Pain 2018, 159, 437–450. [Google Scholar] [CrossRef]
- Pedersen, B.K. Muscular interleukin-6 and its role as an energy sensor. Med. Sci. Sports Exerc. 2012, 44, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Calegari, L.; Nunes, R.B.; Mozzaquattro, B.B.; Rossato, D.D.; Dal Lago, P. Exercise training improves the IL-10/TNF-alpha cytokine balance in the gastrocnemius of rats with heart failure. Braz. J. Phys. Ther. 2018, 22, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Mee-Inta, O.; Zhao, Z.W.; Kuo, Y.M. Physical Exercise Inhibits Inflammation and Microglial Activation. Cells 2019, 8, 691. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Luo, Y.; Liang, X.; Tang, J.; Wang, J.; Xiao, Q.; Qi, Y.; Li, Y.; Zhu, P.; Yang, H.; et al. Beneficial effects of running exercise on hippocampal microglia and neuroinflammation in chronic unpredictable stress-induced depression model rats. Transl. Psychiatry 2021, 11, 461. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Wu, S.; Tang, W.; Li, L.; Huang, L.; Mo, D.; Liu, C.; Song, T.; Wang, S.; Wang, J.; et al. Alleviative effects of foraging exercise on depressive-like behaviors in chronic mild stress-induced ischemic rat model. Brain Inj. 2022, 36, 127–136. [Google Scholar] [CrossRef]
- Koga, S.; Kojima, A.; Ishikawa, C.; Kuwabara, S.; Arai, K.; Yoshiyama, Y. Effects of diet-induced obesity and voluntary exercise in a tauopathy mouse model: Implications of persistent hyperleptinemia and enhanced astrocytic leptin receptor expression. Neurobiol. Dis. 2014, 71, 180–192. [Google Scholar] [CrossRef]
- Liu, M.X.; Luo, L.; Fu, J.H.; He, J.Y.; Chen, M.Y.; He, Z.J.; Jia, J. Exercise-induced neuroprotection against cerebral ischemia/reperfusion injury is mediated via alleviating inflammasome-induced pyroptosis. Exp. Neurol. 2022, 349, 113952. [Google Scholar] [CrossRef]
- Pin-Barre, C.; Constans, A.; Brisswalter, J.; Pellegrino, C.; Laurin, J. Effects of High- Versus Moderate-Intensity Training on Neuroplasticity and Functional Recovery after Focal Ischemia. Stroke 2017, 48, 2855–2864. [Google Scholar] [CrossRef]
- Lu, J.; Wang, J.; Yu, L.; Cui, R.; Zhang, Y.; Ding, H.; Yan, G. Treadmill Exercise Attenuates Cerebral Ischemia-Reperfusion Injury by Promoting Activation of M2 Microglia via Upregulation of Interleukin-4. Front. Cardiovasc. Med. 2021, 8, 735485. [Google Scholar] [CrossRef]
- Ahn, J.H.; Shin, M.C.; Park, J.H.; Kim, I.H.; Cho, J.H.; Lee, T.K.; Lee, J.C.; Chen, B.H.; Shin, B.N.; Tae, H.J.; et al. Effects of long-term post-ischemic treadmill exercise on gliosis in the aged gerbil hippocampus induced by transient cerebral ischemia. Mol. Med. Rep. 2017, 15, 3623–3630. [Google Scholar] [CrossRef]
- Li, C.; Ke, C.; Su, Y.; Wan, C. Exercise Intervention Promotes the Growth of Synapses and Regulates Neuroplasticity in Rats With Ischemic Stroke Through Exosomes. Front. Neurol. 2021, 12, 752595. [Google Scholar] [CrossRef]
- Li, C.; Hu, J.; Liu, W.; Ke, C.; Huang, C.; Bai, Y.; Pan, B.; Wang, J.; Wan, C. Exercise Intervention Modulates Synaptic Plasticity by Inhibiting Excessive Microglial Activation via Exosomes. Front. Cell. Neurosci. 2022, 16, 953640. [Google Scholar] [CrossRef]
- Zagrean, A.M.; Hermann, D.M.; Opris, I.; Zagrean, L.; Popa-Wagner, A. Multicellular Crosstalk between Exosomes and the Neurovascular Unit after Cerebral Ischemia. Therapeutic Implications. Front. Neurosci. 2018, 12, 811. [Google Scholar] [CrossRef]
- Li, H.; Luo, Y.; Zhu, L.; Hua, W.; Zhang, Y.; Zhang, H.; Zhang, L.; Li, Z.; Xing, P.; Zhang, Y.; et al. Glia-derived exosomes: Promising therapeutic targets. Life Sci. 2019, 239, 116951. [Google Scholar] [CrossRef]
- Liebigt, S.; Schlegel, N.; Oberland, J.; Witte, O.W.; Redecker, C.; Keiner, S. Effects of rehabilitative training and anti-inflammatory treatment on functional recovery and cellular reorganization following stroke. Exp. Neurol. 2012, 233, 776–782. [Google Scholar] [CrossRef]
- Keiner, S.; Wurm, F.; Kunze, A.; Witte, O.W.; Redecker, C. Rehabilitative therapies differentially alter proliferation and survival of glial cell populations in the perilesional zone of cortical infarcts. Glia 2008, 56, 516–527. [Google Scholar] [CrossRef]
- Kinoshita, K.; Hamanaka, G.; Ohtomo, R.; Takase, H.; Chung, K.K.; Lok, J.; Lo, E.H.; Katsuki, H.; Arai, K. Mature Adult Mice with Exercise-Preconditioning Show Better Recovery after Intracerebral Hemorrhage. Stroke 2021, 52, 1861–1865. [Google Scholar] [CrossRef]
- Svensson, M.; Lexell, J.; Deierborg, T. Effects of Physical Exercise on Neuroinflammation, Neuroplasticity, Neurodegeneration, and Behavior: What We Can Learn from Animal Models in Clinical Settings. Neurorehabil. Neural Repair 2015, 29, 577–589. [Google Scholar] [CrossRef]
- Svensson, M.; Rosvall, P.; Boza-Serrano, A.; Andersson, E.; Lexell, J.; Deierborg, T. Forced treadmill exercise can induce stress and increase neuronal damage in a mouse model of global cerebral ischemia. Neurobiol. Stress 2016, 5, 8–18. [Google Scholar] [CrossRef]
- Zong, X.; Dong, Y.; Li, Y.; Yang, L.; Li, Y.; Yang, B.; Tucker, L.; Zhao, N.; Brann, D.W.; Yan, X.; et al. Beneficial Effects of Theta-Burst Transcranial Magnetic Stimulation on Stroke Injury via Improving Neuronal Microenvironment and Mitochondrial Integrity. Transl. Stroke Res. 2020, 11, 450–467. [Google Scholar] [CrossRef]
- Zong, X.; Li, Y.; Liu, C.; Qi, W.; Han, D.; Tucker, L.; Dong, Y.; Hu, S.; Yan, X.; Zhang, Q. Theta-burst transcranial magnetic stimulation promotes stroke recovery by vascular protection and neovascularization. Theranostics 2020, 10, 12090–12110. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Liu, M.; Fan, Y.; Zhang, J.; Liu, L.; Li, Y.; Zhang, Q.; Xie, H.; Jiang, C.; Wu, J.; et al. Intermittent theta-burst stimulation improves motor function by inhibiting neuronal pyroptosis and regulating microglial polarization via TLR4/NFkappaB/NLRP3 signaling pathway in cerebral ischemic mice. J. Neuroinflamm. 2022, 19, 141. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Feng, Y.; Li, M.; Yin, M.; Qin, F.; Hu, X. Repetitive Transcranial Magnetic Stimulation Improves Neurological Function and Promotes the Anti-inflammatory Polarization of Microglia in Ischemic Rats. Front. Cell. Neurosci. 2022, 16, 878345. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.Y.; Rui, G.; Zhang, J.P.; Guo, L.; An, G.Z.; Lin, J.J.; He, W.; Ding, G.R. Cathodal tDCS exerts neuroprotective effect in rat brain after acute ischemic stroke. BMC Neurosci. 2020, 21, 21. [Google Scholar] [CrossRef]
- Cherchi, L.; Anni, D.; Buffelli, M.; Cambiaghi, M. Early Application of Ipsilateral Cathodal-tDCS in a Mouse Model of Brain Ischemia Results in Functional Improvement and Perilesional Microglia Modulation. Biomolecules 2022, 12, 588. [Google Scholar] [CrossRef]
- Peruzzotti-Jametti, L.; Cambiaghi, M.; Bacigaluppi, M.; Gallizioli, M.; Gaude, E.; Mari, S.; Sandrone, S.; Cursi, M.; Teneud, L.; Comi, G.; et al. Safety and efficacy of transcranial direct current stimulation in acute experimental ischemic stroke. Stroke 2013, 44, 3166–3174. [Google Scholar] [CrossRef]
- Pikhovych, A.; Stolberg, N.P.; Jessica Flitsch, L.; Walter, H.L.; Graf, R.; Fink, G.R.; Schroeter, M.; Rueger, M.A. Transcranial Direct Current Stimulation Modulates Neurogenesis and Microglia Activation in the Mouse Brain. Stem Cells Int. 2016, 2016, 2715196. [Google Scholar] [CrossRef]
- Walter, H.L.; Pikhovych, A.; Endepols, H.; Rotthues, S.; Barmann, J.; Backes, H.; Hoehn, M.; Wiedermann, D.; Neumaier, B.; Fink, G.R.; et al. Transcranial-Direct-Current-Stimulation Accelerates Motor Recovery after Cortical Infarction in Mice: The Interplay of Structural Cellular Responses and Functional Recovery. Neurorehabil. Neural Repair 2022, 36, 701–714. [Google Scholar] [CrossRef]
- Chen, C.M.; Wu, C.T.; Yang, T.H.; Liu, S.H.; Yang, F.Y. Preventive Effect of Low Intensity Pulsed Ultrasound against Experimental Cerebral Ischemia/Reperfusion Injury via Apoptosis Reduction and Brain-Derived Neurotrophic Factor Induction. Sci. Rep. 2018, 8, 5568. [Google Scholar] [CrossRef]
- Guo, T.; Li, H.; Lv, Y.; Lu, H.; Niu, J.; Sun, J.; Yang, G.Y.; Ren, C.; Tong, S. Pulsed Transcranial Ultrasound Stimulation Immediately after the Ischemic Brain Injury Is Neuroprotective. IEEE Trans. Biomed. Eng. 2015, 62, 2352–2357. [Google Scholar] [CrossRef]
- Wang, J.; Li, G.; Deng, L.; Mamtilahun, M.; Jiang, L.; Qiu, W.; Zheng, H.; Sun, J.; Xie, Q.; Yang, G.Y. Transcranial Focused Ultrasound Stimulation Improves Neurorehabilitation after Middle Cerebral Artery Occlusion in Mice. Aging Dis. 2021, 12, 50–60. [Google Scholar] [CrossRef]
- Siegel, G.; Schafer, R.; Dazzi, F. The immunosuppressive properties of mesenchymal stem cells. Transplantation 2009, 87, S45–S49. [Google Scholar] [CrossRef]
- Wagenaar, N.; de Theije, C.G.M.; de Vries, L.S.; Groenendaal, F.; Benders, M.; Nijboer, C.H.A. Promoting neuroregeneration after perinatal arterial ischemic stroke: Neurotrophic factors and mesenchymal stem cells. Pediatr. Res. 2018, 83, 372–384. [Google Scholar] [CrossRef]
- Ohtaki, H.; Ylostalo, J.H.; Foraker, J.E.; Robinson, A.P.; Reger, R.L.; Shioda, S.; Prockop, D.J. Stem/progenitor cells from bone marrow decrease neuronal death in global ischemia by modulation of inflammatory/immune responses. Proc. Natl. Acad. Sci. USA 2008, 105, 14638–14643. [Google Scholar] [CrossRef]
- Tang, W.; Lv, X.; Huang, J.; Wang, B.; Lin, L.; Shen, Y.; Yao, Y. Neuroprotective Effect of Stroke Pretreated Mesenchymal Stem Cells Against Cerebral Ischemia/Reperfusion Injury in Rats. World Neurosurg. 2022, 165, e1–e11. [Google Scholar] [CrossRef] [PubMed]
- Maacha, S.; Sidahmed, H.; Jacob, S.; Gentilcore, G.; Calzone, R.; Grivel, J.C.; Cugno, C. Paracrine Mechanisms of Mesenchymal Stromal Cells in Angiogenesis. Stem Cells Int. 2020, 2020, 4356359. [Google Scholar] [CrossRef] [PubMed]
- Jha, K.A.; Pentecost, M.; Lenin, R.; Gentry, J.; Klaic, L.; Del Mar, N.; Reiner, A.; Yang, C.H.; Pfeffer, L.M.; Sohl, N.; et al. TSG-6 in conditioned media from adipose mesenchymal stem cells protects against visual deficits in mild traumatic brain injury model through neurovascular modulation. Stem Cell Res. Ther. 2019, 10, 318. [Google Scholar] [CrossRef] [PubMed]
- Orczykowski, M.E.; Calderazzo, S.M.; Shobin, E.; Pessina, M.A.; Oblak, A.L.; Finklestein, S.P.; Kramer, B.C.; Mortazavi, F.; Rosene, D.L.; Moore, T.L. Cell based therapy reduces secondary damage and increases extent of microglial activation following cortical injury. Brain Res. 2019, 1717, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Nagai, A.; Sheikh, A.M.; Mitaki, S.; Wakabayashi, K.; Kim, S.U.; Kobayashi, S.; Yamaguchi, S. A human neural stem cell line provides neuroprotection and improves neurological performance by early intervention of neuroinflammatory system. Brain Res. 2016, 1631, 194–203. [Google Scholar] [CrossRef]
- Boese, A.C.; Le, Q.E.; Pham, D.; Hamblin, M.H.; Lee, J.P. Neural stem cell therapy for subacute and chronic ischemic stroke. Stem Cell Res. Ther. 2018, 9, 154. [Google Scholar] [CrossRef]
- Gnecchi, M.; Danieli, P.; Malpasso, G.; Ciuffreda, M.C. Paracrine Mechanisms of Mesenchymal Stem Cells in Tissue Repair. Methods Mol. Biol. 2016, 1416, 123–146. [Google Scholar] [CrossRef] [PubMed]
- Salikhova, D.; Bukharova, T.; Cherkashova, E.; Namestnikova, D.; Leonov, G.; Nikitina, M.; Gubskiy, I.; Akopyan, G.; Elchaninov, A.; Midiber, K.; et al. Therapeutic Effects of hiPSC-Derived Glial and Neuronal Progenitor Cells-Conditioned Medium in Experimental Ischemic Stroke in Rats. Int. J. Mol. Sci. 2021, 22, 4694. [Google Scholar] [CrossRef]
- Parajuli, B.; Saito, H.; Shinozaki, Y.; Shigetomi, E.; Miwa, H.; Yoneda, S.; Tanimura, M.; Omachi, S.; Asaki, T.; Takahashi, K.; et al. Transnasal transplantation of human induced pluripotent stem cell-derived microglia to the brain of immunocompetent mice. Glia 2021, 69, 2332–2348. [Google Scholar] [CrossRef]
- Imai, F.; Suzuki, H.; Oda, J.; Ninomiya, T.; Ono, K.; Sano, H.; Sawada, M. Neuroprotective effect of exogenous microglia in global brain ischemia. J. Cereb. Blood Flow Metab. 2007, 27, 488–500. [Google Scholar] [CrossRef]
- Hayashi, Y.; Tomimatsu, Y.; Suzuki, H.; Yamada, J.; Wu, Z.; Yao, H.; Kagamiishi, Y.; Tateishi, N.; Sawada, M.; Nakanishi, H. The intra-arterial injection of microglia protects hippocampal CA1 neurons against global ischemia-induced functional deficits in rats. Neuroscience 2006, 142, 87–96. [Google Scholar] [CrossRef]
- Ferreira, L.M.R.; Muller, Y.D.; Bluestone, J.A.; Tang, Q. Next-generation regulatory T cell therapy. Nat. Rev. Drug Discov. 2019, 18, 749–769. [Google Scholar] [CrossRef]
- Shi, L.; Sun, Z.; Su, W.; Xu, F.; Xie, D.; Zhang, Q.; Dai, X.; Iyer, K.; Hitchens, T.K.; Foley, L.M.; et al. Treg cell-derived osteopontin promotes microglia-mediated white matter repair after ischemic stroke. Immunity 2021, 54, 1527–1542.e8. [Google Scholar] [CrossRef]
- Ito, M.; Komai, K.; Mise-Omata, S.; Iizuka-Koga, M.; Noguchi, Y.; Kondo, T.; Sakai, R.; Matsuo, K.; Nakayama, T.; Yoshie, O.; et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 2019, 565, 246–250. [Google Scholar] [CrossRef]
- Zhang, H.; Xia, Y.; Ye, Q.; Yu, F.; Zhu, W.; Li, P.; Wei, Z.; Yang, Y.; Shi, Y.; Thomson, A.W.; et al. In Vivo Expansion of Regulatory T Cells with IL-2/IL-2 Antibody Complex Protects against Transient Ischemic Stroke. J. Neurosci. 2018, 38, 10168–10179. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, J.; Shou, J.; Zhang, W.; Chen, C. Diverse functions and mechanisms of regulatory T cell in ischemic stroke. Exp. Neurol. 2021, 343, 113782. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Gan, Y.; Sun, B.L.; Zhang, F.; Lu, B.; Gao, Y.; Liang, W.; Thomson, A.W.; Chen, J.; Hu, X. Adoptive regulatory T-cell therapy protects against cerebral ischemia. Ann. Neurol. 2013, 74, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Lefaucheur, J.P.; Aleman, A.; Baeken, C.; Benninger, D.H.; Brunelin, J.; Di Lazzaro, V.; Filipovic, S.R.; Grefkes, C.; Hasan, A.; Hummel, F.C.; et al. Evidence-based guidelines on the therapeutic use of repetitive transcranial magnetic stimulation (rTMS): An update (2014–2018). Clin. Neurophysiol. 2020, 131, 474–528. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Jin, J.; Lv, R.; Luo, Y.; Dai, W.; Li, W.; Tang, Y.; Wang, Y.; Ye, X.; Lin, W.J. Repetitive transcranial magnetic stimulation increases the brain’s drainage efficiency in a mouse model of Alzheimer’s disease. Acta Neuropathol. Commun. 2021, 9, 102. [Google Scholar] [CrossRef] [PubMed]
- Ljubisavljevic, M.R.; Javid, A.; Oommen, J.; Parekh, K.; Nagelkerke, N.; Shehab, S.; Adrian, T.E. The Effects of Different Repetitive Transcranial Magnetic Stimulation (rTMS) Protocols on Cortical Gene Expression in a Rat Model of Cerebral Ischemic-Reperfusion Injury. PLoS ONE 2015, 10, e0139892. [Google Scholar] [CrossRef]
- Cullen, C.L.; Young, K.M. How Does Transcranial Magnetic Stimulation Influence Glial Cells in the Central Nervous System? Front. Neural Circuits 2016, 10, 26. [Google Scholar] [CrossRef]
- Li, H.; Sun, J.; Zhang, D.; Omire-Mayor, D.; Lewin, P.A.; Tong, S. Low-intensity (400 mW/cm2, 500 kHz) pulsed transcranial ultrasound preconditioning may mitigate focal cerebral ischemia in rats. Brain Stimul. 2017, 10, 695–702. [Google Scholar] [CrossRef]
- Zhu, S.; Meng, B.; Jiang, J.; Wang, X.; Luo, N.; Liu, N.; Shen, H.; Wang, L.; Li, Q. The Updated Role of Transcranial Ultrasound Neuromodulation in Ischemic Stroke: From Clinical and Basic Research. Front. Cell. Neurosci. 2022, 16, 839023. [Google Scholar] [CrossRef]
- Bobola, M.S.; Chen, L.; Ezeokeke, C.K.; Olmstead, T.A.; Nguyen, C.; Sahota, A.; Williams, R.G.; Mourad, P.D. Transcranial focused ultrasound, pulsed at 40 Hz, activates microglia acutely and reduces Abeta load chronically, as demonstrated in vivo. Brain Stimul. 2020, 13, 1014–1023. [Google Scholar] [CrossRef]
- Yang, F.Y.; Lu, W.W.; Lin, W.T.; Chang, C.W.; Huang, S.L. Enhancement of Neurotrophic Factors in Astrocyte for Neuroprotective Effects in Brain Disorders Using Low-intensity Pulsed Ultrasound Stimulation. Brain Stimul. 2015, 8, 465–473. [Google Scholar] [CrossRef]
- Yoon, K.J.; Oh, B.M.; Kim, D.Y. Functional improvement and neuroplastic effects of anodal transcranial direct current stimulation (tDCS) delivered 1 day vs. 1 week after cerebral ischemia in rats. Brain Res. 2012, 1452, 61–72. [Google Scholar] [CrossRef]
- Boonswang, N.A.; Chicchi, M.; Lukachek, A.; Curtiss, D. A new treatment protocol using photobiomodulation and muscle/bone/joint recovery techniques having a dramatic effect on a stroke patient’s recovery: A new weapon for clinicians. BMJ Case Rep. 2012, 2012, bcr0820114689. [Google Scholar] [CrossRef]
- Salehpour, F.; Mahmoudi, J.; Kamari, F.; Sadigh-Eteghad, S.; Rasta, S.H.; Hamblin, M.R. Brain Photobiomodulation Therapy: A Narrative Review. Mol. Neurobiol. 2018, 55, 6601–6636. [Google Scholar] [CrossRef]
- Vogel, D.D.S.; Ortiz-Villatoro, N.N.; Araujo, N.S.; Marques, M.J.G.; Aimbire, F.; Scorza, F.A.; Scorza, C.A.; Albertini, R. Transcranial low-level laser therapy in an in vivo model of stroke: Relevance to the brain infarct, microglia activation and neuroinflammation. J. Biophotonics 2021, 14, e202000500. [Google Scholar] [CrossRef]
- Terstappen, G.C.; Meyer, A.H.; Bell, R.D.; Zhang, W. Strategies for delivering therapeutics across the blood-brain barrier. Nat. Rev. Drug Discov. 2021, 20, 362–383. [Google Scholar] [CrossRef]
- Steinberg, G.K.; Kondziolka, D.; Wechsler, L.R.; Lunsford, L.D.; Coburn, M.L.; Billigen, J.B.; Kim, A.S.; Johnson, J.N.; Bates, D.; King, B.; et al. Clinical Outcomes of Transplanted Modified Bone Marrow-Derived Mesenchymal Stem Cells in Stroke: A Phase 1/2a Study. Stroke 2016, 47, 1817–1824. [Google Scholar] [CrossRef]
- Chung, J.W.; Chang, W.H.; Bang, O.Y.; Moon, G.J.; Kim, S.J.; Kim, S.K.; Lee, J.S.; Sohn, S.I.; Kim, Y.H.; Starting-2 Collaborators. Efficacy and Safety of Intravenous Mesenchymal Stem Cells for Ischemic Stroke. Neurology 2021, 96, e1012–e1023. [Google Scholar] [CrossRef]
- Kawai, H.; Yamashita, T.; Ohta, Y.; Deguchi, K.; Nagotani, S.; Zhang, X.; Ikeda, Y.; Matsuura, T.; Abe, K. Tridermal tumorigenesis of induced pluripotent stem cells transplanted in ischemic brain. J. Cereb. Blood Flow Metab. 2010, 30, 1487–1493. [Google Scholar] [CrossRef]
- Anastasiadis, P.; Gandhi, D.; Guo, Y.; Ahmed, A.K.; Bentzen, S.M.; Arvanitis, C.; Woodworth, G.F. Localized blood-brain barrier opening in infiltrating gliomas with MRI-guided acoustic emissions-controlled focused ultrasound. Proc. Natl. Acad. Sci. USA 2021, 118, e2103280118. [Google Scholar] [CrossRef]



| Acute Phase of Stroke | Subacute Phase of Stroke | Chronic Phase of Stroke | |
|---|---|---|---|
| Time course | Minutes to days | Days to weeks | Weeks to mouths |
| Pathophysiological mechanisms | Infiltration of peripheral immune cells; activation of resident glial cells; disturbance of ionic homeostasis; oxidative stress; BBB destruction; mitochondrial dysfunction; DNA damage | Amplification of immune responses; increased ROS production; cell edema and ion imbalances | Decrease in excitotoxicity; protective inflammatory response |
| Axonal sprouting, dendrite remodeling; neurogenesis, angiogenesis; increased levels of growth factors | |||
| Consequences | Cell injury or death | Onset of neuroplasticity | Tissue repair |
| Therapeutic targets | Neuroprotection; reducing reperfusion injury | Functional rehabilitation | |
| Species | Model | Microglia Subset | Gene | Protocol | Reference |
|---|---|---|---|---|---|
| CD-1 mice | In embryonic development | The subsets during CNS development and homeostasis in the adult brain | Ctsb, Ctsd, Lamp1, Apoe, Tmsb4x, Eef1a1, Rpl4, Cst3 | SMART-seq2 | [101] |
| In postnatal development | Tmem119, Selplg, Slc2a5, Malat1 | ||||
| C57BL/6J mice | Aging brain (P540) | Aging clusters (OA) 2 | Lgals3, Cst7, chemokines Ccl4, Ccl3, Il1b, Id2, Atf3 | 10 × Genomics | [102] |
| Aging clusters (OA) 3 | Ifitm3, Rtp4, Oasl2 | ||||
| Heterozygous 5XFAD transgenic mice | AD | Microglia type associated with neurodegenerative diseases (DAM) | Apoe, Lpl, Cst7 ↑ P2ry12, Cx3cr1, Tmem119 ↓ | MARS-seq | [103] |
| Frozen human post-mortem midbrain tissue sections | Idiopathic PD | Microglia cluster involving inflammatory response | IL1B, GPNMB, HSP90AA1 | 10 × Genomics | [104] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiao, C.; Liu, Z.; Qie, S. The Implications of Microglial Regulation in Neuroplasticity-Dependent Stroke Recovery. Biomolecules 2023, 13, 571. https://doi.org/10.3390/biom13030571
Qiao C, Liu Z, Qie S. The Implications of Microglial Regulation in Neuroplasticity-Dependent Stroke Recovery. Biomolecules. 2023; 13(3):571. https://doi.org/10.3390/biom13030571
Chicago/Turabian StyleQiao, Chenye, Zongjian Liu, and Shuyan Qie. 2023. "The Implications of Microglial Regulation in Neuroplasticity-Dependent Stroke Recovery" Biomolecules 13, no. 3: 571. https://doi.org/10.3390/biom13030571
APA StyleQiao, C., Liu, Z., & Qie, S. (2023). The Implications of Microglial Regulation in Neuroplasticity-Dependent Stroke Recovery. Biomolecules, 13(3), 571. https://doi.org/10.3390/biom13030571